
Abstract
Circular RNAs are a recently (re-)discovered abundant RNA species with presumed function as miRNA sponges, thus part of the competing endogenous RNA network. We analysed the expression of circular and linear RNAs and proliferation in matched normal colon mucosa and tumour tissues. We predicted >1,800 circular RNAs and proved the existence of five randomly chosen examples using RT-qPCR. Interestingly, the ratio of circular to linear RNA isoforms was always lower in tumour compared to normal colon samples and even lower in colorectal cancer cell lines. Furthermore, this ratio correlated negatively with the proliferation index. The correlation of global circular RNA abundance (the circRNA index) and proliferation was validated in a non-cancerous proliferative disease, idiopathic pulmonary fibrosis, ovarian cancer cells compared to cultured normal ovarian epithelial cells and 13 normal human tissues. We are the first to report a global reduction of circular RNA abundance in colorectal cancer cell lines and cancer compared to normal tissues and discovered a negative correlation of global circular RNA abundance and proliferation. This negative correlation seems to be a general principle in human tissues as validated with three different settings. Finally, we present a simple model how circular RNAs could accumulate in non-proliferating cells.
Similar content being viewed by others
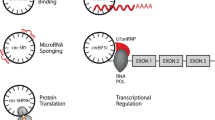

Introduction
Circular RNAs (circRNAs) are RNA molecules with covalently joined 3′- and 5′-ends formed by back-splice events thus presenting as covalently closed continuous loops. Hence, circRNAs are characterized by scrambled exons, which were already reported more than 20 years ago1, but mostly misinterpreted as splicing errors. Only in 2012/2013, circRNAs were rediscovered from RNA sequencing (RNA-seq) data and shown to be widespread and diverse in eukaryotic cells. They can contribute substantially to total RNA: about 2,000 different circRNA species were reported in human and mouse tissues and 700 in Caenorhabditis elegans; typically circular isoforms account for 5–10% of total transcripts of their corresponding coding gene, but certain circRNAs are up to 200 times more abundant than their linear counterparts2,3,4,5,6. They typically comprise one to several coding exons of otherwise linear messenger RNAs (mRNAs) and range between few hundreds and thousands of nucleotides in length7. But also circRNAs which align with antisense, intronic (albeit for these it has been proposed that they represent degraded lariat structures with a 5′-2′ linkage instead of a 5′-3′ linkage8), or intergenic sequences or untranslated regions have been described. They are typically non-polyadenylated and non-translated. One important feature of circRNAs is – due to the lack of accessible ends – resistance to exonucleases rendering them more stable than linear RNA isoforms3.
Neither the formation mechanism, nor their cellular function has been completely understood. For decades circRNAs were mostly disregarded as rare isoforms that result from splicing artefacts or gene rearrangements. But recent studies reporting high abundance, great diversity and tissue- as well as development-specific expression of circRNAs indicate that they originate from non-random back-splice events3,6. Such back-splice events are characterized by the covalent binding of the upstream 5′ splice acceptor to the downstream 3′ splice donor and are guided by specific sequences around the splice sites. So far only little is known about these specific sequences and the concrete mechanism guiding back-splice events remains to be elucidated. However, flanking introns are believed to be essential: they are longer than average introns, contain ALU repeats2 and seem to determine the production rate of circRNAs9. For intronic lariat-originated circRNAs a consensus motif containing a seven-nucleotide GU-rich element at the 5′ splice site and an eleven-nucleotide C-rich element upstream the branchpoint has been reported8.
Concerning their function, it is speculated that circRNAs serve as epigenetic microRNA (miRNA) sponges3,5. MiRNAs are 18 to 25 nucleotides long, belong to the group of non-coding RNAs and regulate a variety of essential biological functions such as cell proliferation, development, apoptosis and pathologies. They either block translation of mRNAs, if they bind imperfectly, or, if they have perfect complementarity with their targets, lead to their degradation. Furthermore, miRNAs are known to regulate expression in the nucleus by binding to promoters10. More than 2,588 miRNAs have been described for humans each targeting hundreds of targets (miRBase v21)11. Thanks to high throughput sequencing of small RNAs and computational methods to predict miRNAs and their targets, more and more are being reported. However, mechanisms concerning regulation of miRNAs are still poorly understood. CircRNAs in their proposed function as miRNA sponges are believed to negatively regulate miRNAs, thus contributing substantially to the competing endogenous RNA (ceRNA) network.
Memczak et al.3 claim that conserved miRNA and AGO protein binding sites are enriched in circRNAs. For at least one specific circRNA, ciRS-75 or also called CDR1as3, that has more than 70 binding sites for miR-7, it has been shown in vivo that the circRNA impairs the regulatory effect of miR-7. Furthermore, evidence for the miRNA sponge function of circRNAs comes from a study reporting a depletion of single nucleotide polymorphisms at miRNA binding sites in circRNAs12. However, whether or not all circRNAs function as miRNA sponges is still not clear. An additional proposed function of circRNAs is the transport of miRNAs. Summarizing, available experimental data are not very comprehensive, especially for their global regulation and function.
Colorectal cancer (CRC) is the third most common cancer in men and women with varying prognosis dependent on tumour stage at first presentation13. CRC is one of the best understood and described cancer entities in mankind. As most cancer entities, CRC is characterized by genetic and epigenetic modifications during cancerogenesis, including changes in miRNA expression14. Hamfjord et al. showed that at least 37 miRNAs were differentially expressed between eight matched pairs of colorectal cancer tissues and normal colon mucosa15.
Based on these findings and the theory of circRNAs tightly interacting with miRNAs, we thought to analyse and define the ‘Circulom’, the entirety of all expressed circRNAs, in CRC samples and matched normal colon mucosa. Additionally, circRNA data was correlated with cell proliferation.
Modelled after the protocol of Memczak et al.3, we predicted circRNAs in silico, verified their existence with RT-qPCR for CRC tissues and studied the effect of RNase R treatment on circRNAs. Moreover, we compared the ratio of circRNAs to their corresponding linear RNAs (linRNAs) in CRC tissue samples with their matched normal control tissues and CRC cell lines and validated our findings with independent RNA-seq data sets.
