
Abstract
Adzuki bean (Vigna angularis var. angularis) is a dietary legume crop in East Asia. The presumed progenitor (Vigna angularis var. nipponensis) is widely found in East Asia, suggesting speciation and domestication in these temperate climate regions. Here, we report a draft genome sequence of adzuki bean. The genome assembly covers 75% of the estimated genome and was mapped to 11 pseudo-chromosomes. Gene prediction revealed 26,857 high confidence protein-coding genes evidenced by RNAseq of different tissues. Comparative gene expression analysis with V. radiata showed that the tissue specificity of orthologous genes was highly conserved. Additional re-sequencing of wild adzuki bean, V. angularis var. nipponensis and V. nepalensis, was performed to analyze the variations between cultivated and wild adzuki bean. The determined divergence time of adzuki bean and the wild species predated archaeology-based domestication time. The present genome assembly will accelerate the genomics-assisted breeding of adzuki bean.
Similar content being viewed by others

Introduction
Adzuki bean (Vigna angularis var. angularis) is a diploid legume crop (2n = 2x = 22) with an estimated genome size of 538 mega bases (Mb)1. It is one of the Asian Vigna in the Ceratotropis subgenus, under the papilionoid subfamily of the Fabaceae2. Adzuki bean is widely cultivated in East Asian countries like China, Japan and Korea as an ingredient for traditional dessert cuisines due to its sweet taste, as well as its nutritious protein and starch contents. The annual cultivation area for adzuki bean in China, Japan, Korean peninsula and Taiwan is estimated to be 670,000, 120,000, 30,000 and 20,000 ha, respectively3. The wild species of adzuki bean such as V. angularis var. nipponensis, V. nakashimae and V. nepalensis, are widely distributed across East Asia and Himalayan countries2. However, archaeological evidences suggested multiple domestication origins in northeast Asia4.
Several important legume crops and model plants have been sequenced. This includes warm-season legumes such as Glycine max, Phaseolus vulgaris, Cajanus cajan5,6,7 and Vigna radiata8. V. angularis var. angularis is a close relative of V. radiata and is adapted to sub-tropical and temperate climate zone. In spite of its economic importance and the demands for improved V. angularis var. angularis variety, genomic studies for agriculturally important traits and efficient breeding methods for this species have been lacking. Elucidation of the genome sequence of V. angularis var. angularis could reveal the general genome structure and evolution of this legume species in comparison to closely related genomes and greatly assist comparative genomics of V. angularis var. angularis and other well-studied legume genomes. In addition, the re-sequencing efforts of cultivated and wild adzuki beans will facilitate the measurement of genetic diversity of each locus and the development of useful markers for putative domestication-related loci.
Here, we assembled a draft genome of adzuki bean into pseudo-chromosomes using sequence data from next generation sequencing. This adzuki bean genome was compared to other warm-season legumes to study genome evolution and speciation. Well established quantitative trait loci (QTLs) of G. max were translated into the adzuki bean genome using gene order conservation. In addition, we sequenced two wild adzuki bean species, Vigna angularis var. nipponensis and Vigna nepalensis and identified the putative loci related to domestication in order to develop SNP markers for more efficient breeding. Our genome sequence of V. angularis var. angularis and QTL-associated genetic markers will boost genomics of warm season legumes and breeding programs of adzuki bean.
Results
Genome assembly
V. angularis var. angularis has a diploid genome. Based on flow cytometry analysis the genome size was estimated to be 612 Mb (Supplementary Table S1) which is higher than the previous estimate of 538 Mb1. The selected domesticated line for sequencing was Gyeongwon, a widely grown variety in Korea, which was developed by the Rural Development Administration (RDA) in Korea to reduce root lodging and to improve grain quality.
For de novo genome assembly, we prepared two paired-end libraries with 180 bp insert size, along with two 5 kb mate pair and one 10 kb mate-pair libraries for 100 bp short read sequencing using the Illumina HiSeq 2000 (Supplementary Table S2). A single linear library was also constructed for sequencing using the Roche GS-FLX+ producing total 1,288,628 reads with average read length, 458 bp. Approximately 291-fold sequence read coverage of the estimated genome size was generated by the two sequencing platforms. For reads generated by the Illumina HiSeq 2000, ALLPATHS-LG assembler was used for de novo assembly9. The long reads generated by GS FLX+ were assembled using Newbler software and the resulting contigs were transformed into paired end reads with 180 bp insert and used as input for ALLPATHS-LG assembly. Using Jellyfish10 software at 22 k-mer frequency, we estimated the genome size to be 591 Mb which is close to the size calculated using flow cytometry analysis (Supplementary Fig. S1). The assembly produced 3,883 scaffolds with proper read coverage statistics of sequencing libraries including the pseudo-library from Newbler assembly and the N50 length of the scaffolds was 703 kb (Supplementary Table S3). The sum of the scaffold length was approximately 443 Mb covering 75 percent of the estimated genome size (Supplementary Table S4).
