
Abstract
Bulk crystals and thin films of PbTi1−xFexO3−δ (PTFO) are multiferroic, exhibiting ferroelectricity and ferromagnetism at room temperature. Here we report that the Ruddlesden-Popper phase Pbn+1(Ti1−xFex)nO3n+1−δ forms spontaneously during pulsed laser deposition of PTFO on LaAlO3 substrates. High-resolution transmission electron microscopy, x-ray diffraction and x-ray photoemission spectroscopy were utilised to perform a structural and compositional analysis, demonstrating that  and
and  . The complex dielectric function of the films was determined from far-infrared to ultraviolet energies using a combination of terahertz time-domain spectroscopy, Fourier transform spectroscopy and spectroscopic ellipsometry. The simultaneous Raman and infrared activity of phonon modes and the observation of second harmonic generation establishes a non-centrosymmetric point group for Pbn+1(Ti0.5Fe0.5)nO3n+1−δ, a prerequisite for (but not proof of) ferroelectricity. No evidence of macroscopic ferromagnetism was found in SQUID magnetometry. The ultrafast optical response exhibited coherent magnon oscillations compatible with local magnetic order and additionally was used to study photocarrier cooling on picosecond timescales. An optical gap smaller than that of BiFeO3 and long photocarrier lifetimes may make this system interesting as a ferroelectric photovoltaic.
. The complex dielectric function of the films was determined from far-infrared to ultraviolet energies using a combination of terahertz time-domain spectroscopy, Fourier transform spectroscopy and spectroscopic ellipsometry. The simultaneous Raman and infrared activity of phonon modes and the observation of second harmonic generation establishes a non-centrosymmetric point group for Pbn+1(Ti0.5Fe0.5)nO3n+1−δ, a prerequisite for (but not proof of) ferroelectricity. No evidence of macroscopic ferromagnetism was found in SQUID magnetometry. The ultrafast optical response exhibited coherent magnon oscillations compatible with local magnetic order and additionally was used to study photocarrier cooling on picosecond timescales. An optical gap smaller than that of BiFeO3 and long photocarrier lifetimes may make this system interesting as a ferroelectric photovoltaic.
Similar content being viewed by others


Introduction
In magnetoelectric multiferroics, where magnetisation and electrical polarisation are strongly coupled1,2,3,4, there is potential for application as capacitors, transducers and actuators5 and in magnetic data storage and spintronic devices6. Coupled ferroelectric and ferromagnetic orders provide additional possibilities for device design, such as magnetic control of ferroelectric polarisation (and vice versa), or the development of 4-state resistive memory7. Ferroelectric photovoltaics are an emerging area of interest as, unlike traditional semiconductor p-n junctions, photovoltages in ferroelectrics are not limited by their band gap8.
Oxides with the ABO3 perovskite structure are remarkably capable in accomodating defects, owing to their high dielectric constants and structural stability. While a small deviation from the 1:1:3 stoichiometric ratio produces point defects, larger deviations can lead to extended (planar) defect formation. Non-conservative crystallographic shear planes can account for oxygen deficiency9,10 in AnBnO3n−2, while the Ruddlesden-Popper (RP) homologous series An+1BnO3n+1 has extra AO planes11. The unit cell of the RP phase consists in the z-direction of n ABO3 layers, a planar stacking fault [a/2, b/2, d] (where  ), another n ABO3 layers and a second stacking fault. The epitaxial growth of strained RP phases can result in novel phases where n can be used to tune functional properties. For instance, ferroelectricity has been reported in the RP series Srn+1TinO3n+1 only when n ≥ 4 and under tensile biaxial strain12. Conversely, out-of-phase boundaries (e.g. antiphase boundaries), where adjacent regions are offset by a fraction of a unit cell13, may be detrimental to long-range ferroic order.
), another n ABO3 layers and a second stacking fault. The epitaxial growth of strained RP phases can result in novel phases where n can be used to tune functional properties. For instance, ferroelectricity has been reported in the RP series Srn+1TinO3n+1 only when n ≥ 4 and under tensile biaxial strain12. Conversely, out-of-phase boundaries (e.g. antiphase boundaries), where adjacent regions are offset by a fraction of a unit cell13, may be detrimental to long-range ferroic order.
Room temperature multiferroics are relatively rare, as common mechanisms for ferroelectricity and ferromagnetism are mutually exclusive: ferroelectricity generally requires empty d-orbitals, while ferromagnetic ordering often needs partial d-orbital occupancy14. Bismuth ferrite (BiFeO3) is one of the few room temperature multiferroics and as such has been studied extensively15. Recently, PbTi1−xFexO3−δ has been reported to be multiferroic at room temperature16. PbTiO3 is a displacive ferroelectric17 that exhibits a tetragonal bulk phase with space group P4mm18,19. Ferroelectric polarisation arises from the displacement of Ti4+ ions with respect to the surrounding oxygens. The partial substitution of Fe on the B site allows magnetism to be induced, but causes a reduction in the tetragonal distortion c/a and a concommitant decrease in the remnant polarization18,20,21,22. Similar to other multiferroics the use of thin films23 and nanoparticles20,21,24 may enhance ferroelectricity and ferromagnetism in comparison to the bulk phase. Negligible leakage current and unsaturated ferroelectric hysteresis loops have been reported for thin films25. Evidence of magneto-electric coupling in PTFO has been observed with electrically written domains visible in magnetic force microscopy images23.
In this article we report a study of the structural, optical and vibrational properties of compressively strained thin films of the Ruddlesden-Popper phase Pbn+1(Ti0.5Fe0.5)nO3n+1−δ grown on LaAlO3 substrates. A structural characterization via X-ray diffraction and transmission electron microscopy (TEM) demonstrates the spontaneous formation of a Ruddlesden-Popper superstructure with  , with a complex microstructure that exhibits a modulated atomic density in the growth direction. X-ray photoelectron spectroscopy (XPS) was used to determine the films' stoichiometry and chemical environments. The simultaneous Raman and infrared activity of phonon modes, along with the observation of second harmonic generation, identify a polar point group compatible with ferroelectricity. The dielectric function was extracted from the far-infrared to the UV using THz time-domain spectroscopy, Fourier transform infrared (FTIR) spectroscopy and UV-visible ellipsometry.
, with a complex microstructure that exhibits a modulated atomic density in the growth direction. X-ray photoelectron spectroscopy (XPS) was used to determine the films' stoichiometry and chemical environments. The simultaneous Raman and infrared activity of phonon modes, along with the observation of second harmonic generation, identify a polar point group compatible with ferroelectricity. The dielectric function was extracted from the far-infrared to the UV using THz time-domain spectroscopy, Fourier transform infrared (FTIR) spectroscopy and UV-visible ellipsometry.