Results
CircRNA prediction, quantification and differential expression
Several recent papers about circRNAs prompted us to bioinformatically analyse a publicly available RNA-seq data set of rRNA depleted RNAs from twelve matched normal colon mucosa samples versus tumour tissues of CRC patients16 to predict circRNA using the pipeline described by Memczak et al.3. With this algorithm the terminal parts of all unmapped reads are aligned independently to the genome. Reads are considered as back-splice junctions from circRNAs, if the alignments of the two terminal parts (i) can be extended to obtain the original read sequence, (ii) are flanked by canonical splice signals, (iii) are on the same chromosome and not too wide apart from each other and (iv) are in a reversed orientation (read two upstream of read one) on the genome. 1,812 circRNAs were predicted by at least two supporting reads and quantified (Supplementary Table 1). Differential gene expression analysis between normal colon mucosa and CRC samples revealed 39 significantly differentially expressed circRNAs (FDR 20%, Supplementary Fig. 1), eleven of them up-regulated in cancer and 28 down-regulated (Supplementary Table 1). Given this over-representation of significantly down-regulated compared to up-regulated circRNAs, 28 versus eleven and seen also from non-significantly down-regulated circRNAs (cf. blue arrow in Supplementary Fig. 1) we next compared the global circRNA expression between CRC tumour tissues and normal mucosa tissues. We first extracted only those circRNAs which were biuniquely annotated to exactly one linear gene (n = 865), accounting for the following observation: Checking predicted circRNAs in the UCSC genome browser revealed some predicted circRNAs spanning several annotated genes, often with the same name-prefixes. This indicates gene duplications on the chromosome and (presumably) could lead to artefacts using the prediction algorithm by Memczak et al.3 We further filtered circRNAs which were covered by at least ten reads over all samples (n = 85). Finally, the normalised log2 values of these 85 circRNAs were averaged for each, tumour and normal, samples and are shown in Figure 1a. Interestingly, the majority of circRNAs were reduced in tumour samples compared to normal colon mucosa samples. The mean log2 difference was -1.91 (95% confidence interval (CI) [-2.23; -1.59], p<2.2 e-16). In linear space, this difference corresponds to a 73.4% reduction (95% CI [66.8%; 78.7%]) of circRNAs in CRC samples compared to normal colon mucosa. Only eleven out of these 85 (12.9%) circRNAs were higher expressed in tumour compared to normal colon mucosa samples (Supplementary Table 1).

Circular RNA expression from RNA-seq data of normal colon mucosa (Normal) and colorectal carcinoma (Carcinoma) samples.
(a) Expression levels of all biuniquely annotatable and reliably quantifiable 85 circular RNAs in twelve matched normal and carcinoma samples; normalised log2 values were averaged among the samples (p<2.2e-16). (b) Expression levels of reliably quantifiable 260 circular RNAs in pooled normal and carcinoma samples; log2 values were normalised to total sequencing read numbers (p<2.2e-16). Significance was assessed by paired student's t-tests (2.2e-16 is the minimum value of the used test in R).
Similarly, RNA-seq data of RNA pools of each 31 normal colon mucosa and of 31 CRC tumour samples showed higher expression of circRNAs in the normal compared to the tumour sample pool (Fig. 1b, for more details see section RNA-seq of RNase R digested pooled colorectal cancer tissue and normal mucosa samples).
Validation of circRNA and corresponding linRNA expression by RT-qPCR
To validate the global down-regulation of circRNAs in CRC tissues, we randomly selected five circRNAs, regardless of whether they were differentially expressed or which difference between normal and cancer samples they showed. Four of the five circRNAs comprised exons of protein coding genes that are also expressed as linear transcripts, whereas for the fifth circRNA, circ7780, no linear transcript is annotated. Four of the five circRNAs were lower expressed (one significant, circ6229) and one up-regulated (circ7374) in cancer compared to normal colon mucosa tissues as revealed from RNA-seq data. For circRNAs, specific TaqMan RT-qPCR systems with either one primer or the probe covering the back-splice junction were designed. For the corresponding linear transcripts Assay-on-Demand systems from Life Technologies were used which were not covered (completely) by the predicted circRNA but covered one normal, forward-splice event outside the circRNA borders (cf. Table 1, Supplementary Fig. 2 and Supplementary Table 2). The specificity and linearity of the TaqMan systems for circRNAs were validated by RT-qPCR of pooled normal and cancer RNAs from 31 CRC patients before and after RNase R digestion. All five circRNAs as well as the four corresponding linear transcripts were shown to be expressed and thus to exist. As expected, circRNAs were more resistant to RNase R treatment compared to the corresponding linear transcripts, i.e. they were 45- to 130,000-fold enriched (Fig. 2). Interestingly, three circRNAs, circ0817, circ3204 and circ6229, which are similarly long (629-706 nucleotides), showed a similar enrichment of 45- to 70-fold, while the circ7374 which is much shorter (288 nucleotides) was much more enriched (16,000 to 130,000-fold) indicating higher in vitro stability of shorter circRNAs. We think this is mainly due to artefacts during RNase R digestion and subsequent RNA purification. In the case of circ7780 (317 nucleotides) without linear counterpart, the effect of RNase R treatment was compared to depletion of housekeeping genes by RNase R. Circ7780 showed an intermediate enrichment of 570 to 830-fold.

Resistance of circular RNAs to RNase R treatment.
RNase R enriched circular compared to linear isoforms in pooled normal colon mucosa and tumour tissue samples of colorectal cancer patients. Enrichment correlates negatively with length of the circular RNA (cf. Table 1). CircRNA 7780 was normalised to housekeeping genes due to lack of the corresponding linear isoform. (Y-axis log2 scale of enrichment of the circular compared to the linear isoform).
For differential gene expression of circRNAs between cancer and normal tissues, the individual 31 matched normal versus CRC samples were used for RT-qPCR analysis together with two housekeeping genes as normalisers. As inferred from RNA-seq data, the ratios of circRNAs to their corresponding linear transcripts (circRNA/linRNA) were reduced in CRC tumour tissues compared to normal colon mucosa tissues, i.e. mean log2 difference over all ratios circRNA/linRNA was -0.89 between normal and CRC tumour tissues, corresponding to a 46.0% reduction in the latter samples in linear space, irrespective of whether the linear transcripts were up- or down-regulated in cancer versus normal tissues (Fig. 3). Expression of the five circRNAs and the correlation to their linear counterparts (if present) was also assessed in eleven CRC cell lines with RT-qPCR. Cancer cell lines have some different characteristics compared to cancer tissues: usually they have a higher proliferation rate (if grown under optimal culture conditions) and are pure cancer cells, sometimes even monoclonal, without tumour stroma or microenvironmental cells. Interestingly, ratios of circRNA/linRNA were even smaller in CRC cell lines compared to colorectal cancer tissues, i.e. mean log2 difference over all ratios circRNA/linRNA was -1.97 between CRC tissues and CRC cell lines, corresponding to a 74.4% reduction in the latter samples in linear space (Fig. 3). The multiple comparison of normal mucosa versus CRC tumour tissue versus CRC cell lines resulted in highly significant p-values for all four ratios of circRNA/linRNA (circ0817/CUL5: p = 3.7e-08; circ3204/USP3: p = 6.4e-10; circ6229/METTL3: p = 2.2e-09; circ7374/TNS4: p = 1.7e-05). Also, all pairwise comparisons of the four ratios of circRNA/linRNA between normal mucosa, CRC tumour tissue and CRC cell lines reached significance (post-hoc tests, Fig. 3).