To link the scaffolds into super-scaffolds, we utilized the synteny relationship between V. angularis and closely related legume genomes such as Phaseolus vulgaris7 and V. radiata var. radiata. This was based on the assumption that the gene order among closely-related warm season legume species is highly conserved. Identification of the synteny relationship and calculation of the Ks values for each orthologous or paralogous gene pair was performed using MCSCANX software11. We retrieved the synteny blocks from the most recent peak in the Ks frequency plot (Supplementary Fig. S2) and extracted conserved genomic blocks that showed multi-species collinearity among the three legume genomes to be used as bridges for super-scaffolding (Supplementary Fig. S3). The synteny-based scaffolding strategy improved the N50 length from 704 kb to 1.5 Mb and the maximum length of scaffolds from 4.4 Mb to 11.1 Mb (Supplementary Table S5).
To assemble the pseudo-molecules, we implemented the genotyping by sequencing (GBS) method12 to construct a high-density genetic map of adzuki bean. The mapping population comprised of 133 F4 lines generated by single-seed descent from a cross between V. angularis var. angularis (Gyeongwon) and the wild species V. nakashimae (IT178530). GBS short reads were mapped to genomic regions flanked by ApeKI restriction sites using Bowtie2 software13. A total of 4,524 segregating SNP sites were identified. Possible co-segregating sites within 1 kb regions were merged reducing the total SNP number to 2,347. However, more than half of the SNPs showed segregation distortions that are probably due to interspecific crossing (Supplementary Table S6). Removal of the distorted SNPs resulted in a high-density genetic map with 814 SNPs in 11 linkage groups (Supplementary Fig. S4 and Supplementary Table S7). In total, 158 scaffolds could be anchored to the genetic map to construct the 11 pseudo-chromosomes. The sum of the anchored scaffolds was 210 Mb and the length of N50 was 25 Mb (Supplementary Table S8). However, 78 scaffolds and super-scaffolds were anchored without orientation information, because only a single marker could be used to anchor them to the map (Supplementary Table S4). Total 45 super-scaffolds were anchored to genetic map and 43 super-scaffolds were consistent with our genetic map suggesting the reliability of the synteny-based scaffolding method (Supplementary Fig. S3 and Supplementary Table S9).
Prediction of genes and repetitive sequences in adzuki bean
After masking the identified repetitive sequences, we implemented the structural and homology-based gene prediction procedure according to the MAKER pipeline14,15. In order to obtain direct evidence of gene expression, we extracted mRNA from adzuki bean flower, pod, leaf and root tissues (Supplementary Table S10). The mRNA samples were sequenced using the Illumina HiSeq 2000 for subsequent de novo assembly using Trinity software16. The assembled contigs were pooled and supplied to the MAKER pipeline as the evidence of transcription and 26,857 high-confident genes were predicted. Using CEGMA pipeline, more than 86 percent of 248 core eukaryotic genes (CEG) could be completely matched to our genome assembly and the 99 percent of 248 CEGs matched to the predicted proteins using BLASTP algorithm with E-value 1e-10 (Supplementary Table S4)17. To test the reliability of the predicted V. angularis gene set, we compared the sequence length distributions of the genes, coding DNA sequences (CDS) and introns to the gene models of P. vulgaris, G. max and V. radiata. The density plot showed consistent distributions of CDS and intron length among the three legume genomes. However, the proportion of short genes (~250 bp) was higher in V. angularis (Supplementary Fig. S5). Of the 26,857 high-confident genes, 15,976 were located on pseudo chromosomes (Fig. 1a, 1b and Supplementary Table S4). Clustering analysis was performed on the protein sets of V. angularis var. angularis and the protein sequences of A. thaliana, M. truncatula, O. sativa, and G. max using OrthoMCL software18 and identified 6,643 gene clusters that are shared among all five species and 1,163 clusters that are specific to V. angularis var. angularis (Fig. 1c, Supplementary Table S11). We could assign functional annotations to 21,532 genes using InterProScan and BLAST against Arabidopsis proteins (Supplementary Table S4).

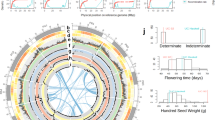
Summary of genome assembly of cultivated adzuki bean and analyses of genes, repetitive sequences and predicted QTLs.
(A) Circular map showing predicted QTL regions of V. angularis var. angularis based on the synteny relationship with G. max and the WGD regions displayed by the grey ribbons at most inner part of circle. From outer to inner layer, four types of QTLs are depicted (disease resistance, flowering time, pod maturity and seed weight, respectively). Highlighted region of each layers represent the candidate physical position of the related QTLs. The red dots on the chromosomes represent the loci of NBS-LRR domain containing genes. (B) The distribution of the predicted genes and the repetitive sequences in adzuki bean genome plotted by 300 kb bin length. The colored bars represent the counts of genes and repeated elements in each bin; LTR/Gypsy (green), LTR/Copia (blue), LINE (blue) and genes (red). (C) Venn diagram depicting the clustering analysis of the five protein sets from A. thaliana, M. truncatula, G. max, O. sativa and V. angularis. The numbers of homolog clusters were indicated for each species and species intersection.
