
Abstract
β2-AR activation increases the risk of sudden cardiac death (SCD) in heart failure (HF) patients. Non-selective β-AR blockers have greater benefits on survival than selective β1-AR blockers in chronic HF patients, indicating that β2-AR activation contributes to SCD in HF. This study investigated the role of β2-AR activation on repolarization and ventricular arrhythmia (VA) in the experimental HF model. The guinea pig HF was induced by descending aortic banding. The effective refractoriness period (ERP), corrected QT (QTc) and the incidence of VA were examined using Langendorff and programmed electrical stimulation. Ikr and APD were recorded by the whole cell patch clamp. Selective β2-AR agonist salbutamol significantly increased the incidence of VA, prolonged QTc and shortened ERP. These effects could be prevented by the selective β2-AR antagonist, ICI118551. Salbutamol prolonged APD90 and reduced Ikr in guinea pig HF myocytes. The antagonists of cAMP (Rp-cAMP) and PKA (KT5720) attenuated Ikr inhibition and APD prolongation induced by salbutamol. However, the antagonists of Gi protein (PTX) and PDE III (amrinone) showed opposite effects. This study indicates that β2-AR activation increases the incidence of VA in the experimental HF model via activation of Gs/cAMP/PKA and/or inhibition of Gi/PDE pathways.
Similar content being viewed by others


Introduction
The prevalence of heart failure (HF) is approximately 2.6% in Western societies and annual incidences approach 5–10 per 1000 persons, making it a major health problem1. Approximately 50% of HF patients die suddenly predominantly due to lethal ventricular arrhythmias2. However, the underlying mechanism for the occurrence of lethal ventricular arrhythmias in HF is still unclear.
The sympathetic nervous system plays a vital role in the pathophysiology of HF. High cardiac sympathetic activities have been reported to be correlated with the risk of death from arrhythmias3. β adrenergic receptors (β-ARs), a superfamily of G-protein-coupled receptors, contribute to the cardiac sympathetic activity. β1-ARs are primarily expressed in the heart and comprise 75–80% of cardiac β-ARs. However, non-selective β-AR blockers have shown greater benefits in survival than selective β1-AR blockers in patients with chronic HF4, indicating that β2-AR or β3-AR activation might contribute to SCD in HF instead of β1-AR. Contrary to β3-AR, which is primarily expressed in adipose tissue and minimally in the heart, β2-AR is expressed in the heart and accounts for 20–25% of cardiac β-ARs. In addition, β2-AR genetic variants increase the risk of sudden cardiac death (SCD) in human5. Abnormal augmentation of cardiac responsiveness to β2-AR is also a risk factor for ventricular fibrillation6. These facts indicate that β2-AR activation might be responsible for SCD in HF.
The rapid component of delayed rectifier potassium current (Ikr), mediated by the human ether-a-go-go-related gene (hERG)7, is a major contributor to phase 3 repolarization of the action potential8. Its reduction often increases action potential duration (APD) and leads to the increase of the incidence of early afterdepolarizations (EADs), which can induce ventricular arrhythmias and sudden cardiac death9,10. Our previous study revealed that Ikr was significantly lower in HF myocytes and increased response to β2-AR stimulation augments inhibition of IKr in HF myocytes11. Thus, β2-AR activation may delay repolarization and induce ventricular arrhythmia in HF by Ikr suppression and APD prolongation.
β2-AR, a G protein-coupled receptor, regulates cardiac function upon binding to catecholamine and leads to the activation of heterotrimeric G-proteins12. β2-AR couples dually to Gs proteins and pertussis toxin (PTX)-sensitive Gi proteins13, leading to the activation of AC and production of cAMP14. Phosphodiesterases (PDE) regulates cAMP production through AC activation and cAMP breakdown to modulate PKA activity and myocardial contractility15. However the possible roles of β2-AR activation in repolarization and ventricular arrhythmia in HF have not yet been determined.
Here we hypothesized that β2-AR activation could induce abnormal repolarization and ventricular arrhythmia through Gs/cAMP/PKA and Gi/PDE. To address this hypothesis, we used β2-AR agonist and antagonist to determine cardiac electrophysiological changes in guinea pigs HF model and investigated the effects of β2-AR activation on Ikr and APD in HF left ventricular myocytes using the whole cell patch clamp technique. From this study, we preliminary clarified the mechanisms of β2-AR activation governing cardiac repolarization and arrhythmogenesis in heart failure.
Results
β2-AR stimulation increases the incidence of ventricular arrhythmia from isolated failing heart
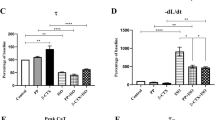
In the control hearts, VA was induced in only one in six hearts perfused with salbutamol and no VA was evoked by an extrastimulus application in the other two groups. Incidence of PES-induced VA dramatically increased in the HF groups (Fig. 1). PES significantly increased the incidence of VA in the HF hearts compared to the control preparations after perfusion of salbutamol (83.3% vs. 16.7%, n = 6, p < 0.05) (Fig. 1B). ICI118551 significantly decreased inducibility of PES-evoked VA in the HF hearts exposed to salbutamol (16.7% vs. 83.3%, n = 6, p < 0.05) (Fig. 1B).