Ratio of circular to corresponding linear RNAs in 31 matched normal mucosa (No) and colorectal tumour (Ca) tissues and in eleven colorectal cancer cell lines (CL).
Expression of (i) five exemplarily chosen circular RNAs (circ0817, circ3204, circ6229, circ7374 and circ7780), (ii) four corresponding linear RNAs (CUL5, USP3, METTL3 and TNS4) and (iii) ratios of circular to linear RNAs (circRNA/linRNA); determined by RT-qPCR. Circ7780 was not detectable (n.d.) in cancer cell lines. Y-axes represent relative expression values normalised to housekeeping genes for the upper two panels and the ratios of circular RNAs to linear RNAs in the lower panel. Significance for multiple comparison of No versus Ca versus CL was assessed by Kruskal-Wallis rank sum tests (given p-values) followed by pairwise comparisons applying the post-hoc Nemenyi test with Chi-squared approximation (significant pairs indicated by blue brackets).
RNA-seq of RNase R digested pooled colorectal cancer tissue and normal mucosa samples
To further validate the globally reduced circRNA expression in CRC compared to normal mucosa samples, the pooled RNA samples of CRC tumour and normal tissues were rRNA depleted and RNase R digested to enrich for circRNAs. Stranded libraries were prepared and sequenced for 50 bp paired end reads. 57 and 61 million read pairs for normal and tumour libraries were obtained, respectively. Reads were mapped with Segemehl v1.7, a back-splice aware mapper; back-splice events were called by the function testrealign. In total, 21,653 different back-splice events were called, 10,758 of them with at least two supporting reads. The majority of back-splice junctions were only covered by one read indicating that we probably missed a lot of low expressed circRNAs, indicating that the real number of expressed circRNAs has to be much higher (Supplementary Fig. 3a). 5,746 out of these 10,758 back-splice events were present in the pooled cancer sample and 6,746 in the pooled normal mucosa sample (Supplementary Fig. 3b); only 1,734 back-splice junctions were found in both samples. However, the relatively low number of overlapping back-splice junctions (16.1% of all back-splice junctions) were supported by 39.6% of all back-splice supporting reads (16,321 out of 41,221, Supplementary Fig. 3c), showing that these are the higher expressed ones. The sum of all circRNA supporting reads in the normal and the cancer pool was 23,563 and 17,658 (in total 41,221) reads, respectively, indicating less back-splice events in CRC tumour tissues compared to normal mucosa tissues (-25.1%). Corrected for sequencing depth (i.e. 57 versus 61 million reads for normal and tumour libraries, respectively), the difference between normal and cancer samples was even more pronounced (-30.0%). If a back-splice junction was called at least five times in each sample (n = 260), it was used for further analysis (Supplementary Table 3). In agreement with the RT-qPCR data of these samples, the RNA-seq data showed higher expression of circRNAs in the normal compared to the tumour sample (Fig. 1b): the mean log2 difference between circRNAs in normal and CRC tumour tissues was -0.47 (95% CI [-0.57; -0.38], p<2.2 e-16). In linear space, this difference corresponds to a 27.8% (95% CI [23.2%; 32.6%]) reduced expression of circRNAs in the CRC tumour tissue compared to the normal mucosa pool. Only 57 out of 260 (21.9%) circRNA expressions were higher in normal colon mucosa compared to CRC tumour samples (Supplementary Table 3).
In Supplementary Figure 4 genome viewer pictures depicting the coverage of circRNAs in the two RNA-seq data sets, 12 tumour versus 12 normal colon tissue samples and the pooled 31 tumour and normal colon samples, are shown. CircRNAs used for RT-qPCR analysis, deregulated circRNAs in one of the two data sets (up or down in tumour samples), or circRNAs from literature were selected and validated: (i) two (out of the five) circRNAs selected for RT-qPCR (circ6229/METTL3 and circ7780); (ii) the most up- and down-regulated circRNA from the 12 versus 12 data set (circ_002583/IKBKB and circ_002223/RNF13, respectively); (iii) the most up- and the second most down-regulated (the most down-regulated was circ_006229/METTL3, already selected in (i)) from the pooled 31 tumour and normal samples (chr7:100410369/EPHB4 and chr12:56094683/ITGA7, respectively) and (iv) one of the best known circRNAs from literature (ciRS-75, also known as CDR1as3). All regulations found in one setting were positively validated in the other setting(s) with one exception: the up-regulation of circ_002583 found in the 12 versus 12 data set was not clearly up-regulated in the pooled tumour and normal data set. Interestingly, the well described circRNA, ciRS-7, which is expressed antisense to the gene CDR1, was clearly up-regulated in tumour tissues compared to normal mucosa in the pooled data set, with 130 sequenced fragments in the tumour tissues and 41 fragments in the normal mucosa (normalised to sequencing depth, 2.13 versus 0.74, respectively, an increase of 187.8% in tumour samples). Expression of the gene CDR1 was not detectable from the stranded libraries, which is not surprising as linear transcripts were depleted by RNase R digestion.
Correlation of circRNA/linRNA ratio and the proliferation index
Given the successive expression reduction of circRNA to linRNA ratios from normal colon mucosa to CRC tumour tissues and finally to CRC cell lines, i.e. 46.0% and 74.4% reduction, respectively and since one major difference between normal tissues, cancer tissues and cancer cell lines is the proliferation rate, we were prompted to assess the percentage of Ki-67 positive cells (proliferation index) of the 31 matched pairs of colon tissues. FFPE tissue sections were immunohistochemically stained for Ki-67. The proliferation indices were assessed and correlated to the circRNA/linRNA ratios. As expected, proliferation indices were higher in tumour than in the normal colon tissues: median 25.2 (7.1–50.2) and 8.4 (3.1–20.5), respectively. For two circRNA-linRNA pairs a significant negative correlation between proliferation and the ratio of circRNA/linRNA was found (circ6229/METTL3: ρ = -0.42, p<0.01; circ7374/TNS4: ρ = -0.35, p<0.05). For the other two circRNA-linRNA pairs only trends (p<0.1) in the same direction were observed (circ0817/CUL5: ρ = -0.39; circ3204/USP3: ρ = -0.35, Fig. 4). Circ7780 showed a non-significant negative correlation of ρ = -0.30 with proliferation (Fig. 4).