Results
Structure and composition
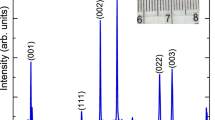
Thin films of PTFO with nominal thicknesses of 100 nm, 200 nm and 300 nm were deposited on (001) LaAlO3 (LAO) by pulsed laser deposition (see Methods) and are referred to herein as PTFO-100, PTFO-200 and PTFO-300. High-resolution 2θ − ω X-ray diffraction scans were taken to examine the crystal structure of the films. In Figure 1a the pseudocubic (002) peak of the LaAlO3 substrate (space group  , a = 3.789Å, α = 90.12°)26 is visible at 2θ = 48°. For each film a sequence of diffraction peaks at lower 2θ (larger c) than the substrate peak is evident. The wider range 2θ − ω scan in Figure 1b indicates diffraction peaks up to the substrate's (004) peak for the PTFO-100 sample. While the (002) film peaks are at lower 2θ than the LAO peak, those for (001) straddle the substrate peak (23.5°) with one of the four at a higher angle. The data resemble the diffraction pattern of a superlattice owing to their regular spacing in 2θ.
, a = 3.789Å, α = 90.12°)26 is visible at 2θ = 48°. For each film a sequence of diffraction peaks at lower 2θ (larger c) than the substrate peak is evident. The wider range 2θ − ω scan in Figure 1b indicates diffraction peaks up to the substrate's (004) peak for the PTFO-100 sample. While the (002) film peaks are at lower 2θ than the LAO peak, those for (001) straddle the substrate peak (23.5°) with one of the four at a higher angle. The data resemble the diffraction pattern of a superlattice owing to their regular spacing in 2θ.

X-ray characterisation of Pbn+1(Ti0.5Fe0.5)nO3n+1−δ films: (a) 2θ scans around the LaAlO3 substrate's (002) peak for PTFO-100, -200 and -300 films (solid lines). The asterisk marks the PTFO's Bragg peak corresponding to  . The dashed line is the model described in the text. (b) Wide angle 2θ scan for PTFO-100. (c) Shallow angle x-ray reflectivity for PTFO-100 from experiment (solid line) and simulation (dashed line). (d) A symmetric 2θ − ω scan for PTFO-100 around the (003) substrate peak, at ϕ = 0. (e), (f) Reciprocal space maps for PTFO-100 around the (103) and (113) LaAlO3 substrate peaks (indicated by the dashed lines), respectively. qx, q∥ and qz denote the scattering wavevectors in the [100], [110] and [001] directions of the substrate.
. The dashed line is the model described in the text. (b) Wide angle 2θ scan for PTFO-100. (c) Shallow angle x-ray reflectivity for PTFO-100 from experiment (solid line) and simulation (dashed line). (d) A symmetric 2θ − ω scan for PTFO-100 around the (003) substrate peak, at ϕ = 0. (e), (f) Reciprocal space maps for PTFO-100 around the (103) and (113) LaAlO3 substrate peaks (indicated by the dashed lines), respectively. qx, q∥ and qz denote the scattering wavevectors in the [100], [110] and [001] directions of the substrate.
Conventional TEM and ADF-STEM images (see Methods) of the atomic structure of the PTFO-100 film are reported in Fig. 2. Bright and dark wave-like patterns can be seen in the low-magnification image of Fig. 2a, with a periodicity in the growth direction of about 4 nm. The ADF image, showing the Pb columns most clearly (Ti atomic columns give weaker contrast), demonstrates that the “waves” consist of areas that appear perovskite-like (labelled 1 and 1′) and others that appear rock-salt-like (areas 2). The A- and B- cation site positions can be seen to reverse on opposite sides of the rock-salt-like layers: rather than lying on the solid white vertical lines in Fig. 2b, which run through the perovskite A-sites in area 1, the Pb atoms in areas 1′ are instead found on the dashed lines (B-sites of area 1). EELS spectra taken in areas 1, 2 and 1′ showed no difference in Fe or Ti composition (Supplementary Fig. S.1), implying that the different regions have similar stoichiometry. Further, TEM-EDX maps over regions up to 75 nm wide showed no variation in composition across each film (Supplementary Fig. S.2).

Transmission electron microscopy of 100 nm PTFO film.
(a) Conventional TEM showing ‘wave’ patterns. (b) ADF-STEM image showing that ‘waves’ consist of a perovskite-like areas (labelled 1 and 1′, blue and red squares) and a rock-salt-like areas (labelled 2, yellow squares). Cyan lines mark the boundaries between these regions. The vertical white solid lines run through the perovskite A site in area 1, becoming the B site in area 1′. The horizontal white dashed lines show atomic planes separated by a stacking fault with  . (c) ADF-STEM image showing a change in c, present in region 1 and 1′. (d) Schematic of structure in (010) plane (left, corresponding to plane of TEM image) and (100) plane (right). A Ruddlesden-Popper unit cell for n = 8 is shaded in blue, while another, offset by an antiphase boundary (see text), is shaded in red. Projection of the right-hand cartoon along [010] yields the left-hand schematic, creating areas where Pb ions appear to be on both the A and B sites.
. (c) ADF-STEM image showing a change in c, present in region 1 and 1′. (d) Schematic of structure in (010) plane (left, corresponding to plane of TEM image) and (100) plane (right). A Ruddlesden-Popper unit cell for n = 8 is shaded in blue, while another, offset by an antiphase boundary (see text), is shaded in red. Projection of the right-hand cartoon along [010] yields the left-hand schematic, creating areas where Pb ions appear to be on both the A and B sites.
The structure can be understood as consisting of a RP phase with  in the [001] growth direction, as pictured schematically in Fig. 2d. The left-hand panel pictures the (010) plane, corresponding to the plane of the TEM specimen, while the right-hand panel depicts the (100) plane. Two RP unit cells (dimensions a,a,C) are shaded in blue and red, where red atoms have been shifted by C/2 in the [001] direction (with respect to the blue atoms) by an antiphase boundary. Projecting the right-hand panel along the [010] direction results in the left-hand panel, which contains rock-salt-like regions (areas 2), as observed in TEM, with Pb ions on the A and B sites. The wave-like structures in Fig. 2a and b may result from either (a) a sharp rock-salt-like planar fault, lying on a plane inclined to the electron beam or (b) a diffuse fault with an extended rock-salt structure. Since TEM images are a projection of the three-dimensional structure it is hard to distinguish between these two possibilities.
in the [001] growth direction, as pictured schematically in Fig. 2d. The left-hand panel pictures the (010) plane, corresponding to the plane of the TEM specimen, while the right-hand panel depicts the (100) plane. Two RP unit cells (dimensions a,a,C) are shaded in blue and red, where red atoms have been shifted by C/2 in the [001] direction (with respect to the blue atoms) by an antiphase boundary. Projecting the right-hand panel along the [010] direction results in the left-hand panel, which contains rock-salt-like regions (areas 2), as observed in TEM, with Pb ions on the A and B sites. The wave-like structures in Fig. 2a and b may result from either (a) a sharp rock-salt-like planar fault, lying on a plane inclined to the electron beam or (b) a diffuse fault with an extended rock-salt structure. Since TEM images are a projection of the three-dimensional structure it is hard to distinguish between these two possibilities.
Angle-dependent X-ray photoelectron spectroscopy (XPS) on the PTFO films identified two contributions to the Pb 4f peaks, as reported in Supplementary Fig. S.3a. Component peaks of a surface lead oxide, with enhanced contributions at a shallower take-off-angle, are present at 0.6–0.7 eV higher binding energy than the principal peaks from the perovskite-like PTFO film. As lead is volatile, lead oxide-rich surface regions can form spontaneously during PLD growth27. The observed binding energy is consistent with Pb3O4. Detailed fits to the XPS Fe 2p3/2, 2p1/2 and ‘shake-up’ satellite spectra were consistent with Fe3+ ions (Supplementary Fig. S.3b).