Salbutamol (Sal) significantly increases the incidence of ventricular arrhythmia (VA) in HF guinea pigs.
(A) Representative ECG traces from programmed LV stimulation for VA non-inducibility (upper) and inducibility (lower). (B) Bar graphs summarize the incidence of VA from Con ( ) and HF (
) and HF ( ) guinea pigs before and after administration of Sal (10 μM) or Sal plus ICI (1 μM) (*p < 0.05, HF + Sal vs. Con + Sal; #p < 0.05, HF + Sal vs. HF + Sal + ICI, n = 6).
) guinea pigs before and after administration of Sal (10 μM) or Sal plus ICI (1 μM) (*p < 0.05, HF + Sal vs. Con + Sal; #p < 0.05, HF + Sal vs. HF + Sal + ICI, n = 6).
β2-AR activation prolongs QTc from isolated failing heart
In the control hearts, the selective β2-AR agonist salbutamol slightly prolonged QTc compared to the control group (262.2 ± 7.29 ms vs 240.8 ± 6.07 ms, n = 6, p < 0.05) (Fig. 2B). However, salbutamol significantly broadened QTc of HF hearts compared to HF hearts perfused without salbutamol (293.5 ± 6.60 ms vs. 258.7 ± 6.65 ms, n = 6, p < 0.01) (Fig. 2B). The percentage increase in QTc was significantly larger in the HF hearts than in the control hearts (13.52 ± 1.72% vs. 8.87 ± 0.84%, n = 6, p < 0.05) (Fig. 2C). To determine if β2-AR mediated QTc broadening was induced by salbutamol, the hearts were perfused with selective β2-AR antagonist ICI118551 for 30 min prior to salbutamol. ICI118551 significantly limited QTc prolongation induced by salbutamol in the control (239.7 ± 7.29 ms vs. 262.2 ± 7.29 ms, n = 6, p < 0.05) (Fig. 2B) and HF groups (264.5 ± 9.29 ms vs 293.5 ± 6.60 ms, n = 6, p < 0.05) (Fig. 2B).

Salbutamol (Sal) significantly prolonges QTc in HF guinea pigs.
(A) Representative ECG traces from control (left), salbutamol (10 μM, middle) and salbutamol plus ICI118551 (ICI, 1 μM, right) treated hearts from Control (upper) and HF guinea pigs (lower). (B) Bar graphs summarize the rate-corrected QT from Con and HF guinea pigs before and after administration of Sal (10 μM) or Sal plus ICI (1 μM) (*p < 0.05, HF vs. Con, n = 6). (C) Bar graphs show the Sal-induced percentage difference of prolongation of QTc from Control ( ) and HF (
) and HF ( ) guinea pigs (*p < 0.05, n = 6).
) guinea pigs (*p < 0.05, n = 6).
β2-AR stimulation shortens effective refractory period of LV in HF model
In this study, the effective refractory period was examined by extrasystolic stimulation and dynamic pacing from the isolated heart twelve weeks after procedure. HF dramatically prolonged LV ERP compared to the control group (82.50 ± 4.79 ms vs. 68.33 ± 3.07 ms, n = 6, p < 0.05) (Fig. 3B). In the control hearts, no significant difference was observed in ERP in the presence or absence of salbutamol (62.33 ± 3.36 ms vs. 68.33 ± 3.07 ms, n = 6, p > 0.05). However, salbutamol significantly shortened ERP in HF group (67.25 ± 3.35 ms vs. 82.50 ± 4.79 ms, n = 6, p < 0.05) and salbutamol-induced ERP shortening was inhibited by ICI118551 (82.00 ± 4.55 ms vs. 67.25 ± 3.35 ms, n = 6, p < 0.05) (Fig. 3C).

Changes of effective refractoriness period (ERP) in HF.
(A) Representative extrasystolic stimulation ECG and for stimulation protocol (S1: S2 8:1) for ERP: loss of capture (upper) and capture (lower). (B) Summarized data from ERP in Control (Con,  , n = 6) and HF (HF,
, n = 6) and HF (HF,  , n = 6) isolated hearts (*p < 0.05, HF vs. Con). (C) Bar graphs summarize ERP from Con and HF guinea pigs before and after administration of Sal (salbutamol, 10 μM) or Sal plus ICI (ICI, 1 μM) (*p < 0.05, n = 6).
, n = 6) isolated hearts (*p < 0.05, HF vs. Con). (C) Bar graphs summarize ERP from Con and HF guinea pigs before and after administration of Sal (salbutamol, 10 μM) or Sal plus ICI (ICI, 1 μM) (*p < 0.05, n = 6).