Correlation of ratios circRNA/linRNA to cell proliferation.
The histograms in each of the subpanels show the distribution of Ki-67 proliferation indices (upper left) and the distribution of (a) the ratios of circRNA/linRNA and of (b) relative circ7780 expression (lower right) in matched normal mucosa and colorectal tumour tissues. The dot plots illustrate the correlation between the Ki-67 index (x-axes) and the (a) ratios of circRNA/linRNA and the (b) relative circ7780 expression (y-axes) together with linear (dashed) and loess regression (continuous) lines. The axes of the dot plots apply also to the x-axes of the histograms: the x-axes of the dot plots correspond to the Ki-67 histograms; the y-axes of the dot plots correspond to the histograms of (a) circRNA/linRNA ratios or of (b) relative circ7780 expression. The corresponding Spearman's rank correlation values are depicted in the upper right. Significance: °p<0.1, *p<0.05, **p<0.01, ***p<0.001. (c) Representative Ki-67 immunohistochemical stainings of two example tissues, quantified with an automated analysis pipeline using the ImmunoRatio ImageJ plugin (lower panel). Scale bar, 100 µm.
Validation of negative correlation of global circRNA abundance with proliferation
To validate the correlation of global circRNA abundance (the circRNA index) and proliferation we used four rRNA depleted RNA-seq datasets (unfortunately most of publicly available RNA-seq data are poly(A) enriched and not rRNA depleted): (i) one from a non-cancerous neo-proliferative disease, idiopathic pulmonary fibrosis (IPF) (Supplementary Table 4a); (ii) one from serous ovarian cancer cell samples and three immortalised normal ovarian surface epithelial (IOSE) cell lines and one normal ovarian surface epithelial (OSE) cell line (Supplementary methods and Supplementary Table 4b). In the following sections, the term (immortalised) normal ovarian surface epithelial ((I)OSE) stands for the summary of IOSE and OSE cell lines; (iii) one from twelve normal human tissues together with one from isolated monocytes (Supplementary Tables 4a–c). Proliferation was determined by MKI67 expression obtained directly from the RNA-seq data. Percentage of circRNAs (the circRNA index) is given as percentage of the sum of all back-splice reads based on the sum of all normal, forward-splice reads.
-
i
In this study, MKI67 expression values were inconsistent in normal lung tissues; in three samples they were higher compared to the MKI67 expression values from IPF tissues and in four they were lower (as expected). We therefore divided normal tissues in normal low (N_low, n = 4) and normal high (N_high, n = 3) according to the observed MKI67 expression. It is unclear why normal lung tissues showed such a heterogeneous MKI67 expression in this dataset. However, it has to be considered that MKI67 mRNA expression – in contrast to Ki-67 nuclear IHC staining of tissue sections – is not the optimal marker for proliferation. From literature it is known that IPF is a proliferative disease17,18 and that proliferation indices in IPF tissues should (always) be higher compared to normal lung tissues. Nevertheless, percentages of circRNA reads from IPF tissues were lower compared to both normal tissue samples: the median difference of circRNA indices was -34.7% and -15.4% between IPF and MKI67 low expressing normal tissues and between IPF and MKI67 high expressing normal tissues, respectively (Fig. 5a).
Figure 5 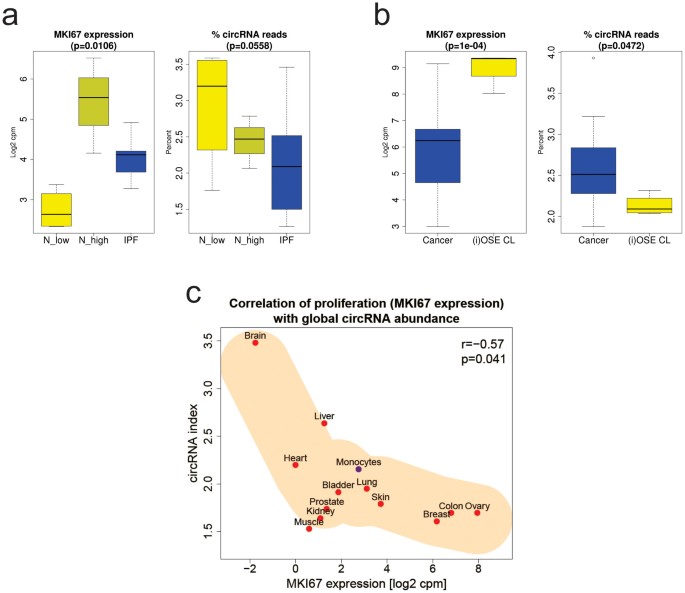
Validation of the correlation of the circRNA index with MKI67 expression.
(a) Normal lung tissues (four with low MKI67 expression, N_low and three with high MKI67 expression, N_high) and tissues from idiopathic pulmonary fibrosis (IPF), a non-cancerous neo-proliferative disease (Supplementary Table 4a) are compared concerning MKI67 expression (p = 0.011, Kruskal-Wallis rank sum test) and percent circRNA reads (trend p-value = 0.056, Kruskal-Wallis rank sum test, R-package compareGroups). (b) Ascitic ovarian cancer cells (Cancer) and (immortalised) normal ovarian surface epithelium cell lines ((i)OSE CL) (Supplementary Table 4b) are compared concerning MKI67 expression (p = 1e-04) and percent circRNA reads (p = 0.047, both Kruskal-Wallis rank sum tests). Yellow indicates normal tissues or cell lines and blue indicates tissues or cells from the diseased state. (c) Correlation of MKI67 expression and global circRNA abundance (the circRNA index) in 13 human tissues (red, twelve human tissues from study SRP033095 and blue, monocytes from study E-MTAB-2399). A significant strong negative correlation (Pearson correlation coefficient ρ = -0.573, p = 0.041) is seen (Supplementary Table 4c).
-
ii
To validate the reduced circRNA abundance in cell lines compared to primary tissues and to test whether malignancy is the driving factor for this reduction, we compared 16 epithelial ovarian cancer cell samples from ascites of eight ovarian cancer patients (each aggregates of cancer cells and single cancer cells) with non-malignant (I)OSE cells. Consistently with the reduced circRNA abundance in CRC cell lines compared to tumour tissues, (I)OSE cell lines showed a higher MKI67 expression and a lower circRNA index (median reduction -16.8%), indicating proliferation as driving factor for global circRNA abundance reduction and not malignancy (Fig. 5b).