The A:B:O ratio from XPS was (1.2 ± 0.3):1:(2.4 ± 0.3) relative to the B-site composition, after the contribution from the surface lead oxide was discluded. This is consistent with an oxygen-deficient RP phase in the limit of large n, when the RP phase tends to the ABO3 perovskite structure. Local EDX spectra yielded the composition Pb1.75±0.18Ti0.53±0.05Fe0.5O3.44±0.34 relative to Fe. The relative excess of Pb and O in the EDX composition may arise from lead oxide surface layers, which (unlike the XPS analysis) could not be resolved separately by EDX and which may form during TEM specimen preparation. The Ti/Fe ratio determined from EDX was 1.06 ± 0.1, consistent with the 1:1 stoichiometry of the target. Considering ionic oxidation states only (Pb2+, Ti4+, Fe3+, O2−), charge balance requires that δ = 2 when n = 8 for Pbn+1(Ti0.5Fe0.5)nO3n+1−δ. An oxygen deficiency can be readily accomodated in oxide thin films by planar defects such as non-conservative boundaries9,10.
Returning to the XRD results, the position of each film's diffraction peaks can be assigned to the Bragg and superstructure peaks as follows. The * symbols in Fig. 1b denote the calculated Bragg angles for the PTFO-100 film assuming the perovskite cell has c = 4.437 Å. The adjacent satellite peaks are at angles consistent with a superlattice period Λ = 35.8 Å as determined using28:

where λ = 1.540598 Å is the X-ray wavelength and θm and θn are the angular positions of adjacent satellite peaks, with orders m and n29. Table 1 lists c and Λ for the PTFO films assuming that m − n = 1. This results in  , which is half the unit cell dimension C of the RP phase observed in TEM. The dashed line in Fig. 1a is a model calculation of the X-ray diffraction pattern of an RP phase on LAO (including Pb and La atoms only), assuming that n = 8, c = 4.43 Å and that the stacking fault had d = 0.12 Å. The model does not include disorder (such as variations in c or n) and hence has narrower diffraction peaks than observed experimentally. Figure 1d is a symmetric 2θ − ω scan for the PTFO-100 film around (003) at ϕ = 0, while panels e and f are reciprocal space maps around the (103) and (113) LAO substrate peaks respectively (indicated by the dashed lines). The RSMs show that the film's a = 3.90 Å lattice constant is slightly larger than that of the LAO substrate (a = 3.789 Å) and that there is negligible polycrystalline texture.
, which is half the unit cell dimension C of the RP phase observed in TEM. The dashed line in Fig. 1a is a model calculation of the X-ray diffraction pattern of an RP phase on LAO (including Pb and La atoms only), assuming that n = 8, c = 4.43 Å and that the stacking fault had d = 0.12 Å. The model does not include disorder (such as variations in c or n) and hence has narrower diffraction peaks than observed experimentally. Figure 1d is a symmetric 2θ − ω scan for the PTFO-100 film around (003) at ϕ = 0, while panels e and f are reciprocal space maps around the (103) and (113) LAO substrate peaks respectively (indicated by the dashed lines). The RSMs show that the film's a = 3.90 Å lattice constant is slightly larger than that of the LAO substrate (a = 3.789 Å) and that there is negligible polycrystalline texture.
The ADF-STEM image of Fig. 2c indicates that areas of the perovskite-like layers 1 and 1′ have a reduced tetragonal distortion c/a. This changes the atomic density in these layers, creating a periodic modulation in the refractive index that can be seen in shallow-angle X-ray reflectivity scans [solid line in Fig. 1c]. The experimental data for the PTFO-100 sample exhibits a weak peak at 2θ = 2.45° indicative of a superlattice with period Λ = 37.6 Å (dashed line shows model, see Methods), consistent with the superlattice period calculated directly from the diffraction peaks (Table 1). Also evident in Fig. 2c are tilts of the local perovskite cell, which may result in the wave-like structure of Fig. 2a.
Linear and non-linear optics in the visible-UV
The UV-visible absorption coefficient α at normal incidence and room temperature (see Methods) is reported in Figure 3(a). The absorption coefficient varies with the film thickness and an increase in the absorption edge (~2.5 eV) is evident with reduced film thickness. The shift is most pronounced for the PTFO-100 film and coincides with the increased tetragonal distortion for thinner films [Fig. 1(a) and Table 1]. In the region below the band gap, before the film begins to absorb strongly, the reported absorption coefficient exhibits oscillations that are artefacts caused by thin film interference.

Linear and non-linear optics of PTFO films: (a) UV-visible absorption coefficient α from transmission at normal incidence (solid lines) and from ellipsometry at various angles of incidence (dashed lines). Vertical lines indicate pump and probe energies used for ultrafast Pump-Probe (PP) spectroscopy and pump energy for Raman. (b) Real ( , solid lines) and imaginary (
, solid lines) and imaginary ( , dashed) parts of complex dielectric function in the UV-visible range, as determined from ellipsometry. Shaded areas show the contribution of each mode to
, dashed) parts of complex dielectric function in the UV-visible range, as determined from ellipsometry. Shaded areas show the contribution of each mode to  . (c), (d) and (e) show the SHG radiation patterns for PTFO-100, -200 and -300 respectively, as a function of sample azimuthal angle ϕ (ϕ = 0 corresponding to p-polarised input) for θ = 45°. Data are shown for p-polarised (“p-out”) and s-polarised (“s-out”) detection. In (c) and (d) the dashed black lines show the SHG intensities expected for a tetragonal phase from the model described in the text. (f) SHG (blue) can only be detected from the “detection volumes” (blue shaded areas) defined by the film's finite absorption depth at the second harmonic.
. (c), (d) and (e) show the SHG radiation patterns for PTFO-100, -200 and -300 respectively, as a function of sample azimuthal angle ϕ (ϕ = 0 corresponding to p-polarised input) for θ = 45°. Data are shown for p-polarised (“p-out”) and s-polarised (“s-out”) detection. In (c) and (d) the dashed black lines show the SHG intensities expected for a tetragonal phase from the model described in the text. (f) SHG (blue) can only be detected from the “detection volumes” (blue shaded areas) defined by the film's finite absorption depth at the second harmonic.
The real and the imaginary part of the dielectric function ( ) for PTFO-100, -200 and -300 extracted from ellipsometry measurements are shown in Fig. 3(b). The imaginary part was calculated using the Tauc-Lorentz model30, which has previously been applied to amorphous semiconductors30 and to the multiferroic BiFeO331. The Tauc-Lorentz model accounts for a finite density of states below the bandgap, making it sensitive to the trap levels that play an important role in the bulk photovoltaic effect in ferroelectrics8,32,33.