β2-AR stimulation prolongs APD90 in myocytes from isolated failing heart
APD at 90% repolarization (APD90) was prolonged to a greater extent by salbutamol in the HF group compared to the control group (14.11 ± 1.10 ms vs. 7.04 ± 1.59 ms, n = 6 p < 0.01) (Fig. 4B). However, no significant difference was observed in change of 50% repolarization (APD50) between the HF and control groups (9.44 ± 1.85 ms vs. 4.61 ± 2.65 ms, n = 6, p > 0.05) (Fig. 4B).

Effects of β2-AR stimulation on APD in myocytes from isolated failing heart.
(A) Representative AP traces stimulated by salbutamol (Sal) in left ventricular myocytes from control (Con, left) and HF guinea pigs (HF, right). (B) Summarized data from change of APD90 ( ) and APD50 (
) and APD50 ( ) in control (Con, left) and HF (HF, right) myocytes (**p < 0.01, HF vs. Con, n = 6 cells, 6 hearts). Test pulses were applied at various voltages from −40 to +40 mV (step width 10 mV, step duration 200 ms) before returning to −40 mV for tail current recording.
) in control (Con, left) and HF (HF, right) myocytes (**p < 0.01, HF vs. Con, n = 6 cells, 6 hearts). Test pulses were applied at various voltages from −40 to +40 mV (step width 10 mV, step duration 200 ms) before returning to −40 mV for tail current recording.
β2-AR activation inhibits Ikr in myocytes from isolated failing heart
Twelve weeks after aortic constriction, left ventricular myocytes isolated from the hearts of the control and HF groups were used to measure Ikr and APD stimulated by salbutamol by the whole-cell patch-clamp technique (Fig. 5). The mean current densities of Ikr in the control and HF groups were shown in Fig. 5B. The Ikr tail current density in the HF group was significantly decreased by salbutamol compared to control group (36.5 ± 3.35% vs. 22.84 ± 3.97%, n = 6, p < 0.05).

Effects of β2-AR stimulation on Ikr in myocytes from isolated failing heart.
(A) Representative tail traces of IKr inhibited by salbutamol (Sal) in LV myocytes isolated from control (Con, left) and HF guinea pigs (HF, right). (B) Summarized data from decreased percentage of Ikr in control ( ) and HF (
) and HF ( ) myocytes (*p < 0.05, HF vs. Con, n = 6 cells, 6 hearts).
) myocytes (*p < 0.05, HF vs. Con, n = 6 cells, 6 hearts).
Gs proteins promote the prolongation effect of β2-AR stimulation on APD in ventricular myocytes
Salbutamol slightly increased APD90 and APD50 in the control cells, but no significant difference was observed in these cells in the presence or absence of Rp-cAMP (4.08 ± 1.67% vs. 7.53 ± 1.66%, n = 6, p > 0.05; 3.09 ± 3.00% vs. 4.70 ± 2.67%, n = 6, p > 0.05) (Fig. 6A). In the HF cells, Rp-cAMP significantly reduced the percentages of prolongation of APD90 and APD50 by salbutamol compared to salbutamol only (3.99 ± 0.80% vs. 14.22 ± 1.18%, n = 6, p < 0.01; 3.64 ± 1.07% vs. 11.84 ± 2.13%, n = 6, p < 0.05) (Fig. 6A).

Effects of cAMP inhibitor, PKA inhibitor, Gi inhibitor and PDE inhibitor on APD90 and APD50 response to β2-AR stimulation.
(A) Summarized data for percentage prolongation in APD90 ( ) and APD50 (
) and APD50 ( ) evoked by Sal (10 μM) alone, Rp-cAMP (cAMP inhibitor, 100 μM) plus Sal and KT5720 (PKA inhibitor, 2.5 μM) plus Sal in Con and HF myocytes (*p < 0.05, **p < 0.01, n = 6 cells, 6 hearts). (B) Summarized data for percentage prolongation in APD90 (
) evoked by Sal (10 μM) alone, Rp-cAMP (cAMP inhibitor, 100 μM) plus Sal and KT5720 (PKA inhibitor, 2.5 μM) plus Sal in Con and HF myocytes (*p < 0.05, **p < 0.01, n = 6 cells, 6 hearts). (B) Summarized data for percentage prolongation in APD90 ( ) and APD50 (
) and APD50 ( ) evoked by Sal (10 μM) alone, PTX (Gi inhibitor, 400 ng/mL) plus Sal and amrinone (PDE inhibitor, 30 μM) plus Sal in Con and HF myocytes (*p < 0.05, **p < 0.01, n = 6 cells, 6 hearts).
) evoked by Sal (10 μM) alone, PTX (Gi inhibitor, 400 ng/mL) plus Sal and amrinone (PDE inhibitor, 30 μM) plus Sal in Con and HF myocytes (*p < 0.05, **p < 0.01, n = 6 cells, 6 hearts).