-
iii
To provide an overview of global circRNA abundances in different normal human tissues an RNA-seq dataset comprised of twelve different tissues and one of isolated monocytes was analysed for MKI67 expression and the circRNA index (Fig. 5c). As expected, a strong negative correlation of MKI67 expression (indicative for proliferation) and the circRNA index was seen (ρ = -0.573; p = 0.041), with brain tissue as extreme circRNA rich and low proliferating. There seems to be a lower limit of the circRNA index, i.e. approximately 1.5%, presumably deduced from the synthesis rate and leading to a flattening of the correlation for the very high proliferating tissues, breast, colon and ovary.

Discussion
Circular RNAs – a newly identified member of the ceRNA network – are an RNA species with yet unknown impact on the cellular (epigenetic) regulatory RNA network. They are abundant, evolutionary old and (at least partly) cell type specific2,6. Several studies have described between hundreds and thousands of different circRNA species mostly comprising one to several exons of protein coding genes. We analysed circRNAs in clinical normal mucosa and tumour tissue samples from CRC patients with RNA-seq and RT-qPCR with regard to proliferation.
We predicted >1,800 circular RNAs in human normal colon mucosa and tumour samples of CRC patients, a number which is in line with other reports about circRNAs2,3,4. However, most probably we underestimated the number of circRNAs, because the power of the detection algorithm is limited by the fact that only reads spanning the back-splice sequence can be used for detection. As expected, enrichment of circRNAs in pools of 31 normal and tumour samples from CRC patients and subsequent deep sequencing resulted in many more different back-splice junctions (i.e. 21,653; 10,758 of them with at least two supporting reads). Moreover, the relatively small overlap of identified back-splice junctions from the CRC tumour and normal mucosa samples (16.1% of all back-splice junctions) corresponding to almost 40% of reads (Supplementary Fig. 3) revealed that even RNAse R enrichment and relatively deep sequencing (~60 million paired end reads) could not ensure the detection of most circular RNAs. The high number of circular RNAs covered by only one supporting read indicates clearly that the actual number of different circRNA species has to be much higher. In CRC tumour tissue samples compared to normal mucosa samples read numbers supporting back-splice events are globally reduced (normalised -30.0%). Regarding the 260 highest expressed circRNAs, a mean reduction of circRNA expression of 27.8% in tumour compared to normal was observed, with 78.1% (n = 203) of circRNAs lower expressed in tumour samples (Fig. 1b). For this analysis it has to be taken into account that if transcription of a gene is up-regulated in tumour tissues, the circRNA is also up-regulated (presuming a constant ratio of back- and forward-splicing). Therefore the number of down-regulated circRNAs could be even underestimated. An example of such a constellation is shown in Figure 3 (circ7374 and TNS4).
Specific RT-qPCR systems for four pairs of circRNAs and corresponding linear transcripts revealed a reduction of the ratios circRNA/linRNA in all four cases, regardless of whether the linRNAs or circRNAs alone were lower or higher expressed in tumour compared to normal samples. The one circRNA without annotated linear transcript (circ7780) was also lower expressed in tumour samples compared to normal samples. All ratios circRNA/linRNA were further reduced in eleven CRC cell lines and circ7780 was not even detectable (Fig. 3). Assuming that proliferation is highest in CRC cell lines (cultured under optimal conditions in vitro), followed by CRC tumour tissues and finally normal mucosa tissues (both in vivo), we determined the proliferation index of the matched pairs of tumour and normal tissues of CRC patients and correlated these proliferation indices with the ratios of the four circRNA-linRNA pairs and the expression of circ7780. All four ratios and the expression of circ7780 correlated negatively with the proliferation index, two ratios significantly (p<0.05) and two ratios and circ7780 expression as trend (Fig. 4). In contrast, one of the best known circRNAs, ciRS-7 (also known as CDR1as) acting as sponge for miR-73,5, was higher expressed in tumour samples compared to normal colon mucosa (Supplementary Fig. 4). As miR-7 regulates the expression of several oncogenes (miR-7 is believed to have tumour-suppressive function by down-regulating oncogenic factors like YY119, EGF-R, IRS-1, IRS-2, Pak1, Raf1, Ack1 and PIK3CD and is the most reduced miRNA in cancer stem cells20), the up-regulation of the corresponding sponge, ciRS-7, in tumour samples seems plausible. As for ciRS-7 no linear transcript (from the same strand) is known, our model of global circRNA regulation (see below) is not applicable for this specific circRNA. But specific regulatory mechanisms have to be supposed.
Taken together, we have reliable evidence that circRNAs are globally reduced in tumour tissues from CRC patients compared to matched normal tissues and are even more reduced in CRC cell lines. The following mechanisms could explain our data: (a) the back-splice machinery is compromised in malignant tissues; (b) the lower amount of circRNAs is a result of increasing degradation by miRNAs which are deregulated in tumour tissue; even simpler (c) circRNAs get passively thinned out by cell proliferation or vice versa accumulate in non-proliferating (starving, dormant or finally differentiated) cells (schematically outlined in Fig. 6); or (d) of course, also yet unknown mechanisms could be the reason for the results described above. For the proliferation scenario (c) the following three assumptions have to be fulfilled: (i) CircRNAs and linRNAs are processed in a gene (and cell type and condition) specific ratio. (ii) Linear transcripts are regulated by transcription and degradation and are therefore in a cell type and condition specific steady state. Although they are also – just as their circular counterparts – passively distributed to the daughter cells, thus putatively also thinned out by cell proliferation, their level is very rigorously controlled. Assuming constant environmental signals, the required levels of mRNAs are adjusted very quickly to the level before cell division also resulting in circRNA synthesis in a gene and condition specific ratio. This leads to daughter cells with the same amount (and ratio) of linear and circular RNAs compared to their mother cell. (iii) CircRNAs are more stable than linRNAs in cells, i.e. are not regulated by (general exonucleatic) degradation.

Schematic model of circular and linear RNAs in proliferating and non-proliferating cells.
Assumption: Circular and linear RNAs are synthesized by specific splice events in a gene (cell type and condition) dependent ratio. While linear RNAs are in a steady state of regulated synthesis and degradation, circular RNAs are much more stable. During proliferation both RNA species are evenly distributed to daughter cells. The steady state level of linear transcripts is accurately controlled and maintained by active transcription and degradation also leading to new expression of circular RNAs and resulting in constant ratios of circular to linear isoforms (lower panel). In contrast, in non-proliferating cells stable circular RNAs accumulate whereas linear transcripts are in a regulated steady state of transcription and degradation (upper panel).