) for PTFO-100, -200 and -300 extracted from ellipsometry measurements are shown in Fig. 3(b). The imaginary part was calculated using the Tauc-Lorentz model30, which has previously been applied to amorphous semiconductors30 and to the multiferroic BiFeO331. The Tauc-Lorentz model accounts for a finite density of states below the bandgap, making it sensitive to the trap levels that play an important role in the bulk photovoltaic effect in ferroelectrics8,32,33.
Two Tauc-Lorentz oscillators with a common energy gap Eg were used for each film. The determined film thicknesses were in good agreement with their nominal values, as summarised in Table 2, which also gives the parameters for the Tauc-Lorentz oscillators. Oscillators are represented graphically in Figure 3(b) by the shaded curves. As the film thickness is increased the central energy of the individual oscillators is blue-shifted. The dashed data in Figure 3(a) is the absorption coefficient as determined from ellipsometry. Note that no oscillatory artefact in the absorption is present below the band gap in the ellipsometry results (unlike α from UV-visible spectroscopy) because thin film interference is taken into consideration. Clear differences between the UV-visible transmission results (obtained at normal incidence) and the ellipsometry findings (at large angles) above the absorption edge may arise from the film's optical anisotropy in the growth (z) direction. A tetragonal phase will have a dielectric tensor with  and this was not taken into account in the analysis of the ellipsometry data.
and this was not taken into account in the analysis of the ellipsometry data.
The second harmonic generation (SHG) intensity Ip,s(ϕ) was obtained as a function of the polarisation angle ϕ of the incident 800 nm pump pulse (Methods), for p-polarised and s-polarised detection, as reported in Figs. 3(c)–(e) for the PTFO-100, -200 and -300 films. The clear SHG signal may arise from either (a) a non-centrosymmetric crystal structure at room temperature for the films, or (b) an interfacial contribution due to the break in translational symmetry (surface SHG). The peak SHG intensity Ip(ϕ) was found to be in the ratio 1.0:8.0:0.7 for the PTFO-100, -200 and -300 films. The strong increase in SHG intensity between the PTFO-100 and PTFO-200 film indicates that any contribution from surface SHG is negligible in comparison to the SHG signal from the polar film, as a surface SHG contribution should not vary with film thickness. The observed intensity ratio can be qualitatively understood with reference to Fig. 3(f). The second harmonic (blue dashed lines) is generated throughout the film by the fundamental (red dashed lines) as the absorption coefficient at 800 nm is negligible. While the SHG intensity should increase with the length squared for a transparent medium, absorption in the films at 3.1 eV (400 nm) means that not all of the generated SHG is detected. Rather, the detected second harmonic comes from the region close to the film/air interface and for beams propagating in the correct direction, labelled the “detection volume” in Fig. 3(f). The depth of the detection volume can therefore be limited to the absorption depth, which is 1/α = 200 nm, 111 nm and 91 nm for the PTFO-100, -200 and -300 films [from Fig. 3(a)]. Thus, for the PTFO-100 film the SHG signal can come from the whole film thickness, while for the PTFO-300 film only the top 91 nm contributes. Since the thickest film exhibits smaller tetragonality c/a and strain relaxation in the detection volume (as evidenced by the XRD results), the SHG intensity is therefore reduced for PTFO-300.
The SHG ϕ-plots can be qualitatively understood as follows. Since the sample is rotated around the vertical axis by θ = 45° there is a maximum component of the electric field of the incident beam along the c-axis (spontaneous polarisation direction) for a p-polarised fundamental, leading to maxima in the SHG intensity at ϕ = 0 and ϕ = 180° and a double-lobed shape. Quantitatively, for a tetragonal phase with point group 4 mm, the ϕ-dependence of the detected SHG intensity is Ip(ϕ) = (A cos2 ϕ + B sin2 ϕ)2 and Is(ϕ) = C sin2 2ϕ, where the constants A, B and C are linked to the SHG coefficients d31 and d15, as described by Kumar et al.34 for tetragonal-phase BiFeO3. The black dashed lines in Figs. 3(c)–(d) show such fits, which are in good agreement with experiment for the two thinner films. For the PTFO-300 Ip,s(ϕ) varied somewhat between different spots on the sample [solid and dashed lines in Fig. 3(e)] and the maximum intensity was away from p-polarised incident light for p-polarised detection. The distortion of the crystal structure due to strain relaxation near the surface (in the detection volume), such that the spontaneous polarisation is no longer along [001], may explain shape of the Ip,s(ϕ) for the PTFO-300 film. Similar results were obtained on polydomain 4 mm (Ba,Sr)TiO3 films with a deviation in ferroelectric polarisation from the c-axis35. Alternatively, anisotropy of the dielectric function at the second harmonic's wavelength may modify the polarisation state detected experimentally from the predicted pattern.
Ultrafast optical response
Photoinduced changes in reflectivity ΔR result from a modified refractive index Δn (at the probe wavelength) caused by changes in the occupancy of electronic states. Time-resolved reflectivity thus permits electronic generation and relaxation processes to be examined. Further, coherent oscillations in ΔR can result from magnons in multiferroics36,37. The observed transients for the PTFO films (Fig. 4) comprise an electronic component with a sharp rise and a non-exponential decay for both films. This electronic component is described in more detail in the Discussion.

Pump probe spectroscopy of PbTiFeO3 films: (a) Raw pump probe for PTFO-300 (blue) and PTFO-200 (green). (b) Extracted oscillation for PTFO-300 with (i) no oscillation, (ii) oscillation and (iii) 180° phase shifted oscillation. (c) FFT of oscillation in b(ii). (d) Schematic diagram of carrier density model.
After numerical removal of the electonic contribution to ΔR a distinct oscillatory component superimposed on the reflectivity transient is observed for PTFO-300, as reported in Fig. 4(b), at a frequency of around 75 GHz [Fig. 4(c)]. The different curves (i)–(iii) correspond to different locations on the sample. While ΔR appeared as in curve (ii) in the majority of positions, in a smaller number of cases either no oscillations [curve (i)] or oscillations with a 180° phase shift [curve (iii)] were observed. Acoustic phonons can contribute to ΔR for oxide thin films37. The oscillation frequency depends upon the acoustic phonon velocity and thus changes when the acoustic pulse propagates from the film to the substrate. Acoustic phonons in the substrate may create the peaks at low frequency (and from long time delays) in Fig. 4(c). However, the high frequency (75 GHz) feature cannot result from the acoustic phonon mechanism, as the oscillation is present at all time delays. In a previous study of magnons in BiFeO3 thin films a mode at a comparable frequency was found to disappear above the Néel temperature37. Both the PTFO films reported here and the BiFeO3 films previously studied37 had LaAlO3 substrates and therefore have comparable in-plane lattice constants and Fe-Fe distances (assuming that there are regions of the PTFO samples with Fe ions in adjacent cells). It is thus plausible that the oscillation here is also a magnon. To confirm this, demonstrating that it disappears above the Néel temperature is desirable and will be the subject of further study. The occasional absence of the magnon and the 180° phase shift could be attributed to ferromagnetic domains or sample inhomogeneity.