The predicted gene content of V. angularis var. angularis showed extensive synteny relationship with closely-related warm season legumes including G. max, P. vulgaris and V. radiata (Fig. 2 and Supplementary Fig. S6). We examined the tissue specificity of gene expressions in V. angularis var. angularis and V. radiata var. radiata using RNA-Seq data from four different tissues (Supplementary Table S10 and Supplementary Fig. S7). There are 9,196 orthologs between V. angularis var. angularis and V. radiata var. radiata that showed persistent tissue specificity, suggesting that gene functions were extensively retained even after speciation (Supplementary Table S12).

Visualization of the synteny relationships among the closely related warm season legumes, V. angularis, V. radiata, P. vulgaris and G. max.
The x-axis indicates chromosomal locations of genes in synteny relationship and the y-axis indicates Ks value of the corresponding gene pair, showing both conservation of gene order as well as chromosomal rearrangements of synteny blocks.
Using the Pfam annotations of each protein, we classified transcription factors according to the rules described in Lang et al.19. In total, 2,669 genes encoding transcription factors (TFs) were identified in the adzuki bean genome. We compared the relative TF abundance with that of other plant genomes (Supplementary Table S13) and found the overall proportions of TF gene families to be similar in these plant genomes. However, bZIP2 consistently makes up less than 1% of the total TFs in legume genomes. This contrasts with non-legume genomes such as A. thaliana, Z. mays, O. sativa and B. distachyon where bZIP2 represented more than 3% of all TFs, suggesting a possible gene loss event of this TF family in the common ancestor of legume plants (Supplementary Table S13).
We surveyed the repetitive sequences within the scaffold sequences to examine the abundance and distribution of transposable elements, which are known as major drivers of genome evolution20. Homology- and structure-based analysis revealed approximately 43.1% of sequenced adzuki bean genome as repetitive sequences (Supplementary Table S14). As in the other legumes and in that of the closely related V. radiata, the predominant repetitive sequences were annotated as long terminal repeat (LTR) retrotransposons5,6,21,22. Among the LTR retrotransposons, Gypsy and Copia constituted 19% and 10% of the adzuki bean genome sequences, respectively (Supplementary Table S14). DNA transposons, CACTA, Mutator, PIF-Harbinger, hAT, Helitron, MULE-MuDR and Tc1-Mariner, were also detected, comprising approximately 2.7% of the adzuki bean genome sequences.
Domestication traces of adzuki bean
Human selection activities on crops have inflicted major effects on crop genomes, which ultimately result in domestication syndrome, which is marked by loss of seed shattering, minimization of seed dormancy and an increase in both seed size and number23. To identify the domestication traces within the genome of V. angularis var. angularis, we sequenced wild relatives of adzuki bean, V. nepalensis (AusTRCF85148) and V. angularis var. nipponensis (IT241912) (Supplementary Table S2). Single paired-end libraries from each genotype were sequenced using Illumina HiSeq 2000 generating about 50 Gb of short reads which represents more than 85-fold coverage of the adzuki bean genome. We also analyzed short read sequences of wild adzuki bean, Vigna nakashimae (IT178530), which was generated in our previous study24.
The 22-base k-mer frequency analysis of these short read sequences revealed a variation of genome size among wild adzuki beans (Supplementary Fig. S1). This is especially true for V. nakashimae, which showed a much larger genome size than V. angularis var. angularis and the rest of the wild adzuki beans.
We mapped the short read sequences onto our adzuki bean reference genome using Burrows-Wheeler Aligner software (Supplementary Table S15 and S16)25. V. angularis var. nipponensis had 667,097 SNPs compared with the reference genome and the SNP frequency to total mapped regions (SNPs per kb) was 1.82. 75,476 of the identified SNPs are located within coding regions and 3,840 of those SNPs could potentially cause non-synonymous protein sequence changes in 1,421 gene products. We also identified 97, 932 insertions and deletions (INDELs) of which 14,033 were in coding regions.
V. nepalensis showed much higher polymorphism than V. angularis var. nipponensis as demonstrated by the presence of 3,511,378 SNPs (10.24 SNPs per kb). The number of SNPs in coding region was 433,210 and there were 18,034 non-synonymous SNPs that affected 6,464 genes. A total of 410,232 INDELs were identified and 71,337 were within coding regions.
V. nakashimae showed 3,342,795 SNPs and 624,856 intragenic SNPs. However, the ratio of aligned reads to total reads and the predicted number of INDELs were notably lower than those of other wild adzuki beans. This could be due to the difficulty of read mapping between the reference species (V. angularis) and the highly diverged species (V. nakashimae) showing notable genome size discrepancy (Supplementary Fig. S1).