The first assumption is plausible, but was not shown so far, only the competitive generation of linear and circular transcripts from one pre-transcript was shown recently9, the second assumption is the current view of gene expression and regulation and the third assumption was shown by Memczak et al. by transcription inhibition experiments, which showed that circRNAs are tremendously more stable in cells compared to linear transcripts3.
To further validate the reduced circRNAs index in faster proliferating cells, we showed similar correlations in a non-cancerous neo-proliferative disease, idiopathic pulmonary fibrosis and with malignant ovarian cancer cells isolated from patients compared to normal (I)OSE cells grown in vitro. In both cases, the circRNA index was lower in faster proliferating cells regardless of whether these cells were the diseased (IPF) or the normal ones ((I)OSE cell lines), which is in line with the negative correlation of the circRNA index and proliferation observed in colon tissues and cell lines. Furthermore, the circRNA index of 13 normal human tissues is provided and compared to their MKI67 expression values – which also showed a significant strong negative correlation. Negative correlation of the circRNA index with proliferation seems to be a general principle in human tissues and cells.
The consequences of a higher circRNA index in a starving, resting, dormant, or finally differentiated cell are not completely clear yet, but will presumably have a tremendous impact on epigenetic regulation. Given a possible miRNA sponge function of circRNAs, circRNAs would suppress miRNAs activity by binding newly synthesized mature miRNAs. MiRNAs sponged by circRNAs are believed to bind imperfectly to the circRNA sponge, thus do not induce miRNA-mediated degradation of the circRNA3. Therefore miRNAs would be less disposable for binding transcripts or promoters, i.e. regulating translation or transcription or mediating transcript degradation. Thus, more circRNAs would lead to a more stable situation regarding epigenetic regulation by miRNAs. Cleavage of circRNAs is believed to be mediated by perfect miRNA binding and to cause the release of all imperfectly bound miRNAs, a scenario which was proposed for at least ciRS-7 by Hansen et al.20,21. This mechanism seems to be very efficient, since few circRNA-specific miRNAs lead to the release of many (other) miRNAs, thus are then active for regulation.
The biological raison d'être of such a more stable situation regarding miRNA regulation in differentiated cells is easily understandable as differentiated cells should not react to (weak) signals from outside with strong regulatory activities. In contrast, proliferating cells should be more susceptible to autocrine and paracrine regulation and react faster to environmental signals than circRNA-buffered, non-proliferating cells.
Furthermore, this tremendously reduced amount of circRNAs in in vitro cultured cell lines compared to corresponding tissues in vivo could compromise the translation of results from siRNA/miRNA in vitro cell line experiments to the in vivo situation. Some of the observed discrepancies between in vitro and in vivo experiments concerning miRNAs and the ceRNA network could therefore be attributed to differences in the total number of circRNAs, simply driven by proliferation.
Summarized, we show for the first time a global reduction of circRNAs in cancer cell lines and cancer tissues compared to normal mucosa for CRC, negatively correlated to proliferation. Moreover, we propose a simple passive mechanism for this reduction in proliferating cells (Fig. 6). This correlation was validated in three additional settings, involving a non-cancerous disease, normal cell lines and normal human tissues.
If this general regulation mechanism – proposed to be a general principle at least in human cells – will be positively validated in other cancer entities and tissues, the impact on the understanding of epigenetic regulation via miRNAs and the competing endogenous RNA network cannot be overestimated. Increased caution has to be taken in translating results from cell lines to the in vivo situation especially if the ceRNA network is involved.
Methods
RNA-seq data sets
Publicly available RNA-seq data of ribosomal RNA (rRNA) depleted RNAs (RiboMinus, Life Technologies) from twelve matched CRC tissues and normal colon mucosa (SRA043728, experiment SRP00758416, downloaded from the Sequence Read Archive (SRA) and comprised of 71.7 million short sequencing reads (Illumina GAII, 72 bp paired end reads)) were mapped using RNA Seq Unified Mapper (RUM) and circRNAs predicted by the pipeline published by Memczak et al.3 and http://circrna.org (find_circ.tar.gz, version 1).
For validation four independent datasets of rRNA depleted RNA-seq were used: a publicly available RNA-seq dataset from idiopathic pulmonary fibrosis (IPF), comprised of seven normal lung tissues and eight IPF tissues (SRP033095, Supplementary Table 4a), a publicly available normal human tissue dataset, comprised of twelve different human tissues, together with an RNA-seq dataset of isolated monocytes (Supplementary Table 4b) and a dataset from our group, comprised of 16 samples of ovarian cancer cells from ascites of eight individuals (each aggregates of cancer cells and EpCAM positive single cancer cells, see Supplementary methods) and four (I)OSE cell lines (three IOSE and one OSE cell line) (Supplementary Table 4c).
Clinical samples and cell lines
Tumour tissues and matched normal colon mucosa from 31 CRC patients were collected according to standardised procedures. Ovarian cancer samples are described in Supplementary Table 4c and Supplementary methods. The study inclucing all experimental protocols was approved by the Ethics Committee of the Medical University of Vienna and all patients gave pre-operative written informed consent. The methods were carried out in accordance with the approved guidelines.
The patient cohort was typical for CRC patients: 57% of patients were male, the median age of patients was 71.5 years (23.2 to 88.7 years), 39.3% were rectal cancer, 60% of low grade and 32% of high grade (8% unknown) and 14% presented with distant metastases.
CRC cell lines (Caco-2, COLO 205, COLO 320HSR, DLD-1, HCT-15, HCT-8, HT-29, LS 174T, SW1116, SW480, SW620) were cultured in the according cell culture medium at 5% CO2 at 37°C. Cell lines were authenticated by STR DNA profiling analysis. Non-malignant human immortalised ovarian surface epithelial cells (IOSE-80, IOSE-364, IOSE-386; N. Auersperg, Canadian Ovarian Tissue Bank, British Columbia Cancer Research Centre, Canada), revealing extended but finite life span due to exogenous expression of the proto-oncogene SV40 large T antigen (SV40T/t), were grown in medium 199/MCDB105 (1:2) for a maximum of 25 passages22. Human ovarian surface epithelial cells (HOSEpiC) were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in medium OEpiCM according to manufacturer's instructions.
RNA preparation
About 30 mg of the freshly frozen (in liquid N2) tumour and normal tissues were homogenised using a Mikro-Dismembrator U (B. Braun, Biotech International, Germany) and lysed in 600 µl RLT buffer (QIAGEN, Venlo, The Netherlands). One million CRC cancer cells were lysed in 600 µl RLT buffer for cell line samples. Total RNA was isolated with RNeasy Mini Kit (QIAGEN) according to the manufacturer's instructions and quantified spectrophotometrically (NanoDrop, Wilmington, DE, USA). The quality of RNA was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). RNA with an RNA Integrity Number > 6 was used for further analysis.