IR active phonons
Terahertz time domain spectroscopy38,39 was used to examine the dielectric function in the range 1–13 meV. The spectral transmission is reported in Fig. 5(a) and is flat and featureless for the PTFO-100 and PTFO-200 samples in the range below 10 meV. Above 10 meV all samples exhibit a sharp rise in transmission, an artifact which arises from the LaAlO3 substrate, which acts as a half wave plate at these frequencies40. The response for the PTFO-300 film displays a prominent reduction in transmission around 9 meV in Fig. 5(a). The dashed line indicates the transmission calculated for a thin film of PTFO on LaAlO3, assuming that the dielectric function of the film includes a phonon at 9.1 meV (2.2 THz) as described by Equation 2 (Methods) with linewidth Γ = 3.3 meV,  and
and  . Here,
. Here,  was chosen to match
was chosen to match  from the FTIR results.
from the FTIR results.

Infrared active vibrational modes in Pbn+1(Ti0.5Fe0.5)nO3n+1−δ films: (a) Amplitude transmission |T| in the far-infrared (from THz-TDS, see Methods) relative to that through a bare LAO substrate. (b) Power reflectivity from FTIR spectroscopy (solid lines). Models (dashed lines) are described in the text.
FTIR reflectivity spectra are presented in Figure 5(b) (solid lines). The substrate data (black line) displays three Rehstrahlen bands. For the PTFO films the reflectivity lowers substantially in the areas marked by the arrows and this reduction is more pronounced as the film thickness increases. This is a consequence of phonon modes in the film within the Rehstrahlen bands of the substrate. Reflectivity spectra were modelled (Methods) using two Drude-Lorentz oscillators for the films (parameters given in Table 2) and a 6-phonon model for the LaAlO3 substrate (similar to results reported elsewhere)41,42, producing the dashed lines. Oscillators in the PTFO films at 29.5 meV and 64.7 meV (see Table 2) are needed to create a good fit to experiment. There is little shift in the mode frequencies with thickness.
Raman active phonons
Raw Raman spectra for the PTFO samples and a (001)-oriented LaAlO3 reference sample obtained under 442 nm (2.81 eV) excitation are presented in Fig. 6(a). As the films are not strongly absorbing at this wavelength, there is a strong component from the LaAlO3 substrate (black line). In contrast, using 325 nm (3.82 eV) excitation, where the film absorbs strongly, yields a dominant Raman signal from the PTFO films [coloured lines in Figure 6(b)], but over a more limited spectral range (see Methods). For the data obtained at 442 nm the subtraction of the substrate's contribution43 permits the Raman spectra of the PTFO films to be seen [coloured lines in Figure 6(c)]. The narrow gaps correspond to the sharp Raman active modes  of the substrate, at 15.3 meV, 18.9 meV, 25.4 meV, 57.5 meV and 60.3 meV, corresponding well to previous reports44,45.
of the substrate, at 15.3 meV, 18.9 meV, 25.4 meV, 57.5 meV and 60.3 meV, corresponding well to previous reports44,45.

Raman spectra of PTFO films and LAO substrate: (a) Raman intensity I under 442 nm excitation. (b) I for 325 nm excitation. The dashed black line indicates a two-Lorentzian fit to the PTFO-200 spectra. (c) Raman intensity under 442 nm excitation after subtracting the substrate's contribution. The dashed black line is the model for PTFO-200 as described in the text and Table 2, where black arrows indicate individual oscillators. The lengths of the arrows represent the oscillator strengths, while the widths of the arrow heads denote the widths Γ. The thin back lines (bottom) are the data of Sun et al.22 for x = 0 and x = 0.1.
The Raman modes of the films are broader than those previously seen for bulk PTFO (with x = 0.05 and x = 0.1)22, which are reproduced by the solid thin black lines in Fig. 6(c) along with their mode assignments. Further, the Raman active modes of Pbn+1(Ti0.5Fe0.5)nO3n+1−δ are shifted in energy in comparison to those at lower x22. The large changes in c evidenced by TEM will also alter the phonon eigenfrequencies and this disorder will broaden the Raman resonances. As a does not vary substantially, eigenmodes that are IR-active in the plane may not be broadened as significantly. The PTFO-200 data was modelled by a series of Lorentzians (black dashed lines in panels b and c), where the arrows in Fig. 6(c) indicate the position and strength of the individual oscillators for the fits to the substrate-subtracted 442 nm data. For the fit in Fig. 6(b) [325 nm excitation, PTFO-200 film] only two Lorentzians were used, to better highlight the peaks at 65 meV and 97 meV.
Importantly, for ferroelectric media without an inversion centre vibrational modes can be both Raman and infrared active, as for PbTiO346. For the two IR-active modes observed for Pbn+1(Ti0.5Fe0.5)nO3n+1−δ at 29.5 meV and 64.7 meV in the FTIR results there are corresponding Raman active modes at 29 and 65 meV visible in the 442 nm Raman data [Figure 6(c)] and at 65 meV in the 325 nm Raman data [Figure 6(c)]. This discludes a centrosymmetric crystal structure for the Pbn+1(Ti0.5Fe0.5)nO3n+1−δ films, as in a material possessing a center of symmetry no mode can be both infrared and Raman active by the rule of mutual exclusion.
Discussion
Below, we contrast our results for the Ruddlesden-Popper phase Pbn+1(Ti0.5Fe0.5)nO3n+1−δ with literature reports for perovskite phase PTFO and the model multiferroic BFO.
For PTFO powder samples a single perovskite phase is retained only for Fe fractions x ≤ 0.3, while x > 0.3 exhibits secondary phases visible as additional XRD peaks and consisting mainly of PbFe12O1918. Here, the extra peaks visible in the XRD ω − 2θ scans arise from the RP superstructure and no evidence of such secondary phases was observed via XRD or TEM.
To examine whether epitaxial strain or the oxygen-rich growth conditions are responsible for the formation of the RP phase it is desirable to examine films grown on different substrates. The deposition rate and substrate temperature can have a strong influence on the superlattice period, as reported for RP phases of barium stannate47. Crystallographic shear planes that lower oxygen content exhibit wave-like features and kinks in Pb2Sr2Bi2Fe6O16 crystals10, while 2D and 3D RP stacking faults have been witnessed in LaNiO3/LaAlO3 superlattices48 and Srn+1TinO3n+1 ferroelectric films12.
In-plane and out-of-plane magnetisation measurements using a SQUID magnetometer (Methods) on the PTFO-300 film showed no evidence of ferromagnetic order for temperatures from 5 K to 300 K. Diamagnetic contributions from the LAO substrate and the sample holder were seen, as reported in Supplementary Fig. S.4. At low temperatures an increased magnetisation consistent with paramagnetic rare-earth impurity ions in the LAO substrate was observed. The lack of macroscopic ferromagnetic order contrasts with bulk PTFO and thin-films on alternative substrates and may be a consequence of the spontaneous RP phase of these films. Alternatively, the disorder introduced by the variations in c and unit cell tilt throughout the film may disrupt magnetic order, as the strength of the superexchange interaction depends sensitively on Fe-O-Fe bond angles.