The effect of PKA on salbutamol-induced prolongation of APD was similar to cAMP. KT5720 did not significantly decrease the degree of broadened APD90 and APD50 by salbutamol in the control cells (3.69 ± 2.63% vs. 7.53 ± 1.66%, n = 6, p > 0.05; 2.98 ± 5.08% vs. 4.70 ± 2.67%, n = 6, p > 0.05) (Fig. 6A). However, this effect was dramatically prevented by KT5720 in the HF cells. The difference in percentage prolongation of APD90 was significant between salbutamol with and without KT5720 (4.74 ± 2.00% vs. 14.22 ± 1.18%, n = 6, p < 0.01) (Fig. 6A) and the change of APD50 was the same as that in APD90 (3.75 ± 1.88% vs. 11.84 ± 2.13%, n = 6, p < 0.05) (Fig. 6A).
Gi proteins weaken the prolongation effect of β2-AR stimulation on APD in ventricular myocytes
To investigate the effect of Gi protein on β2-AR activation-induced APD prolongation, we studied the effects of PTX on changes in APD in the control and HF myocytes after perfusion with salbutamol. No difference was observed in APD90 and APD50 with or without PTX in the control cells (9.61 ± 0.94% vs 7.53 ± 1.66%, n = 6, p > 0.05; 8.77 ± 1.44% vs 4.70 ± 2.67%, n = 6, p > 0.05, Fig. 6B). After treatment with PTX in the HF cells, the percentages of prolongation of APD90 and APD50 induced by salbutamol were greater than those without PTX administration (18.74 ± 1.11% vs. 14.22 ± 1.18%, n = 6, p < 0.05; 18.3 ± 1.85% vs. 11.84 ± 2.13%, n = 6, p < 0.05) (Fig. 6B).
We further determined the effects of PDE on salbutamol-induced APD prolongation. The results of amrinone were similar to that of PTX. Amrinone had no significant effects on salbutamol-induced APD90 and APD50 broadening in the control cells (10.32 ± 0.51% vs. 7.53 ± 1.66%, n = 6, p > 0.05; 9.78 ± 0.78% vs. 4.70 ± 2.67%, n = 6, p > 0.05) (Fig. 6B). However, APD prolongation was dramatically augmented by amrinone in the HF cells and the percentage prolongation of APD90 and APD50 was greater with salbutamol and amrinone compared to salbutamol with no amrinone (19.95 ± 1.34% vs. 14.22 ± 1.18%, n = 6, p < 0.01, 18.79 ± 1.53% vs 11.84 ± 2.13%, n = 6, p < 0.05) (Fig. 6B).
Gs proteins promote the inhibitory effect of β2-AR stimulation on Ikr in ventricular myocytes
Rp-cAMP reduced the inhibitory effect of salbutamol on Ikr in the control group from 22.78 ± 4.16% to 10.41 ± 2.97% (n = 6, p < 0.05) (Fig. 7A) and dramatically attenuated the inhibitory effect of salbutamol on Ikr in the HF group (from 36.97 ± 3.56% to 11.44 ± 1.75%, n = 6, p < 0.01) (Fig. 7A).

Effects of cAMP inhibitor, PKA inhibitor, Gi inhibitor and PDE inhibitor on Ikr response to β2-AR stimulation.
(A) Summarized data for percentage decrease in the amplitude of IKr tail current evoked by Sal (10 μM) alone, Rp-cAMP (cAMP inhibitor, 100 μM) plus Sal and KT5720 (PKA inhibitor, 2.5 μM) plus Sal in Con and HF myocytes (*p < 0.05, **p < 0.01, n = 6 cells, 6 hearts). (B) Summarized data for percentage decrease in the amplitude of IKr tail current evoked by Sal alone, PTX (Gi inhibitor, 400 ng/mL) plus Sal and amrinone (PDE inhibitor, 30 μM) plus Sal in Con and HF myocytes (*p < 0.05, n = 6 cells, 6 hearts).
To investigate whether PKA correlated with decreased Ikr response to β2-AR, we explored the effects of KT5720 on the reduction of salbutamol stimulated Ikr. Salbutamol-induced inhibition of Ikr was attenuated by KT5720 in the control group (from 22.78 ± 4.16% to 12.73 ± 1.32%, n = 6, p < 0.05) (Fig. 7A) and the HF group (from 36.97 ± 3.56% to 17.70 ± 1.88%, n = 6, p < 0.01) (Fig. 7A).
Gi proteins weaken the inhibitory effect of β2-AR stimulation on Ikr in ventricular myocytes
No significant difference was observed in the reduction of Ikr between cells from the control group with or without PTX (27.88 ± 2.13% vs. 22.78 ± 4.16%, n = 6, p > 0.05) (Fig. 7B). However, the reduction of Ikr was significantly greater in the HF group with the presence of PTX compared to that with the absence of PTX (46.63 ± 2.40% vs. 36.97 ± 3.56%, n = 6, p < 0.05) (Fig. 7B).
This study showed that the effects of PDE on β2-AR activation were similar to those on Gi. In the control cells, amrinone did not further decrease Ikr compared to cells treated with salbutamol only (26.39 ± 1.79% vs. 22.78 ± 4.16%, n = 6, p > 0.05) (Fig. 7B). The effects of β2-AR activation on Ikr were augmented by amrinone in HF cells in contrast to those not pretreated with amrinone (49.25 ± 1.05% vs. 36.97 ± 3.56%, n = 6, p < 0.05) (Fig. 7B).