RNase R digestion, cDNA synthesis and RT-qPCR
400 ng of each normal and tumour RNA sample were pooled and of each pool 11 µg were DNase I (Epicentre, Illumina, San Diego, CA, USA) digested for 15 minutes at 37°C following the manufacturer's protocol. An AMPure bead (Beckman Coulter, Brea, CA, USA) clean-up with a 1.8-fold volume of beads followed according to the manufacturer's instructions. Next, 5 µg of each normal and tumour RNA pool were used for an RNase R (Epicentre, Illumina, San Diego, CA, USA) digestion (16 units RNase R, 15 minutes at 37°C) to deplete linear and enrich circular RNAs. RNase R is a highly processive 3′ to 5′ exoribonuclease. A 10-fold volume (500 µl) of QIAzol Lysis Reagent (QIAGEN) and 3-fold volume of chloroform was added to the RNase R reaction (50 µl). After mixture and phase separation (30 min, 15 krpm, 4°C), the aqueous phase (~300 µl) was extracted with 600 µl chloroform. 1 µl glycogen was added to the aqueous phase and nucleic acids precipitated with a 1-fold volume of isopropanol. The pellet was washed once with 70% ethanol and resuspended in water. A second round of RNase R digestion, QIAzol/chloroform extraction and isopropanol precipitation followed.
This pooled RNase R digested normal and tumour RNA, RNA from matched tumour and normal colon mucosa tissues from 31 CRC patients and RNA of eleven CRC cell lines were used for cDNA synthesis using the Promega Reverse Transcription Kit (Promega, Fitchburg, WI, USA) with 1 µg RNA according to manufacturer's instructions.
TaqMan Gene Expression Assays (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA) were designed to distinguish between circular and the corresponding linear isoforms for four exemplarily chosen pairs of circular and linear RNAs and one circular isoform without annotated linear transcript (for detailed design and sequences see Table 1, Supplementary Fig. 2 and Supplementary Table 2). qPCR with TaqMan systems for these specific circular and linear isoforms and for the two housekeeping genes RPL9 and EEF1A1 (TaqMan probe numbers: Hs01552541_g1 and Hs00265885_g1, respectively; Life Technologies) was performed according to the manufacturer's instructions. Housekeeping genes were selected from five candidates (i.e. RPL9, Hs01552541_g1; RPL21, Hs03003806_g1; RPL12, Hs02385039_g1; ACTB, Hs01060665_g1; and EEF1A1, Hs00265885_g1) using geNorm and NormFinder methods. Each reaction was performed in duplicates.
RNA sample treatment, library synthesis and Illumina HiSeq 2000 sequencing
Normal and tumour RNA samples were pooled, DNase I digested, RNase R treated and purified as described above with the following modifications: Before the first RNase R treatment (40 units, 30 minutes at 37°C) a rRNA depletion step was included using the RiboZero rRNA Removal Kit (Epicentre), performed according to the manufacturer's instructions followed by an AMPure bead (Beckman Coulter) clean-up. A second RNase R treatment (40 units, 30 minutes at 37°C) with subsequent QIAzol/chloroform extraction and isopropanol precipitation as described above was performed.
NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA, USA) was used for library synthesis with the following specifications: starting material was 8 ng of normal and tumour rRNA depleted and RNase R digested RNA, twelve PCR cycles in the final PCR reaction, two AMPure bead clean-up steps after ligation and two AMPure bead clean-up steps after PCR. Libraries were quality controlled with Agilent's 2100 Bioanalyzer High Sensitivity DNA Analysis Kit (Agilent Technologies) and sequenced with 50 bp paired end to a mean depth of 59 million reads on a HiSeq 2000 (Illumina).
Ki-67 immunohistochemistry (IHC) and analysis
Four µm formalin-fixed paraffin-embedded (FFPE) whole tissue sections of tumour and normal colon mucosa were stained for proliferation marker Ki-67 (MIB-1 mouse monoclonal, dilution 1 : 20; Novocastra) according to standard IHC staining procedures. Slides were scanned with the TissueFAXS system (version 2.0.4.0147, TissueGnostics, Vienna, Austria) using an x20 objective lens. The Ki-67 proliferation index (percentage of stained nuclei) was automatically determined with a picture deconvolution method using the ImmunoRatio23 ImageJ plugin.
Bioinformatics and statistical analysis
Statistical measures and tests were applied according to data distributions: parametric tests for normally distributed data (mean value, student's t-test and Pearson correlation); non-parametric measures and tests for non-normally distributed data (median value, Kruskal Wallis rank sum test and Spearman's rank correlation). Paired tests were used if applicable. For the percent circRNA reads comparison from IPF data (Fig. 5a) a trend p-value is given over all three groups (R-package compareGroups, v2.0.5). If log-transformation was performed to obtain normal distribution of data, the results were also given back-transformed to linear space.
For gene expression analysis raw read pairs were mapped to the human transcriptome and genome (hg19) with RNA-Seq Unified Mapper (RUM v1.1024). Putative reads coming from circRNAs (see below) were filtered and the remaining fragments were counted (htseq-count mode ‘union’ from the HTseq 0.6.1 Python framework25) into the gene model from gencode v1526. Circular RNAs were predicted and filtered from all 24 downloaded libraries combined using the pipeline of Memczak et al.3 with standard parameters. The obtained back-splice junctions (fragments with length of 120 bp with the junction at 60) were used for individual mapping (i.e. for each of the 12 CRC and normal samples; method: Bowtie 2 with a combined index of all known transcripts and the 120 bp back-splice junction fragments and following parameters: “--very-sensitive --mm -R20 --score-min = C,-15,0”27) and counting (htseq-count) of supporting reads. Significant differences of circRNA expressions were analysed by function SAMseq (R-package samr v2.028) using a false discovery rate (FDR) of 20%. Global circRNA expression analysis was performed with all circRNAs which were biuniquely annotated to one linear transcript (n = 865) and supported by at least ten reads over all samples (n = 85). CircRNAs were normalised using the function PS.Est.Depth from R-package PoissonSeq v1.1.228.