The direct optical absorption edge of the Pbn+1(Ti0.5Fe0.5)nO3n+1−δ films, at about 2.5 eV, is comparable to that of rhombohedral BFO (2.7 eV) and below that of tetragonal BFO (3.1 eV)31. The onset of absorption, parameterised by the Tauc bandgap Eg, is lower for Pbn+1(Ti0.5Fe0.5)nO3n+1−δ (<2 eV) than BFO (2.14 eV and 2.30 eV for rhombohedral and tetragonal phase films)31. A detailed comparison between the UV-visible and ellipsometry results on Pbn+1(Ti0.5Fe0.5)nO3n+1−δ reveals that there is a redshift in the absorption edge from UV-visible transmission for increasing film thickness (lower c), while the individual oscillator energies from ellipsometry blue shift. This can be understood as a consequence of the changes to oscillator widths Γ and amplitudes A: a slight increase in oscillator energies is swamped by a marked increase in Γ and A of the lowest mode. The increase in Γ could occur due to a larger variation in c throughout the thicker films.
Similar ultrafast reflectivity transients (with a rapid rise and a non-exponential decay) have been reported in recent studies of bulk BFO49,50 and thin films grown on a variety of substrates37,51,52. The rapid rise in reflectivity after excitation is attributed to the transfer of electrons between states. In BFO this is O2p → Fe3d53; similar transitions to Fe3d or Ti3d are expected here. The electronic response for the PTFO-200 film is reduced in comparison to that of the PTFO-300 sample because of its reduced absorption at the pump energy [Fig. 3(a)].
The subsequent electronic decay can be approximated by the sum of two exponentials,  , with lifetimes τ1 = 113 ps and τ1 = 78 ps for PTFO-200 and -300 respectively and τ0 > 1 ns. However, such a biexponential model would signify that two independent photoexcited electron populations contribute to ΔR. As an alternative, a more physical model that includes carrier cooling between two levels54 was also found to describe the electronic behavior well. In this approach electrons are photoexcited with density N1 into an upper level 1 and relax with rate N1/τ1 (corresponding to the cooling rate of hot electrons) to lower level 0, as illustrated in the schematic in Fig. 4(d). This approach gave τ1 = 89 ps and τ0 = 2150 ps for PTFO-200 and τ1 = 63 ps, τ0 = 1350 ps for PTFO-300. The greater lifetime of the fast component for PTFO-200 can be explained by the closer proximity of the pump energy to the band edge, where cooling rates are lower. The τ0 > 1 ns lifetime for photocarrier recombination makes Pbn+1(Ti0.5Fe0.5)nO3n+1−δ potentially useful in optoelectronic applications that require slow population decay, such as ferroelectric photovoltaics.
, with lifetimes τ1 = 113 ps and τ1 = 78 ps for PTFO-200 and -300 respectively and τ0 > 1 ns. However, such a biexponential model would signify that two independent photoexcited electron populations contribute to ΔR. As an alternative, a more physical model that includes carrier cooling between two levels54 was also found to describe the electronic behavior well. In this approach electrons are photoexcited with density N1 into an upper level 1 and relax with rate N1/τ1 (corresponding to the cooling rate of hot electrons) to lower level 0, as illustrated in the schematic in Fig. 4(d). This approach gave τ1 = 89 ps and τ0 = 2150 ps for PTFO-200 and τ1 = 63 ps, τ0 = 1350 ps for PTFO-300. The greater lifetime of the fast component for PTFO-200 can be explained by the closer proximity of the pump energy to the band edge, where cooling rates are lower. The τ0 > 1 ns lifetime for photocarrier recombination makes Pbn+1(Ti0.5Fe0.5)nO3n+1−δ potentially useful in optoelectronic applications that require slow population decay, such as ferroelectric photovoltaics.
Large dielectric constants  (reported here in the THz range) are often observed in ferroelectrics55 and incipient ferroelectrics. The extra mode seen in THz-TDS for PTFO-300 is not visible for the 100 nm or 200 nm samples. This may be a consequence of the experimental sensitivity, as thinner films produce smaller transmission changes, or be a feature of Pbn+1(Ti0.5Fe0.5)nO3n+1−δ films above a certain thickness.
(reported here in the THz range) are often observed in ferroelectrics55 and incipient ferroelectrics. The extra mode seen in THz-TDS for PTFO-300 is not visible for the 100 nm or 200 nm samples. This may be a consequence of the experimental sensitivity, as thinner films produce smaller transmission changes, or be a feature of Pbn+1(Ti0.5Fe0.5)nO3n+1−δ films above a certain thickness.
Methods
PLD
Pulsed laser deposition (PLD) was used to deposit films onto (001)-oriented LaAlO3 substrates. Target compositions were PbTi1−xFexO3 with x = 0.5. A laser fluence and frequency of 2 J/cm2 and 10 Hz, an oxygen pressure of 0.5 mbar, a substrate temperature of 625°C and a substrate-target distance of 5 cm were employed. The deposition time was varied to get films with different thickness.
TEM
Aberration-corrected transmission electron microscopy (TEM) was performed using an ARM200F system equipped with energy-dispersive X-ray (EDX) and electron energy loss spectroscopy (EELS) capabilities.
XPS
An Omicron XM1000 monochromated Al Kα x-ray source was used to illuminate the sample, with photoelectrons collected in an Omicron SPHERA analyser using an estimated sampling radius of 1.1 mm. Due to the insulating nature of the samples, an Omicron CN10 charge neutraliser was used to prevent surface charging. All binding energies were referenced to the C 1s peak from atmospheric contamination at 284.6 eV. All data were analysed using the CasaXPS package, with compositional analysis facilitated via determination of the analyser transmission function, calculated using polycrystalline Ag, Au and Cu foils.
XRD
A four-circle x-ray diffractometer (PANalytical Xpert Pro MRD) with a Cu source and a 4-bounce hybrid monochromator was used to give pure K-α1 radiation (λ = 1.540598 Å). A Pixcel detector was used in scanning mode to collect the 2θ − ω diffraction scans. The reciprocal space maps were also collected in scanning mode as a collection of 2θ − ω scans, which were then converted into reciprocal space. Shallow-angle reflectivity data were modelled by the Genx software56 assuming the sample comprised a superlattice with two layers, one 2.2 nm layer (with c = 4.68 Å) and another 1.56 nm layer (c = 3.91 Å), with superlattice period 3.76 nm. The density in each bilayer was set by c.
SQUID
The in-plane and out-of-plane magnetisation was measured with a SQUID magnetometer (Quantum Design MPMS-5S) from 5 K to 300 K in a magnetic field of up to B = 5 T.
UV-visible transmission & ellipsometry
Room temperature transmittance spectra T were taken in the range 1–5 eV a using Perkin-Elmer LAMBDA 1050 UV/Vis/NIR Spectrophotometer at normal incidence. The absorption coefficient, α = −ln(T/d), was determined, where d is the film thickness from ellipsometry (see Table 2). Ellipsometry spectra (Δ, Ψ) for 3 angles of incidence (θ = 60°, 65° and 70°) were taken taken in the range 1–5.5 eV using a Horiba Scientific UVISEL Ellipsometer. The experimental spectra in Δ and Ψ were then fitted using a Tauc-Lorentz oscillator model30 to extract the dielectric function,  . The Tauc-Lorentz model is made up of oscillators that share a common Tauc gap, Eg. The real part of the dielectric function,
. The Tauc-Lorentz model is made up of oscillators that share a common Tauc gap, Eg. The real part of the dielectric function,  was calculated from the imaginary part,
was calculated from the imaginary part,  using the Kramers-Kronig relations.
using the Kramers-Kronig relations.