We compared orthologs between cultivated and wild adzuki bean to elucidate domestication-related loci. The orthologous coding sequences of V. angularis var. nipponensis and V. nepalensis were reconstructed by substituting the coding sequences of V. angularis var. angularis with their respective SNP data. Since V. nakashimae short reads were not efficiently mapped onto our reference genome, we performed de novo assembly of the short read sequences using ABySS software26 and implemented gene prediction (Supplementary Table S4). From the ortholog comparison between cultivated and wild adzuki bean, we calculated the ratio of the number of nonsynonymous substitutions per non-synonymous site (Ka) to the number of synonymous substitutions per synonymous site (Ks) to estimate the selective pressure on each gene (Fig. 3a). V. angularis var. nipponensis showed the lowest number of polymorphic genes (1,823) in the Ka/Ks calculation. We observed one consistent peak at Ka/Ks value of 0.2 suggesting purifying selection between cultivated and three wild adzuki beans (Ka/Ks < 1)27 (Fig. 3b). Notably, the Ka/Ks distributions of V. angularis var. angularis x V.angularis var. nipponensis and V. angularis var. angularis x V. nepalensis were highly similar consistently showing three peaks (0.2, 0.5 and 0.7 ~ 0.8). In both comparisons, a total of 307, 152 and 75 genes were commonly found within the three peaks, respectively (Supplementary Table S17). This suggests three different degrees of selection pressure on these loci between cultivated and wild adzuki bean. Even though a Ka/Ks of less than 1 has been interpreted as a signature of purifying selection27, a subset of the genes in each peak can be candidates for explaining the difference between wild and cultivated adzuki bean, such as speciation and domestication (Supplementary Table S17). For example, the homologs to disease related genes in the second (0.4 ~ 0.6) and third peak (0.6 ~ 0.9) such as Vang03g15160, Vang02g14420, Vang0291s00070, Vang0229s00140, Vang02g14420 may possess novel disease resistance alleles in wild adzuki beans which are distinct to those in cultivated adzuki bean (Supplementary Table S17).

Selection pressure for each locus in adzuki bean genome between cultivated (V. angularis var. angularis) and wild adzuki beans (V. angularis var. nipponenesis, V. nepalensis and V. nakashimae).
(A) Circos map displaying Ka/Ks value of each locus (from outer most layer), the positions of common loci in purifying selection and gene density. The most inner ribbons indicate the duplicated synteny blocks (B) Density plot showing the distribution of Ka/Ka values between cultivated and wild adzuki beans.
Marker development and its utilization in breeding program
In order to produce genetic markers that are easily applicable for QTL mapping and marker assisted breeding programs, we identified simple sequence repeat (SSR) markers using MISA software28. A total of 143,113 SSRs were detected and the number of tri-repeat unit SSRs, the preferred type for genotyping, were 1,941 (Supplementary Table S18).
We predicted the associated QTLs to these SSR markers using translational genomics approach29. G. max is a warm-season model legume crop closely related to V. angularis var. angularis showing co-linearity of most of the gene content (Fig. 2). Hence, the predicted QTLs from the comparison between G. max and V. angularis would be useful clue to determine genomic regions related to agriculturally important traits in V. angularis var. angularis genome30. We translated the genomic positions of 2,010 QTL-associated SSR markers of G. max to corresponding genomic positions of V. angularis by 569 orthologous synteny blocks (Supplementary Table S19) and plotted the agriculturally important QTLs such as flowering time, maturity, seed size, yield and disease resistance onto a circular map (Fig. 1a). The disease resistance QTLs were likely to be around the 87 genes that code for nucleotide-binding site (NBS) and leucine-rich repeat (LRR) domains, which are commonly associated with disease resistance (Fig. 1a and Supplementary Table S20)31. The flanking markers of these translated QTLs can be used for breeding programs (Supplementary Table S19). We also constructed a database containing the gene information, genetic markers and associated QTL data in Jbrowse environment, which can be accessed at http://plantgenomics.snu.ac.kr/ (Supplementary Fig. S8).
Genome evolution of adzuki bean
Using 60 orthologs of P. vulgaris, V. radiata, V. nakashimae, V. nepalensis, V. angularis var. nipponensis and V. angularis var. angularis, we constructed a species tree (Fig. 4) (Supplementary Table S21). V. angularis var. angularis formed a distinct clade that included the wild adzuki beans in the species tree. As expected from genome size difference, V. angularis var. nipponensis was closest to V. angularis var. angularis, whereas V. nakashimae was more diverged. We estimated the speciation times, which were calibrated using the divergence time, 8.0 million years ago (MYA), between Phaseolus and Vigna32. For our sampled accessions, the minimum speciation time between cultivated and wild adzuki bean was 0.05 MYA, which predated the archaeological evidence for adzuki bean cultivation (~5,000 years before present)4. The Ks density plot calculated using synteny relationship within the legume genomes revealed the single ancient whole genome duplication at ~53.3 MYA based on the substitution rate, 6.1 × 10−9 33, which is commonly shared among V. angularis, V. radiata and P. vulgaris (Fig. 4). We identified 1,273 tandemly duplicated genes in the adzuki bean genome; these genes are highly enriched in the gene ontology categories of defense response, oxidation reduction and phosphorylation, which is consistent with findings in other plant genomes (Supplementary Table S22 and Supplementary Fig. S9)34,35.

