
Abstract
We describe methods for the assessment of amplified-fragment single nucleotide polymorphism and methylation (AFSM) sites using a quick and simple molecular marker-assisted breeding strategy based on the use of two restriction enzyme pairs (EcoRI-MspI and EcoRI-HpaII) and a next-generation sequencing platform. Two sets of 85 adapter pairs were developed to concurrently identify SNPs, indels and methylation sites for 85 lines of cassava population in this study. In addition to SNPs and indels, the simplicity of the AFSM protocol makes it particularly suitable for high-throughput full methylation and hemi-methylation analyses. To further demonstrate the ease of this approach, a cassava genetic linkage map was constructed. This approach should be widely applicable for genetic mapping in a variety of organisms and will improve the application of crop genomics in assisted breeding.
Similar content being viewed by others


Introduction
DNA methylation can interfere with transcription or the formation of transcriptionally silent heterochromatin and it plays an important role in regulating gene expression in plant genomes1,2,3. The molecular mechanisms underlying DNA methylation and its functions are relatively well understood in several crop species. In terms of gene expression, it may be possible for DNA methylation variations to cause drastic phenotypic changes when the involved genes play large roles in qualitative characteristics, such as those affecting flower symmetry, fertility, or the inhibition of fruit ripening. Methylation variation may also lead to less drastic phenotypic changes associated with continuous complex quantitative traits when they involve small-effect genes, such as those affecting flowering time, plant size, or resistance to pathogens4,5,6,7. However, until recently, available methods for analysing genome-wide methylation, such as Restriction landmark genome scnning (RLGS), methylated DNA immunoprecipitation followed by sequencing (MeDIP-seq) and bisulphite sequencing (BS-seq), have been expensive, cumbersome and time-consuming8,9,10,11,12,13. An economical, simple and efficient method that can be used to detect a large number of samples is urgently needed for the assessment of genome-wide methylation.
The single nucleotide polymorphism (SNP) has become the preferred genetic marker that is widely used in the evaluation of model species with well-developed genomic resources, such as Arabidopsis14,15, rice16, maize17 and soybeans18. Next-generation sequencing technologies significantly reduce the cost of sequencing; however, for large genomes with high levels of heterozygosity, genome-wide sequencing remains expensive. In addition, many studies do not require the high marker densities that are generated by whole-genome sequencing. The restriction-site-associated DNA (RAD), 2b-RAD and genotyping-by-sequencing (GBS) approaches reduce genome complexity and have been shown to be powerful tools that can be used to identify evolutionary origins and to carry out genotyping, genetic mapping and the identification of quantitative trait loci19,20,21,22. The ability to simultaneously discover SNPs and methylated sites across an entire genome, particularly for non-model plants with large genomes and high levels of heterozygosity that lack comprehensive genomic resources, such as reference assemblies, would be optimal.
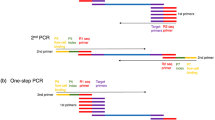
Here, we describe a quick and simple molecular marker-assisted breeding method that is based on two restriction enzyme pairs (EcoRI-MspI and EcoRI-HpaII) and a next-generation sequencing platform, which is termed the amplified-fragment SNP and methylation (AFSM) approach. These enzyme pairs can reduce genome complexity by cleaving target sites, producing methylation-difference tags that are suitable for the sequencing of a large number of samples on next-generation platforms (Figure 1). Cassava (Manihot esculanta Crantz), known as the “king of starch,” is a main food staple for 600 million people worldwide. The cassava genome is large and complex with high levels of heterozygosity, but few pertinent genomic resources have been developed23. To date, there are no reports in the literature describing methylation in cassava. To validate this new method, we utilised the cassava KS population, which was derived from a cross between KU50 and SC124 and consisted of 85 lines to prepare a sequence library. This 85-plex library was sequenced in one lane of an Illumina HiSeq 2500 platform with paired-end sequencing, which generated 16 G of data.

Preparation and sequencing of AFSM tags.
Sample preparation for AFSM genotyping is accomplished by combining two restriction enzyme pairs (EcoRI-MspI and EcoRI-HpaII) to digest genomic DNA and incorporating barcodes for multiplex sequencing. EcoRI is used as a rare cutter and the methylation-retraction enzymes HpaII and MspI are employed as frequent cutters. HpaII and MspI have different sensitivities to methylation of the inner or outer cytosines and can produce different products, reflecting the different methylation states of the cytosines.