SHG and pump-probe reflectivity
A mode-locked Ti:sapphire laser oscillator (4 MHz, 650 nJ, 50 fs) provided the fundamental beam (800 nm or 1.55 eV) for SHG and time-resolved reflectivity. For SHG, a PMT placed after an analyser and short pass filters was used to detect the second harmonic. Samples were at θ = 45° from the normal and with the incident beam polarised at angle ϕ (ϕ = 0 corresponding to p-polarisation). For the pump-probe reflectivity experiments the fundamental was doubled in frequency to 400 nm (3.1 eV) to photoexcite the multiferroic films at a fluence of 60 μJcm−2. The transient optical reflectivity subsequent to excitation was detected using a time-delayed fraction of fundamental beam, which was incident on the sample at close to normal incidence. The absorption depth of the samples for the pump and probe are 65–355 nm at λ = 400 nm and greater than 1.5 μm at λ = 800 nm.
Infrared and THz dielectric function
Terahertz time-domain spectroscopy38,39 was used to examine the dielectric function in the range 1–13 meV, from amplitude and phase resolved transmission measurements. The THz pulse was generated by a 800 nm, 20 fs pulse from a Ti:sapphire laser focused onto a GaAs photoconductive switch and detected by electro-optic sampling. The spectral transmission was calculated from the FFT of the time-domain data, using blank LaAlO3 as the reference. Data were averaged over four possible orientations of the PTFO samples and a blank substrate (VV, VH, HV, HH, where H/V represent horizontal or vertical orientation of the sample and substrate respectively). This removes the influence of substrate anisotropy40.
FTIR reflectivity spectra were taken using a Bruker Vertex 70v spectrometer at an angle of incidence of 11°. A globar light source was used, along with a KBr beamsplitter/DLaTGS detector for the mid-IR range (50–1000 meV) and a Si beamsplitter/DTGS detector for the far-IR range (15–48 meV). The RefFit software (http://optics.unige.ch/alexey/reffit.html) was used to model the reflectivity of thin film PTFO on LaAlO3. The Drude-Lorentz dielectric function was used, with the form:

where  for each mode i.
for each mode i.
Raman spectroscopy
Raman spectra were taken using a Renishaw inVia Reflex Raman microscope with an excitation wavelength of 442 nm (2.81 eV) and focused with a 20× objective. UV-Raman spectra were taken under excitation at 325 nm (3.82 eV) and with a 40× objective. The Raman filters used for the 442 nm and 325 nm lasers cut out all signal below 10 meV and 52 meV, respectively, limiting the spectral range at low energies.
References
Kimura, T. et al. Magnetic control of ferroelectric polarization. Nature 426, 55–58 (2003).
Wang, J. et al. Epitaxial BiFeO3 multiferroic thin film heterostructures. Science 299, 1719–1722 (2003).
Spaldin, N. A. & Fiebig, M. Materials science. The renaissance of magnetoelectric multiferroics. Science 309, 391–2 (2005).
Ramesh, R. & Spaldin, N. A. Multiferroics: progress and prospects in thin films. Nat. Mater. 6, 21–9 (2007).
Scott, J. F. Applications of modern ferroelectrics. Science 315, 954–9 (2007).
Bibes, M. & Barthélémy, A. Multiferroics: towards a magnetoelectric memory. Nat. Mater. 7, 425–6 (2008).
Gajek, M. et al. Tunnel junctions with multiferroic barriers. Nat. Mater. 6, 296–302 (2007).
Bhatnagar, A., Roy Chaudhuri, A., Heon Kim, Y., Hesse, D. & Alexe, M. Role of domain walls in the abnormal photovoltaic effect in BiFeO3 . Nat. Commun. 4, 2835 (2013).
Abakumov, A. M. et al. Slicing the perovskite structure with crystallographic shear planes: the AnBnO3n−2 homologous series. Inorg. Chem. 49, 9508–16 (2010).
Batuk, M. et al. Atomic structure of defects in anion-deficient perovskite-based ferrites with a crystallographic shear structure. Inorg. Chem. 53, 2171–80 (2014).
Ruddlesden, S. N. & Popper, P. The compound Sr3Ti2O7 and its structure. Acta Cryst. 11, 54–55 (1958).
Lee, C.-H. et al. Exploiting dimensionality and defect mitigation to create tunable microwave dielectrics. Nature 502, 532–6 (2013).
Zurbuchen, M. et al. Morphology, structure and nucleation of out-of-phase boundaries (OPBs) in epitaxial films of layered oxides. J. Mat. Res. 22, 1439–1471 (2007).
Hill, N. A. Why are there so few magnetic ferroelectrics? J. Phys. Chem. B 104, 6694–6709 (2000).
Catalan, G. & Scott, J. F. Physics and Applications of Bismuth Ferrite. Adv. Mater. 21, 2463–2485 (2009).
Palkar, V. & Malik, S. Observation of magnetoelectric behavior at room temperature in Pb(FexTi1−x)O3 . Solid State Commun. 134, 783–786 (2005).
Cohen, R. E. Origin of ferroelectricity in perovskite oxides. Nature 358, 136–138 (1992).
Ganegoda, H., Kaduk, J. A. & Segre, C. U. X-ray powder diffraction refinement of PbTi1−xFexO3−δ solid solution series. Powder Diffr. 28, 238–245 (2013).
Nelmes, R. J. & Kuhs, W. F. The crystal structure of tetragonal PbTiO3 at room temperature and at 700 K. Solid State Commun. 54, 721–723 (1985).
Ren, Z. et al. Room-temperature ferromagnetism in Fe-doped PbTiO3 nanocrystals. Appl. Phys. Lett. 91, 063106 (2007).
Verma, K. C., Kotnala, R. K. & Negi, N. S. Improved dielectric and ferromagnetic properties in Fe-doped PbTiO3 nanoparticles at room temperature. Appl. Phys. Lett. 92, 152902 (2008).
Sun, C. et al. Negative thermal expansion in the PbTi1−xFexO3 system. Phys. Status Solidi B 245, 2520–2523 (2008).
Palkar, V. R., Purandare, S. C., Gohil, S., John, J. & Bhattacharya, S. Scanning probe imaging of coexistent ferromagnetism and ferroelectricity at room temperature. Appl. Phys. Lett. 90, 172901 (2007).
Verma, K. C., Kotnala, R. K., Thakur, N., Rangra, V. S. & Negi, N. S. Resistivity dependent dielectric and magnetic properties of Pb(Fe0.012Ti0.988)O3 nanoparticles. J. Appl. Phys. 104, 093908 (2008).
Sun, C. et al. Ferroelectric and ferromagnetic properties of Pb(Ti0.8Fe0.2)O3−δ thin film. Dalton Trans. 39, 9952–5 (2010).
Hayward, S. A. et al. Transformation processes in LaAlO3: Neutron diffraction, dielectric, thermal, optical and Raman studies. Phys. Rev. B 72, 054110 (2005).