Analyses of the evolution of adzuki bean with comparison to closely related warm season legumes.
(A) Estimation of WGD using the density plot of Ks values within each gene set of V. angularis (blue), P. vulgaris (green), V. radiata (purple) and G. max (red). (B) Species tree of cultivated adzuki bean (V. angularis var. angularis), wild adzuki beans (V. angularis var. nipponensis, V. nepalensis and V. nakashimae), V. radiata and P. vulgaris. The divergence times of each nodes were estimated by a Bayesian MCMC method calibrated by the root divergence time (8 MYA) between Phaseolus and Vigna. The posterior probability of each node is depicted in parentheses.
Discussion
Among Asian Vigna, adzuki bean is an economically important grain legume due to its nutritional properties and popular use in dessert foods. A better understanding of adzuki bean genetics is important for more efficient breeding and in light of an increase in biotic and abiotic stresses on crops that may accompany climate change. The adzuki bean reference genome sequence and re-sequencing efforts of two different wild adzuki beans, V. angularis var. angularis and V. nepalensis, presented in this manuscript, are a rich source of genetic markers, loci under degrees of selection pressure and putative candidate genes for several agriculturally important traits that were derived from translational genomics with G. max (Fig 1a, Fig 3a and Supplementary Table S19).
Plant genome complexity and the length of repeats that often exceed the insert size of present mate pair library technology can limit the de novo assembly of NGS reads to a certain saturation point. We largely improved the assembly by implementing the synteny-based scaffolding approach using gene order conservation between closely related legume species (V. angularis var. angularis, V. radiata var. radiata and P. vulgaris). Thus, this method could be used as an alternative to the longer sequencing reads or mate pair library with larger insert size solving the assembly problem. In addition, we anchored super-scaffolds onto the genetic map constructed using the GBS-based genotypes of F4 recombinant inbred lines of V. angularis var. angularis and V. nakashimae. Due to the segregation distortion, relatively small amounts of GBS-derived SNPs were used for constructing the genetic map. The possible cause of this segregation distortion in this population is the genome size difference of parental lines, which may result in ectopic recombination (Supplementary Fig. S1)36.
Even in our limited number of cultivated and wild adzuki bean samples, we could catch a glimpse of the speciation time between V. angularis var. angularis and V. angularis var. nipponensis, which occurred at around 50,000 years ago. As the domestication time evidenced by archaeology was ~5,000 before present4, we could hypothesize that the domestication event by start of human cultivation occurred after speciation between wild and cultivated adzuki bean. Additional population-level re-sequencing efforts of cultivated and wild adzuki beans would reveal the speciation time and also the domestication sites at higher confidence level and also test the previous hypothesis of multiple domestication sites of V. angularis4.
Methods
Plant materials
The cultivated adzuki bean accession, Gyeongwon (V. angularis var. angularis, IT213134) and wild adzuki bean accession (V. angularis var. nipponenesis, IT241912) were provided by the Rural Development Administration (RDA) Genebank Information Center in Korea. The other wild adzuki bean (V. nepalensis, AusTRCF85148) was sourced from the Australian Collections of Plant Genetic Resources in Australia.
Genome assembly and gene prediction
For genome size estimation, we used Jellyfish10 software to observe K mer (22-mer for this study) frequency distribution by “jellyfish count” command with parameter -C, -m 22, -s 50G, -t 10 and -c 6. “jellyfish histo” command created the histogram of K mer frequency and the peak frequency could be observed. The sequencing depth (N) was estimated by the equation: N = M * L/(L − K + 1) where M is peak K mer frequency, L is read length, K is K mer length.
We also estimated the genome size by the flowcytometry analysis using procedure modified from Arumuganathan and Earle et al.37. Briefly, the procedure consists of preparing suspensions of intact nuclei by chopping of 50 mg adzuki bean leaf tissues in MgSO4 buffer mixed with DNA standards and stained with propidium iodide in a solution containing DNAase-free-RNAase. Fluorescence intensities of the stained nuclei are measured by FACScalibur flow cytometer (Becton-Dickinson, San Jose, CA). Values for nuclear DNA content are estimated by comparing fluorescence intensities of the nuclei of the testing sample with those of an appropriate internal DNA standard such as nuclei from Chicken Red blood cells (2.5 pg/2C), Glycine max (2.45 pg./2C), Oryza sativa cv. Nipponbare (0.96 pg/2C), or Arabidopaia thaliana (0.36 pg/2C). For each measurement, the propidium iodide fluorescence area signals (FL2-A) from 1000 nuclei were collected and analyzed by CellQuest software (Becton-Dickinson, San Jose, CA).