Results
A total of 85 barcode adapters, out of 96 developed barcode adapters and two methylation difference (HpaII-methylation and MspI-methylation) adapters were used in this study to simultaneously assess the SNPs, indels and methylation sites in the 85 samples. The barcode adapters were designed using the following criteria: 1) each barcode was 2 or more bp different from all other barcodes; 2) the barcodes could not contain a run of more than 2 of the same nucleotide; and 3) the barcodes could not contain or reform (when ligated) EcoRI, HpaII, or MspI restriction sites. A full set of barcodes was designed to optimise the uniformity of each nucleotide at each position. The barcode adapter terminated with a 5-bp barcode sequence at the 3′ end of its top stand and a 4-bp overhang at the 5′ end of its bottom strand that was complementary to the sticky end that was generated by EcoRI (AATT). The sequences of the two oligonucleotides comprising the barcode adapter were as follows: 5′-TAGCTCGTAGACACCGTCAG[barcode]G-3′ and 5′-AATTC[barcode]CTGACGGTGTCTACGAGCTA-3′ (Figure 2). The full list of barcode adapters for EcoRI is included in Supplementary Table 1. The second HpaII-methylation adapter had the following HpaII-compatible sticky ends: 5′-CGGTGAGATGAGGCATGAC-3′ and 5′- GTCATGCCTCATCTCAC-3′. The third MspI-methylation adapter had the following MspI-compatible sticky ends: 5′-CGGACTAATGAGGCATGAC-3′ and 5′-GTCATGCCTCATTAGTC-3′ (Figure 2). The adapters were designed so that the EcoRI-HpaII and EcoRI-MspI recognition sites did not occur in any adapter sequence and were not regenerated after ligation to genomic DNA. The list of HpaII-methylation and MspI-methylation adapters are included in Supplementary Table 2.

AFSM adapters and primers.
(a, c) Sequences of three different types of double-stranded AFSM adapters. The barcode adapter terminates with a 5-bp barcode at the 3′ end of its top stand and a 5-bp overhang at the 5′ end of its bottom strand that is complementary to the sticky end generated by EcoRI. The HpaII-methylation adapter has a HpaII-compatible sticky end and the MspI-methylation adapter has a MspI-compatible sticky end. (b) PCR primer sequences for EcoRI-HpaII. (d) PCR primer sequences for EcoRI-MspI.
The AFSM method uses EcoRI as a rare cutter and the methylation-sensitive restriction enzymes HpaII and MspI as frequent cutters; the latter two are a pair of isoschizomers that recognise the same tetranucleotide (5′-CCGG) but have differential sensitivities to methylation of their inner or outer cytosines. HpaII is inactive if one or both cytosines are methylated on both DNA strands, but it cleaves when one or both cytosines are methylated on only one strand. MspI, by contrast, cleaves C5mCGG but not 5mCCGG24. Differences in the products that are obtained using EcoRI–HpaII and EcoRI–MspI should thus reflect the different methylation states of the cytosines at the CCGG sites that are recognised by HpaII or MspI, rendering AFSM an efficient method for detecting alterations in cytosine methylation in crops.
Out of 113,472,592 raw reads, 99,446,059 (87.64%) were high-quality (filtered) reads that contained the barcode adapter, EcoRI cleavage site, HpaII-methylation adapter and HpaII cleavage site or the barcode adapter, EcoRI cleavage site, MspI-methylation adapter and MspI cleavage site (Supplementary Table 3). On average, 1,169,954 reads were produced per DNA sample. The female parent KU50 and male parent SC124 generated 3,164,835 and 2,703,331 reads, respectively. The KS population samples generated between 428,131 and 3,164,835 reads. The coefficient of variation (cv = standard deviation/mean) for the number of reads containing the appropriate barcode adapter and EcoRI cleavage site was 45.33% among the KS F1 population samples (Supplementary Figure 1). From a total of 99,446,059 high-quality (filtered) reads, 50,780,017 possessed HpaII-methylation adapters and the HpaII cleavage site and 48,457,678 had MspI-methylation adapters and the MspI cleavage site. Of the 48,253,199 AFSM sequence reads that aligned perfectly to the cassava genome (no mismatches), 12,102,671 (25.08%) mapped to a single genomic location, while 3,033,510 (6.29%) mapped to multiple locations.
A total of 48,253,199 reads from the cassava KS populations were aligned with the AM560 cassava genomic DNA sequences (http://www.phytozome.net/cassava.php) using Bowtie230. On average, 567,685 reads were mapped per DNA sample. The coefficient of variation was 44.44% for the F1 population samples. A total of 1,561,416 reads were mapped for the female parent KU50, while 1,343,187 reads were mapped for the male parent SC124. Regardless of the disproportionate sample representations, we were able to map a minimum of 201,558 reads for the poorest performing F1 samples in the KS population. A total of 25,055,929 reads with HpaII-methylation adapters and the HpaII cleavage site and 24,179,781 reads with the MspI-methylation adapters and MspI cleavage site were mapped.