Vasco, E., Bohme, O., Roman, E. & Zaldo, C. Origin and control of the lead-enriched near-surface region of (Pb, La)TiO3 . Appl. Phys. Lett. 78, 2037 (2001).
Fewster, P. F. X-ray analysis of thin films and multilayers. Rep. Prog. Phys. 59, 1339–1407 (1996).
Fullerton, E., Schuller, I., Vanderstraeten, H. & Bruynseraede, Y. Structural refinement of superlattices from x-ray diffraction. Phys. Rev. B 45, 9292–9310 (1992).
Jellison, G. E. & Modine, F. A. Parameterization of the optical functions of amorphous materials in the interband region. Appl. Phys. Lett. 69, 371 (1996).
Chen, P. et al. Optical properties of quasi-tetragonal BiFeO3 thin films. Appl. Phys. Lett. 96, 131907 (2010).
Alexe, M. Local mapping of generation and recombination lifetime in BiFeO3 single crystals by scanning probe photoinduced transient spectroscopy. Nano Lett. 12, 2193–8 (2012).
Ruppel, W., Von Baltz, R. & Wurfel, P. The origin of the photo-emf in ferroelectric and non-ferroelectric materials. Ferroelectrics 43, 109–123 (1982).
Kumar, A. et al. Probing mixed tetragonal/rhombohedral-like monoclinic phases in strained bismuth ferrite films by optical second harmonic generation. Appl. Phys. Lett. 97 (2010).
Mishina, E. D. et al. Domain orientation in ultrathin (Ba,Sr)TiO3 films measured by optical second harmonic generation. J. Appl. Phys. 93, 6216 (2003).
Talbayev, D. et al. Detection of coherent magnons via ultrafast pump-probe reflectance spectroscopy in multiferroic Ba0.6Sr1.4Zn2Fe12O22 . Phys. Rev. Lett. 101, 097603 (2008).
Doig, K. I. et al. Coherent magnon and acoustic phonon dynamics in tetragonal and rare-earth-doped BiFeO3 multiferroic thin films. Phys. Rev. B 88, 094425 (2013).
Ulbricht, R., Hendry, E., Shan, J., Heinz, T. F. & Bonn, M. Carrier dynamics in semiconductors studied with time-resolved terahertz spectroscopy. Rev. Mod. Phys. 83, 543–586 (2011).
Lloyd-Hughes, J. & Jeon, T.-I. A review of the terahertz conductivity of bulk and nanomaterials. J. Infrared Milli. Terahz. Waves 33, 871 (2012).
Lloyd-Hughes, J., Jones, S. P. P., Castro-Camus, E., Doig, K. I. & MacManus-Driscoll, J. L. Modifying the polarization state of terahertz radiation using anisotropic twin-domains in LaAlO3 . Opt. Lett. 39, 1121–1124 (2014).
Calvani, P. et al. Infrared optical properties of perovskite substrates for high-Tc superconducting films. Physica C 181, 289–295 (1991).
Shimada, T., Kakimoto, K.-I. & Ohsato, H. Microwave dielectric properties of lanthanum aluminate ceramics and single crystal. J. European Ceram. Soc. 25, 2901–2905 (2005).
Iliev, M. N., Abrashev, M. V., Mazumdar, D., Shelke, V. & Gupta, A. Polarized Raman spectroscopy of nearly tetragonal BiFeO3 thin films. Phys. Rev. B 82, 014107 (2010).
Abrashev, M. V., Litvinchuk, A. P., Iliev, M. N. & Meng, R. L. Comparative study of optical phonons in the rhombohedrally distorted perovskites LaAlO3 and LaMnO3 . Phys. Rev. B 59, 4146–4153 (1999).
Saine, M. C., Husson, E., Brusset, H. & Cerez, A. Etude vibrationnelle d'aluminates et de gallates de terres rares-I. Alluminates de structure perovskite. Spectroc. Acta Pt. A-Molec. Biomolec. Spectr. 37, 985–990 (1981).
Foster, C. M., Li, Z., Grimsditch, M., Chan, S. K. & Lam, D. J. Anharmonicity of the lowest-frequency A1(TO) phonon in PbTiO3 . Phys. Rev. B 48, 160–167 (1993).
Takahashi, R. et al. Long-range spontaneous structural ordering in barium stannate thin films. Appl. Phys. Lett. 97, 081906 (2010).
Detemple, E. et al. Ruddlesden-Popper faults in LaNiO3/LaAlO3 superlattices. J. Appl. Phys. 112, 013509 (2012).
Sheu, Y. M. et al. Ultrafast carrier dynamics and radiative recombination in multiferroic BiFeO3 . Appl. Phys. Lett. 100, 242904 (2012).
Ruello, P. et al. Photoexcitation of gigahertz longitudinal and shear acoustic waves in BiFeO3 multiferroic single crystal. Appl. Phys. Lett. 100, 212906 (2012).
Jin, Z. M. et al. Structural dependent ultrafast electron-phonon coupling in multiferroic BiFeO3 films. Appl. Phys. Lett. 100, 071105 (2012).
Chen, L. Y. et al. Ultrafast photoinduced mechanical strain in epitaxial BiFeO3 thin films. Appl. Phys. Lett. 101, 041902 (2012).
Pisarev, R., Moskvin, A., Kalashnikova, A. & Rasing, T. Charge transfer transitions in multiferroic BiFeO3 and related ferrite insulators. Phys. Rev. B 79, 1–16 (2009).
Jepsen, P. U. et al. Ultrafast carrier trapping in microcrystalline silicon observed in optical pump-terahertz probe measurements. Appl. Phys. Lett. 79, 1291 (2001).
Merz, E. F. & Walter, J. Ferroelectricity (North Holland Pub. Co., Amsterdam, 1967).
Björck, M. & Andersson, G. GenX: an extensible X-ray reflectivity refinement program utilizing differential evolution. J. Appl. Cryst. 40, 1174–1178 (2007).
Acknowledgements
The authors would like to thank Dr. T. P. A. Hase (University of Warwick) and Dr. J. Bowen (University of Birmingham) for technical assistance. J.L. would like to thank the EPSRC (UK) for financial support under grant EP/H003444/2. The XRD, TEM, Raman, FTIR equipment and Science City Photoemission Facility used in this research were obtained through the Science City Advanced Materials Project: Creating and Characterizing Next Generation Advanced Materials, with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF).
Author information
Authors and Affiliations
Contributions
K.I.D. performed the XRD, optical, pump-probe and vibrational spectroscopy studies, aided by D.W. for the XRD experiments. J.L. and K.I.D. performed the SHG measurements. J.J.P.P., R.B. and A.M.S. undertook the TEM study. M.W. and C.F.M. performed and analysed the XPS results. M.L. performed the SQUID magnetometry. S.N. and V.R.P. grew the samples. K.I.D. and J.L. wrote the manuscript and all authors discussed the results.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Doig, K., Peters, J., Nawaz, S. et al. Structural, optical and vibrational properties of self-assembled Pbn+1(Ti1−xFex)nO3n+1−δ Ruddlesden-Popper superstructures. Sci Rep 5, 7719 (2015). https://doi.org/10.1038/srep07719
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07719
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