Discussion
The major findings of this study are as follows: (1) β2-AR activation prolongs QTc and APD90, shortens ERP and increases the incidence of VA in isolated failing heart. (2) Inhibition of Gs/cAMP/PKA pathway reduces the incidence of VA and the suppression of Gi/PDE pathway increases the incidence of VA induced by β2-AR activation in chronic HF. To the best of our knowledge, this study for the first time showed that Gs and Gi pathway are involved in the occurrence of VA induced by β2-AR activation.
First, β2-AR activation prolongs repolarization, shortens effective refractory period and increases the incidence of VA in isolated failing heart. β-adrenergic receptors are composed of three subtypes, β1, β2 and β3-adrenergic receptors and stimulation by the sympathetic nervous system or circulating catecholamine is involved in cardiac myocytes13. Previous studies have shown that β1 proteins are significantly reduced and β2 proteins remain unchanged in HF hearts; thus, the proportion of β2 protein increase might indicate the prominence of β2 proteins in the diseased condition11,13.
As early as the 1980s, the β2-adrenoceptor agonist salbutamol was found to have beneficial hemodynamic effects to patients with HF, but also had increased incidence of ventricular tachycardia16. Catecholamine increased repolarization dispersion and induced VA by β2-adrenoceptor activation17. β2-AR antagonist ICI118551 significantly reduced incidence of ventricular fibrillation18. QT interval reflects the total duration of ventricular depolarization and repolarization or integrated APD of heart cells. Accumulating evidence has demonstrated that the major ionic current responsible for QT prolongation is the rapid delayed rectifier K+ current (Ikr)19. Our results showed that APD was significantly broadened in HF ventricular myocytes following Ikr inhibition stimulated by β2-adrenoceptor agonist. Meanwhile, the ERP in LV and QTc was dramatically prolonged by salbutamol in isolated failing hearts. Thus, β2-adrenoceptor activation might participate in VA in HF, which had been suggested as a more refined assessment for the risk of SCD in HF.
Second, the inhibition of Gs/cAMP/PKA and Gi/PDE pathway shows opposite effects on the incidence of VA induced by β2-AR activation in chronic HF. Cardiac β2-AR couples dually to Gs and Gi proteins20,21. Ikr is a target for cAMP/PKA regulation. A-kinase anchoring proteins (AKAPs) target PKA to hERG channels, contributing to the acute regulation of Ikr by cAMP22. Phosphorylation of hERG resulted from activation of cAMP-dependent PKA brings about a rapid reduction in current amplitude23. Our results further demonstrated that cAMP/PKA was involved in the inhibitory effect of β2-AR activation on Ikr. The β2AR-Gs signaling had positive effects on AC activity, cAMP synthesis and PKA activation, but negative responses mediated by Gi13. It has been reported that myocyte apoptosis was induced when the coupling of β2AR to Gi was inhibited24. In addition, PDE attenuated contractility and cAMP production mediated by β2-AR in rat ventricular myocardium; Gi protein also limited isotropic effects of salbutamol mediated by β2-AR25. In our studies, Gi and PDE inhibitors enhanced Ikr reduction and APD prolongation induced by β2-AR agonist in HF myocytes. These results demonstrated that Gi protein contributed to β2-AR agonist induced Ikr inhibition and APD prolongation, oppositely to Gs protein. However, the precise mechanism for β2-AR activation-induced inhibition of IKr for HF myocytes remains unclear.
Third, phosphodiesterase inhibitors affected the incidence of VA in chronic heart failure. Mirinone and amrinone inhibit phosphodiesterase and suppress degradation of cAMP26. PDE3 inhibitors improve cardiac contractility by cAMP pathways. However, chronic treatment with PDE3 inhibitors deteriorates HF and lead to sudden death27. Milrinone has been used as an independent risk factor for clinically significant tachyarrhythmia in the early postoperative period after congenital heart surgery28. Moreover, PDE5 inhibitors have already been reported to have a concentration-dependent inhibitory effect on Ikr and prolongation of cardiac repolarization29. Interestingly, most PDE inhibitors decreases hERG current directly or indirectly while two PDE3 inhibitors (amrinone and milrinone) fail to exhibit any hERG inhibitory effects30. In this study, amrinone did not inhibit Ikr current in the control and HF myocytes, consistent with previous reports30. However, our studies further showed that Ikr currents were inhibited by salbutamol after pretreatment with PDE3 inhibitor and APD was prolonged more. Thus, PDE3 inhibitor combined with β2-AR activation might prolong cardiac repolarization by blocking Ikr and improving the incidence of VA in HF patients. Low-dose β-blockers in combination with milrinone has also been reported to improve cardiac function in acute decompensated HF patients with tachycardia31, with the addition of low-dose β-blockers preventing hemodynamic deterioration due to tachycardia.