For genome assembly of cultivated adzuki bean, we used the Illumina HiSeq 2000 using types of sequencing libraries such as two fragment libraries and two 5 kb and one 10 kb mate pair libraries. One additional single linear library was sequenced using the Roche GS-FLX+ (Supplementary Table S2 and Supplementary Table S23). The assembles of Illumina HiSeq 2000 and GS-FLX+ read sequences were implemented using ALLPATHS-LG9 and Newbler, respectively. ALLPATH-LG was run with default parameter and Newbler was run with option -large and -trim for “runProject” command. The contigs generated by Newbler were chopped into pseudo fragment library reads and supplied to ALLPATHS-LG again. Software ABYSS was used for the assembly of V. nakahsimae using the option k = 84 and q = 20 for “abyss-pe” command. The traces of repetitive sequences were searched using LTR-harvest38 and TransposonPSI (http://transposonpsi.sourceforge.net/) with default parameters. The captured repetitive sequence blocks were annotated by LTR-digest39 using a set of hmm signatures: PF03078.8, PF00385.17, PF01393.12, PF04094.7, PF07253.4, PF00552.14, PF05380.6, PF00077.13, PF08284.4, PF00078.20, PF07727.7, PF06815.6, PF06817.7, PF03732.10, PF00075.17, PF01021.12, PF04195.5, PF00692.12, PF00692.12 and PF00098. The hmm signature of AP_ty1copia and AP_ty3gypsy elements was built using their alignment information from GyDB40. We further utilized the G. max TE classification based on sequence homology41. The predicted and annotated repetitive sequence blocks were supplied to RepeatMasker software as adzuki bean repetitive sequence library14.
Adzuki bean gene contents were predicted following MAKER pipeline15. RNA-Seq using Illumina HiSeq 2000 were implemented on mRNAs of the four different tissues (leaf, flower, root and pod) of adzuki bean and de novo assembled using Trinity software with default parameter16. Contigs were pooled from de novo assembly and redundant sequences were removed by the CD-HIT-EST algorithm in CD-HIT software42. The non-redundant transcriptome assemblies were supplied into the MAKER pipeline along with G. max protein sequences and the complete protein sequences of Arabidopsis from Uniprot as evidence for the homology based gene prediction. For ab initio gene prediction, we used AUGUSTUS software43 with mungbean (V. radiata) training set. The resulting protein sequences were annotated using InterProScan544.
Using the Pfam annotations from InterProScan result, transcription factors (TF) of adzuki bean proteins from V. angularis var. angularis and V. nakashimae were classified based on the TF classification rules described in Lang et al19. For comparative analysis of TF contents, we further classified the TF from the protein sequences of 8 plant genomes (A. thaliana, G. max, M. truncatula, C. cajan, C. arietinum, B. distachyon, Z. mays, O. sativa).
Using Pfam45 HMM profile of LRR domain (PF00560, PF07725, PF12799, PF13504, PF13516, PF13855 and PF14580) and NB-ARC domain (PF00931), we performed a genome-wide scan to find NBS and LRR domain containing proteins. The sequence blocks matched as LRR and NBS domain were re-aligned to construct V. angularis var. angularis specific hmm profiles of LRR and NBS using the HMMER software package with default parameter for “hmmbuild” command46. Using the V. angularis-specific hmm profiles, proteins with NBS and/or LRR domain were identified using HMMER software package with default parameter for “hmmscan” command. The putative functions of NBS-LRR were annotated using the blast result against Uniprot database47.
We further linked the scaffolds based on the gene order conservation (synteny) of P. vulgaris, V. radiata and V. angularis. The pairwise synteny relationships and the Ks values of gene pairs were calculated using MCSCANX software11. The synteny blocks between V. radiata and P. vulgaris were used as links for scaffolding. We retrieved every link that could be deduced from synteny blocks and concatenated each other to build consensus links (Supplementary Fig. S3). The orientation of the scaffold was determined by the orientation of the gene orders of the synteny blocks used for links.
Genetic map construction
To construct a genetic map of adzuki bean, we genotyped 133 F4 lines derived from the cross between Gyeongwon and V. nakashimae (IT178530) using genotyping by sequencing (GBS)12 (Supplementary Table S23). The genomic DNA of each line was extracted and fragmented by the ApeKI restriction enzyme. Validated PCR fragments using the Agilent Technologies Bioanalyzer 2100 were constructed into GBS sequence library and were sequenced using the Illumina HiSeq 2000. We retrieved the sequences flanked with ApeKI restriction site from de novo assembled scaffolds. The short reads were mapped onto the ApeKI flanking sequences using software Bowtie213. The genotypes of 133 RILs were collected from the genomic positions showing over 10 read depth and we determined homo- and heterozygous genotypes following SAMtools genotyping statistics with default parameter48. If the arrays of paternal or maternal genotypes of 133 individuals within 1 Kb are consistent each other we regarded these sites as co-segregating block and we used one representative site with lowest ‘N’ to construct genetic map. The genotypes of 133 F4 lines were supplied to JoinMap 4 and we constructed 11 linkage groups using regression mapping algorithm with Kosambi mapping function.