RT-qPCR results from experiments with the pooled, RNase R digested tumour and normal RNAs circRNA expressions were analysed with respect to their linear counterparts or – in the case of circ7780 – normalised to geometric mean of the housekeeping genes RPL9 and EEF1A1 (in default of the corresponding linear transcript). RT-qPCR results from the 31 matched CRC and normal samples were analysed by the ΔCq method and normalised to the geometric mean of both housekeeping genes. Expression levels of (i) circRNAs, (ii) of linRNAs and (iii) the ratios of circRNA to linRNA were compared between normal colon mucosa and CRC tumour tissue samples and CRC cell lines with the Kruskal-Wallis rank sum test followed by pairwise comparison applying the post-hoc Nemenyi test with Chi-squared approximation. This analysis was performed with the R package The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR), v1.0.
CircRNA prediction and quantification of pooled, RiboZero depleted and RNase R digested sequencing reads was performed with mapper Segemehl 0.1.7 (parameters: “-D 2 -T –S”29) and the function testrealign.x (Segemehl 0.1.7) allowing only splice sites annotated as ‘C’ for circular and ‘P’ for passed, indicating that there was only one corresponding acceptor-donor pair. For normalization total sequencing read numbers of the corresponding libraries were used.
For the validation datasets (cf. Supplementary Tables 4a–c and Supplementary methods), reads were mapped with Segemehl and reads indicative for circRNAs (i.e. back-splice reads) and normal (i.e. forward-splice) reads were determined as described above. MKI67 expression was determined from each library by counting all fragments (read pairs) which mapped uniquely to the gene MKI67 (featureCounts from the subread package, v1.4.5-p130) and normalised to millions of all uniquely mapped fragments and is given as log2 counts per million (cpm). Percentage of circRNA supportive reads (the circRNA index) was calculated as follows: sum of all back-splice reads divided by sum of all forward-splice and back-splice reads multiplied by 100.
Statistical analysis was performed with R and R-packages described above (version 3.0.1, R Core Team, R: A Language and Environment for Statistical Computing. (2014), available at: http://www.R-project.org, date of access:18/11/2014)).
References
Nigro, J. M. et al. Scrambled exons. Cell 64, 607–613 (1991).
Jeck, W. R. et al. Circular RNAs are abundant, conserved and associated with ALU repeats. RNA 19, 141–157 (2013).
Memczak, S. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338 (2013).
Salzman, J., Gawad, C., Wang, P. L., Lacayo, N. & Brown, P. O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7, e30733 (2012).
Hansen, T. B. et al. Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388 (2013).
Salzman, J., Chen, R. E., Olsen, M. N., Wang, P. L. & Brown, P. O. Cell-type specific features of circular RNA expression. PLoS Genet 9, e1003777 (2013).
Guo, J. U., Agarwal, V., Guo, H. & Bartel, D. P. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15, 409 (2014).
Zhang, Y. et al. Circular intronic long noncoding RNAs. Mol Cell 51, 792–806 (2013).
Ashwal-Fluss, R. et al. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol Cell 56, 55–66 (2014).
Salmanidis, M., Pillman, K., Goodall, G. & Bracken, C. Direct transcriptional regulation by nuclear microRNAs. Int J Biochem Cell Biol 54, 304–311 (2014).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42, D68–73 (2014).
Thomas, L. F. & Saetrom, P. Circular RNAs are depleted of polymorphisms at microRNA binding sites. Bioinformatics 30, 2243–2246 (2014).
Stewart, B. W. & Wild, C. P. [Colorectal cancer]. World cancer report 2014. [Stewart, B. W. & Wild, C. P. (ed.)] [392–403] (IARC Press, Lyon 2014).
Pritchard, C. C. & Grady, W. M. Colorectal cancer molecular biology moves into clinical practice. Gut 60, 116–129 (2011).
Hamfjord, J. et al. Differential expression of miRNAs in colorectal cancer: comparison of paired tumor tissue and adjacent normal mucosa using high-throughput sequencing. PLoS One 7, e34150 (2012).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22, 299–306 (2012).
Ramos, C. et al. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol 24, 591–598 (2001).
Vancheri, C. Idiopathic pulmonary fibrosis: an altered fibroblast proliferation linked to cancer biology. Proc Am Thorac Soc 9, 153–157 (2012).
Zhang, N. et al. microRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene 32, 5078–5088 (2013).
Hansen, T. B., Kjems, J. & Damgaard, C. K. Circular RNA and miR-7 in cancer. Cancer Res 73, 5609–5612 (2013).
Hansen, T. B. et al. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 30, 4414–4422 (2011).
Li, P. et al. Multiple roles of the candidate oncogene ZNF217 in ovarian epithelial neoplastic progression. Int J Cancer 120, 1863–1873 (2007).
Tuominen, V. J., Ruotoistenmaki, S., Viitanen, A., Jumppanen, M. & Isola, J. ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR) and Ki-67. Breast Cancer Res 12, R56 (2010).
Grant, G. R. et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM). Bioinformatics 27, 2518–2528 (2011).
Anders, S., Pyl, P. T. & Huber, W. HTSeq - A Python framework to work with high-throughput sequencing data. Bioinformatics; 10.1093/bioinformatics/btu638 (2014).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Li, J. & Tibshirani, R. Finding consistent patterns: a nonparametric approach for identifying differential expression in RNA-Seq data. Stat Methods Med Res 22, 519–536 (2013).
Hoffmann, S. et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol 15, R34 (2014).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Acknowledgements
We would like to thank the Vienna Campus Support Facility (CSF) NGS Unit for performing the Illumina Next Generation Sequencing. Furthermore, our thank goes to Theresia Thalhammer and Erika Bajna (Medical University of Vienna, Department of Pathophysiology) for their support with the TissueFAXS system, which we used for the digitalisation of immunohistochemical stainings. We also thank Jagdeep Singh for collecting clinical information of CRC patients.
Author information
Authors and Affiliations
Contributions
A.B. and A.R. drafted the manuscript and designed the study. A.R. carried out the RNA purification and library synthesis. A.B. performed the RNase R digestion and RT-PCR analysis. K.A. and N.S. carried out the computerised detection and quantification of immunohistochemical staining and participated in the study design. S.A. provided ovarian cancer tissues and clinical data. A.B., K.A. and S.A. prepared ovarian cancer samples. T.B. collected patients' materials and clinical and patients' information. I.M. performed the pathological examination and evaluated the immunohistochemical stainings. T.G. provided and cultured (I)OSE cells. R.Z. participated in the design and coordination of the study. D.P. performed the data interpretation and bioinformatical and statistical analyses, drafted the manuscript and designed the study. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary Table 1
Supplementary Information
Supplementary Table 3
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Bachmayr-Heyda, A., Reiner, A., Auer, K. et al. Correlation of circular RNA abundance with proliferation – exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis and normal human tissues. Sci Rep 5, 8057 (2015). https://doi.org/10.1038/srep08057
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08057
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