In summary, our study elucidated the potential mechanism of β2-AR activation in the inhibition of Ikr and prolongation of APD. β2-AR stimulation induced VA in failing hearts through activating the Gs/cAMP/PKA pathway. Gi/PDE was involved in cardiac repolarization prolongation induced by β2-AR activation, but the effects were opposite to Gs/cAMP/PKA; Inhibition of Gs/cAMP/PKA pathway could reduce the incidence of ventricular arrhythmias induced by β2-AR activation in chronic HF. In addition, suppression of Gi/PDE pathway can increase incidence of VA. These findings showed that Gs/cAMP/PKA and Gi/PDE could be important therapeutic targets for preventing the occurrence of VA in HF.
Methods
All experiments were performed in accordance with the protocols approved by the Institutional Animal Care and Use Committee and the Guide for the Care and Use of Laboratory Animals of Nanjing Medical University, China.
Establishment of guinea pig heart failure model
Male guinea pigs (250–280 g) were divided into two groups: Control and HF. The animals in HF groups were injected with atropine sulfate (0.1 mg/kg) to inhibit mucus secretion. Thirty minutes later, they were anaesthetized with intraperitoneal pentobarbital (30 mg/kg) and then intubated. A rodent respirator provided positive pressure ventilation (tidal volume of 4–5 mL at 60 strokes/min). Left thoracotomy was performed along the 2nd–3rd intercostal space and the descending aorta was exposed. A metal tube (1 to 2 mm in external diameter and 1 cm in length) was tied to the vessel using a 2-0 surgical ligature and the tube was removed followed by closing the chest incision. Sham animals underwent the same operation, with the exception that the aortae were not banded. Sham-operated animals were used as controls.
Langendorff preparation and ex vivo ECG recordings
The HF and control guinea pigs were anaesthetized with pentobarbital (30 mg/kg) and anti-coagulated with sodium heparin (100 U). After the chest was open, the heart was removed and immediately suspended in a Langendorff apparatus. Heart preparations were perfused via the aorta at a constant pressure of 50 mmHg–60 mmHg with warm (37°C) Tyrode's solution, gassed with 95% O2 and 5% CO2. The Tyrode's solution was composed of the following (in mM/L): NaCl 143, KCl 5.4, NaH2PO4 0.25, HEPES 5, glucose 5.6, CaCl2 1.8 and MgCl2 0.5 (pH 7.3).
An ex vivo electrocardiogram (ECG) was obtained from three electrodes, two of which were placed on the right atrium and the apex of the left ventricle and the third was used for grounding. ECG was recorded by an Animal BioAmp amplifer (Lab/8s, AD Instruments). The QTc was calculated by Bazett's formula where QTc = QT/√RR.
To investigate the different effects of β2-AR activation on QTc, isolated hearts from the HF and control groups were exposed to salbutamol (10 μm) (Selleckchem, Houston, TX, USA). After 10-min stabilization, ECGs were taken until the hearts were perfused with salbutamol for 10 min.
Programmed ventricular stimulation
Heart preparations were stimulated with 2-ms rectangular pulses and twice the diastolic threshold current through a bipolar electrode at the LV apex. Continuous pacing was started with a cycle length of 200 ms (S1–S1 interval) and a premature extrastimulus (S2) was delivered after each 8 beats, while the interval (S1–S2) was progressively shortened by 10 ms until loss capture had occurred. The longest S1–S2 interval which failed to capture the ventricle was the effective refractoriness period (ERP). The programmed electrical stimulation protocol was invoked until ventricular arrhythmia was induced or until the protocol was exhausted.
To investigate the incidence of ventricular arrhythmia (VA), we used the programmed electrical stimulation (PES) to stimulate left ventricle of the isolated heart. The control and HF hearts were both divided into three sub-groups including the control, salbutamol and salbutamol plus ICI118551 groups.
Isolation of cardiac myocytes
The myocytes were isolated from guinea pig hearts by enzymatic technique as described previously11 with minor modification. After the heart was removed, the ascending aorta was mounted onto a Langendorff perfusion system. The preparation was first perfused at 37°C with Tyrode's solution. The heart was then perfused with Ca2+-free Tyrode's solution for 5 min, followed by perfusion with the same buffer with the addition of 0.4 mg/mL collagenase type II and 1% bovine serum albumin. Until the heart was soft, the left ventricular myocytes were gently separated and kept in KB solution (in mM): KOH 85, KCl 30, KH2PO4 30, MgSO4 3, HEPES 10, EGTA 0.5, taurine 20, glucose 10, L-glutamic acid 50 (pH 7.4 with KOH) at room temperature for 60 min for electrophysiological recording.
Cell Electrophysiological recordings
Cells were transferred to a recording chamber continuously perfused with the bath solution. Pipettes had resistances of 3–6 MΩ. Whole-cell patch-clamp currents were measured with an Axopatch 200B (Axon, USA). The flow rate was maintained at 2–3 mL/min.