Resequencing of three wild Vigna species
The sequence reads of V. nakashimae, V. nepalensis and V. angularis var. nipponensis were aligned to our reference genome of V. angularis var. angularis using the BWA-MEM algorithm of BWA software with default options25 (Supplementary Table S23). The resulting read mapping files were supplied to AddOrReplaceReadGroups, MarkDuplicates, FixMateInformation, RealignerTargetCreator, IndelRealigner, BaseRecalibrator, PrintReads modules included in Picard (http://picard.sourceforge.net) and GenomeAnalysisTK (3.1–1 version) to optimize the sequence alignment49. As there is no comprehensive set of high-confidence known variant site for adzuki bean genome, we used the parameter –run_without_dbsnp_potentially_ruining_quality for BaseRecalibrator step. For variation calling process, the UnifiedGenotyper module of GenomeAnalysisTK was used. We set the cut-off for the mapping quality as 30 and the sites lower than the cut-off were ignored for further analyses. Moreover, we regarded the genomic positions showing the read depth over twice of the sequence coverage as duplicated region and discarded for accuracy of variation calling. Additionally, if more than four reads with zero mapping quality mapped on certain genomic position or the ratio of the read with zero mapping quality to total mapped reads exceed 10 percent, the genomic position was excluded.
Species tree construction
We constructed the species tree among the closely related diploid warm season legume species, such as P. vulgaris, V. radiata, V. angularis var. angularis, V. angularis var. nipponensis, V. nakashimae and V. nepalensis based on Bayesian Markov Chain Monte Carlo (MCMC) analysis using the 60 orthologous loci by *BEAST of the software package BEAST version 1.850. To find the high-confident ortholog, we used synteny relationship among four legume species such as, P. vulgaris, V. radiata, V. angularis and M. truncatula. Even though we didn't include the M. truncatula for species tree construction, we used it to narrow down the confident orthologs. Among the retrieved orthologs, 60 highly conserved ones were chosen, that showed the low relative standard deviation (RSD < 0.00001) of protein length to have clear speciation signal. To this confident ortholog set, we also added the corresponding orthologs of V. nepalensis, V. angularis var. nipponensis and V. nakashimae. The coding sequences of the orthologous loci were aligned using Prank software with –translation option51. The starting tree for the analysis was set to random and we implemented four runs of MCMC with the length of chain 50 million and the parameters logged at every 5000 steps. The substitution model was determined using software ProtTest to choose JTT as best model52. The relaxed clock model with log normally distributed uncorrelated rates was used and the divergence time for each node was calibrated using 8 MYA divergence of Vigna and Phaseolus of previous estimation32.
Accession codes
The adzuki bean genome information such as genome assembly, gene prediction and annotation, genetic markers and other related files of this study can be searched and downloaded from http://plantgenomics.snu.ac.kr. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession JRFV00000000.
References
Parida, A., Raina, S. N. & Narayan, R. K. J. Quantitative DNA Variation between and within Chromosome Complements of Vigna Species (Fabaceae). Genetica 82, 125–133 (1990).
Tomooka, N., Vaughan, D. & Moss, H. The Asian Vigna: genus Vigna subgenus Ceratotropis genetic resources. (Kluwer, Dordrecht, 2002).
Rubatzky, V. E., Yamaguchi, M., Rubatzky, V. & Yamaguchi, M. World vegetables: principles, production and nutritive values. (Chapman & Hall New York, 1997).
Lee, G.-A. Archaeological perspectives on the origins of azuki (Vigna angularis). Holocene 23, 453–459 (2013).
Schmutz, J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010).
Varshney, R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30, 83–U128 (2012).
Schmutz, J. et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46, 707–713 (2014).
Kang, Y. J. et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat Commun 5, 5443 (2014).
Gnerre, S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A 108, 1513–1518 (2011).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Wang, Y. P. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. Plos One 6, e19379 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25, 4.10.1–4.10.14 (2009).
Cantarel, B. L. et al. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18, 188–196 (2008).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29, 644–U130 (2011).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genornes. Bioinformatics 23, 1061–1067 (2007).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003).
Lang, D. et al. Genome-Wide Phylogenetic Comparative Analysis of Plant Transcriptional Regulation: A Timeline of Loss, Gain, Expansion and Correlation with Complexity. Genome Biol Evol 2, 488–503 (2010).
Lisch, D. How important are transposons for plant evolution? Nat Rev Genet 14, 49–61 (2013).
Varshney, R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31, 240–246 (2013).