For Ikr recording, pipette solution and bath solution were prepared as our previously described11. Briefly, the slow component of the delayed rectifier potassium currents (Iks) were blocked by 10 μM chromanol (Sigma, St. Louis, MO, USA) and calcium currents were ablated by 10 μM nifedipine (Sigma, St. Louis, MO, USA) in the bath solution. Salbutamol was dissolved in distilled water at a final concentration of 10 mM; chromanol was dissolved in dimethyl sulfoxide (DMSO) to reach a concentration of 10 mM and KT5720 was at the concentration of 1 mM, the solutions were stocked at −20°C until use. Final concentration of DMSO was less than 0.5% in bath, which has been shown without effects on currents11.
To record the Ikr, the following test pulse protocol was used: from a holding potential −40 mV, test pulses were applied at various voltages from −40 mV to +40 mV (step width 10 mV and step duration 200 ms each) prior to returning to −40 mV for tail current recording32.
APs were evoked using whole-cell current-clamp mode by suprathreshold current pulse of 5 ms duration at the frequency of 1 Hz. APD was measured at 90% repolarization (APD90) and 50% repolarization (APD50). APs were generated with the same amplifier and recorded at 37 ± 5°C.
To investigate whether the inhibitory effects of β2-AR activation on APD and Ikr were mediated by cAMP, an inhibitory cAMP analog Rp-cAMP (100 μM) (Sigma, St. Louis, MO, USA) was added to the pipette solution; To investigate whether PKA correlated with change of Ikr and APD response to β2-AR, the ventricular myocytes from the control and HF groups were incubated with KT5720 (2.5 μM) (Sigma, St. Louis, MO, USA) for 1 h. To determine whether the Gi signaling pathway also mediated β2-AR-induced inhibition of Ikr or prolongation of APD, cells were pretreated with Gi protein inhibitor PTX (400 ng/mL) (Tocris, San Francisco, CA, USA) and PDE inhibitor amrinone (30 μM) (Sigma, St. Louis, MO, USA) respectively. After 1-h incubation, Ikr and APD before and after the application of salbutamol in ventricular myocytes were recorded.
Statistical analysis
Measurement data were expressed as mean ± S.E.M. As to the analysis of the data, normality in the repeated measures was tested with Shapiro Wilk Test and statistical analysis was performed with student's T test and one-way ANOVA. The P-value reported was two-sided and value of less than 0.05 was considered statistically significant. All analyses were performed using the SPSS software (Version 13.0, SPSS Inc., Chicago, IL, USA).
References
Go, A. S. et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127, e6–e245 (2013).
Mosterd, A. & Hoes, A. W. Clinical epidemiology of heart failure. Heart 93, 1137–1146 (2007).
Barretto, A. C. et al. Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int. J. Cardiol. 135, 302–307 (2009).
Poole-Wilson, P. A. et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet 362, 7–13 (2003).
Sotoodehnia, N. et al. Beta2-adrenergic receptor genetic variants and risk of sudden cardiac death. Circulation 113, 1842–1848 (2006).
Altschuld, R. A. & Billman, G. E. beta(2)-Adrenoceptors and ventricular fibrillation. Pharmacol. Ther. 88, 1–14 (2000).
Sanguinetti, M. C., Jiang, C., Curran, M. E. & Keating, M. T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307 (1995).
Sanguinetti, M. C. & Jurkiewicz, N. K. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 96, 195–215 (1990).
Szabo, G., Farkas, V., Grunnet, M., Mohacsi, A. & Nanasi, P. P. Enhanced repolarization capacity: new potential antiarrhythmic strategy based on HERG channel activation. Curr. Med. Chem. 18, 3607–3621 (2011).
Seebohm, G. Activators of cation channels: potential in treatment of channelopathies. Mol. Pharmacol. 67, 585–588 (2005).
Wang, H. et al. Increased response to beta(2)-adrenoreceptor stimulation augments inhibition of IKr in heart failure ventricular myocytes. PloS. One. 7, e46186 (2012).
Dilly, K. W. et al. Overexpression of beta2-adrenergic receptors cAMP-dependent protein kinase phosphorylates and modulates slow delayed rectifier potassium channels expressed in murine heart: evidence for receptor/channel co-localization. J Biol. Chem. 279, 40778–40787 (2004).
Woo, A. Y. & Xiao, R. P. beta-Adrenergic receptor subtype signaling in heart: from bench to bedside. Acta. Pharmacol. Sin. 33, 335–341 (2012).
Xiang, Y. & Kobilka, B. K. Myocyte adrenoceptor signaling pathways. Science 300, 1530–1532 (2003).
Gregg, C. J. et al. beta2-adrenergic receptor-coupled phosphoinositide 3-kinase constrains cAMP-dependent increases in cardiac inotropy through phosphodiesterase 4 activation. Anesth. Analg. 111, 870–877 (2010).
Mettauer, B., Rouleau, J. L. & Burgess, J. H. Detrimental arrhythmogenic and sustained beneficial hemodynamic effects of oral salbutamol in patients with chronic congestive heart failure. Am. Heart. J. 109, 840–847 (1985).
Lowe, M. D., Rowland, E., Brown, M. J. & Grace, A. A. Beta(2) adrenergic receptors mediate important electrophysiological effects in human ventricular myocardium. Heart 86, 45–51 (2001).
Billman, G. E., Castillo, L. C., Hensley, J., Hohl, C. M. & Altschuld, R. A. Beta2-adrenergic receptor antagonists protect against ventricular fibrillation: in vivo and in vitro evidence for enhanced sensitivity to beta2-adrenergic stimulation in animals susceptible to sudden death. Circulation 96, 1914–1922 (1997).
Zhang, Y. et al. Ionic mechanisms underlying abnormal QT prolongation and the associated arrhythmias in diabetic rabbits: a role of rapid delayed rectifier K+ current. Cell. Physiol. Biochem. 19, 225–238 (2007).
Zhu, W. et al. Gi-biased beta2AR signaling links GRK2 upregulation to heart failure. Circ. Res. 110, 265–274 (2012).
Xiao, R. P. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci. STKE. 2001, re15 (2001).
Li, Y., Sroubek, J., Krishnan, Y. & McDonald, T. V. A-kinase anchoring protein targeting of protein kinase A and regulation of HERG channels. J. Membr. Biol. 223, 107–116 (2008).
Cui, J., Melman, Y., Palma, E., Fishman, G. I. & McDonald, T. V. Cyclic AMP regulates the HERG K(+) channel by dual pathways. Curr. Biol. 10, 671–674 (2000).
Zhu, W. Z. et al. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc. Natl. Acad. Sci. USA 98, 1607–1612 (2001).
Gonzalez-Munoz, C., Fuente, T. & Hernandez-Cascales, J. Phosphodiesterases inhibition unmask a positive inotropic effect mediated by beta2-adrenoceptors in rat ventricular myocardium. Eur. J. Pharmacol. 607, 151–155 (2009).
Alousi, A. A. & Johnson, D. C. Pharmacology of the bipyridines: amrinone and milrinone. Circulation 73, III10–24 (1986).
Amsallem, E., Kasparian, C., Haddour, G., Boissel, J. P. & Nony, P. Phosphodiesterase III inhibitors for heart failure. Cochrane. Database. Syst. Rev. 1, 1–90, 10.1002/14651858.CD002230.pub2 (2005).
Smith, A. H., Owen, J., Borgman, K. Y., Fish, F. A. & Kannankeril, P. J. Relation of milrinone after surgery for congenital heart disease to significant postoperative tachyarrhythmias. Am. J. Cardiol. 108, 1620–1624 (2011).
Varma, A., Shah, K. B. & Hess, M. L. Phosphodiesterase inhibitors, congestive heart failure and sudden death: time for re-evaluation. Congest. Heart. Fail. 18, 229–233 (2012).
Yunomae, K. et al. Effects of phosphodiesterase (PDE) inhibitors on human ether-a-go-go related gene (hERG) channel activity. J. Appl. Toxicol. 27, 78–85 (2007).
Kobayashi, S. et al. Low-dose beta-blocker in combination with milrinone safely improves cardiac function and eliminates pulsus alternans in patients with acute decompensated heart failure. Circ. J. 76, 1646–1653 (2012).
Karle, C. A. et al. Rapid component I(Kr) of the guinea-pig cardiac delayed rectifier K(+) current is inhibited by beta(1)-adrenoreceptor activation, via cAMP/protein kinase A-dependent pathways. Cardiovasc. Res. 53, 355–362 (2002).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81170162 and No. 81470457), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Graduate Innovation Foundation of Jiangsu Province (JX22013281 and KYLX_0923). The authors acknowledge the help of Professor Junjie Xiao (Shanghai University) and Fang Wang (Department of Cardiology, the First Affiliated Hospital of Nanjing Medical University).
Author information
Authors and Affiliations
Contributions
W.Y. and Y.J. conducted the most patch clamp, electrophysiology, animal models and analyzed the data. Q.Z. conducted part of patch clamp, animal models and analyzed the data. Z.X. and C.Y. conducted animal models and analyzed the data. H.X. conducted electrophysiology experiments. W.Y., Y.J. and Q.Z. conceived the idea. Z.J. designed experiments, developed analysis tool, analyzed the data and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Wang, Y., Yuan, J., Qian, Z. et al. β2 adrenergic receptor activation governs cardiac repolarization and arrhythmogenesis in a guinea pig model of heart failure. Sci Rep 5, 7681 (2015). https://doi.org/10.1038/srep07681
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07681
This article is cited by
-
New drug discovery of cardiac anti-arrhythmic drugs: insights in animal models
Scientific Reports (2023)
-
Assessment of the epi-pericardial fibrotic substrate by collagen-targeted probes
Scientific Reports (2022)
-
cAMP-PKA-CaMKII signaling pathway is involved in aggravated cardiotoxicity during Fuzi and Beimu Combination Treatment of Experimental Pulmonary Hypertension
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