Young, N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011).
Harris, D. R. Domestication of plants in the old world: The origin and spread of cultivated plants in west Asia, Europe and the Nile valley. Agr Hist Rev 49, 226–227 (2001).
Lestari, P. et al. Genome-wide single nucleotide polymorphism discovery and validation in adzuki bean. Mol Breeding 33, 497–501 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Simpson, J. T. et al. ABySS: a parallel assembler for short read sequence data. Genome Res 19, 1117–1123 (2009).
Kimura, M. Preponderance of Synonymous Changes as Evidence for Neutral Theory of Molecular Evolution. Nature 267, 275–276 (1977).
Thiel, T., Michalek, W., Varshney, R. K. & Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106, 411–422 (2003).
Cannon, S. B., May, G. D. & Jackson, S. A. Three Sequenced Legume Genomes and Many Crop Species: Rich Opportunities for Translational Genomics. Plant Physiol 151, 970–977 (2009).
Grant, D., Nelson, R. T., Cannon, S. B. & Shoemaker, R. C. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res 38, 843–846 (2010).
Meyers, B. C., Kozik, A., Griego, A., Kuang, H. H. & Michelmore, R. W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15, 1683–1683 (2003).
Lavin, M., Herendeen, P. & Wojciechowski, M. Evolutionary Rates Analysis of Leguminosae Implicates a Rapid Diversification of Lineages during the Tertiary. Syst Biol 54, 575–594 (2005).
Lynch, M. & Conery, J. S. The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (2000).
Rizzon, C., Ponger, L. & Gaut, B. S. Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. Plos Comput Biol 2, 989–1000 (2006).
Hanada, K., Zou, C., Lehti-Shiu, M. D., Shinozaki, K. & Shiu, S. H. Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol 148, 993–1003 (2008).
Jenczewski, E. et al. Insight on segregation distortions in two intraspecific crosses between annual species of Medicago (Leguminosae). Theor Appl Genet 94, 682–691 (1997).
Arumuganathan, K. & Earle, E. Estimation of nuclear DNA content of plants by flow cytometry. Plant Mol Biol Rep 9, 229–241 (1991).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Steinbiss, S., Willhoeft, U., Gremme, G. & Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res 37, 7002–7013 (2009).
Llorens, C. et al. The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res 39, D70–D74 (2011).
Du, J. C. et al. SoyTEdb: a comprehensive database of transposable elements in the soybean genome. BMC Genomics 11, 113 (2010).
Li, W. Z. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Stanke, M., Tzvetkova, A. & Morgenstern, B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol 7 (Suppl. 1). S11.1–S11.8 (2006).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res 33, W116–W120 (2005).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–230 (2014).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–37 (2011).
Consortium, U. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 40, D71–D75 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7, 214 (2007).
Loytynoja, A. & Goldman, N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A 102, 10557–10562 (2005).
Abascal, F., Zardoya, R. & Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 (2005).
Acknowledgements
The research was supported by a grant from the Next Generation BioGreen 21 Program (Code No. PJ00811701), Rural Development Administration, Republic of Korea.
Author information
Authors and Affiliations
Contributions
Y.K. designed the experiments and pipelines of bioinformatics. D.S. designed and performed the experiments and bioinformatics. S.S. performed the single nucleotide polymorphism (SNP) detection. T.L. annotated the genomes. J.L. performed the comparative genome analyses. W.H., S.K.K., P.L., K.L. conducted the transcriptome analyses. K.K., J.H., M.K., J.-K.M., J.-M.K., Y.L. created the mapping populations. A.C., R.K.V., B.-S.P. performed the genotyping-by-sequencing (GBS) experiments. J.-G.G. provided the experimental materials. S.-H.L. initiated and coordinated the project.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Supplementary Information
Supplementary Table S6
Supplementary Information
Supplementary Table S7
Supplementary Information
Supplementary Table S9
Supplementary Information
Supplementary Table S11
Supplementary Information
Supplementary Table S12
Supplementary Information
Supplementary Table S13
Supplementary Information
Supplementary Table S17
Supplementary Information
Supplementary Table S18
Supplementary Information
Supplementary Table S19
Supplementary Information
Supplementary Table S22
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kang, Y., Satyawan, D., Shim, S. et al. Draft genome sequence of adzuki bean, Vigna angularis. Sci Rep 5, 8069 (2015). https://doi.org/10.1038/srep08069
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08069
This article is cited by
-
Translocations and inversions: major chromosomal rearrangements during Vigna (Leguminosae) evolution
Theoretical and Applied Genetics (2024)
-
Genome-Wide Identification and Analysis of NBS-LRR-Encoding Genes in Mungbean (Vigna radiata L. Wilczek) and Their Expression in Two Wild Non-progenitors Reveal Their Role in MYMIV Resistance
Journal of Plant Growth Regulation (2023)
-
Histological and molecular responses of Vigna angularis to Uromyces vignae infection
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