Of 20,598 total methylated sites, 11,526 polymorphic methylated sites (55.95%) were found in the parents and F1 individuals (Figure 3a, upper right diagonal region) using the AFSM approach. A total of 13,598 filtered CCGG-methylated sites (hemi-methylated or fully methylated sites >2/3) (Figure 3a, lower left diagonal region) included 9193 hemi-methylated and 4405 fully methylated sites. The associated genes were functionally categorised and significant differences were assessed using BGI WEGO31 (P<0.05, x2 test, Figure 3c, d). For the hemi-methylated genes, the GO categories that were enriched in the molecular function sub-ontology included binding (including nucleoside binding, nucleotide binding and ion binding) and hydrolase activity. For the cellular component sub-ontology, the enrichment of protein complex and membrane part were observed. For the biological process sub-ontology, the regulation of metabolic process, metabolic process (including catabolic process and macromolecule metabolic process) and localisation (including macromolecule localisation and cellular localisation) figured prominently in addition to the establishment of protein localisation and the establishment of localisation in the cell. In contrast, for the fully methylated genes, the enrichment of the cell part GO category was observed, particularly in association with protein complex, membrane and membrane part. For molecular function, hydrolase activity, transmembrane transporter activity and protein binding figured prominently.

DNA methylation landscape in the Cassava genome.
(a) Graphical representation of the pattern of methylated sites in the cassava KS population. The y-axis and x-axis indicates the number of individuals, which were detected to have either hemi-methylated or fully methylated sites in the KS cassava population. The lower left diagonal region indicates the distribution of hemi-methylated or fully methylated sites. The blue circles represent the hemi-methylated sites (hemi-methylated sites >2/3), while the red circles depict the fully methylated sites (fully methylated sites >2/3). The upper right diagonal region indicates the distribution of DNA methylation polymorphisms for the cassava KS population. The orange triangles represent the polymorphic methylated sites in the population, while the green triangles represent the monomorphic methylated sites. (b) Relative levels of cytosine methylation at the CCGG sites in the cassava KS population. (c, d) Annotation of hemi-methylated (c) and fully methylated (d) genes with BGI WEGO31. Of the 19,155 genes with GO annotations, 867 hemi-methylated and 1297 fully methylated genes showed significant enrichment differences (P<0.05, x2 test) compared with total analysed cassava genes. Annotations are grouped by cellular component, molecular function or biological process based on the JGI cassava gene GO annotation information. Gene numbers and percentages (on a log scale) are listed for each category.
To analyse the genomic distributions of the filtered hemi-methylated and fully methylated sites, they were mapped to the cassava scaffolds. We observed that 1,354 methylated sites were located in PMGs (genes that were methylated only within their promoter regions, which were defined as the regions 2,000-bp upstream of the transcription start sites), 2,937 were located in GMGs (genes that were methylated within gene regions, including introns and exons, but not within promoter regions) and 1,661 were located in CDSMGs (genes that were methylated within CDS regions but not within promoter regions) (Figure 3b). The total relative full methylation level (32.39%) was lower than the hemi-methylation level (67.61%) and the full methylation and hemi-methylation levels of the promoter regions were similar (51.40% vs. 48.60%, respectively). Interestingly, the full methylation levels were higher than the hemi-methylation levels in the gene and CDS regions (62.82% vs. 37.18% in the gene region and 66.23% vs. 33.77% in the CDS region, respectively) (Figure 3b).
We further used BGI WEGO31 to functionally categorise the CDSMGs, GMGs and PMGs and associated significant differences (P<0.05, x2 test, Supplementary Figure 2). The CDSMGs tended to show enrichment of the cell part GO category, including membrane and protein complex. This is similar to the findings for the GMGs. For molecular function, the binding GO category (including nucleotide binding, nucleoside binding and protein binding) was enriched for the CDSMGs, while catalytic activity (including transferase activity and hydrolase activity), transporter activity (including transmembrane transporter activity and substrate-specific transporter activity) and binding (including nucleotide bind, nucleoside bind and protein binding) figured prominently for the GMGs. For biological process, the CDSMGs exhibited the enrichment of the catabolic process GO category, while metabolic process (including alcohol metabolic process, catabolic process, macromolecule metabolic process, cellular metabolic process and oxidation reduction) predominated for the GMGs. In contrast, the PMGs displayed the enrichment of the organelle (including non-membrane-bounded organelle, intracellular part and intracellular organelle part) and protein complex GO category. For molecular function, the oxidoreductase activity, structural constituent of ribosome and structural molecule activity ontologies were enriched for the PMGs. The PMGs also showed enrichment of the regulation of metabolic process GO category for the biological process sub-ontology.
These 3284 methylated sites in both the parents and F1 individuals could be grouped into four major classes (class A, B, C and D, as shown in Supplementary Table 6). Class A, which consisted of two patterns, included 716 methylated sites with monomorphic methylation (hemi-methylation or full methylation) in the male and female parents and F1 progenies. Class B, which consisted of four patterns, included sites displaying differing methylation patterns in the male and female parents and monomorphic methylation (hemi-methylation or fully methylation) in the F1 individuals. Class C, which consisted of two patterns, was comprised of sites with the same methylation patterns in the parents but different patterns in the F1 progenies. No sites were grouped into class B or class C. Class D, which consisted of four patterns, included 2568 methylated sites showing differing methylation patterns in the F1 progenies and 2038 methylated sites showing disparate methylation patterns between the parents (Supplementary Table 6).
Besides methylation and indel markers, 278,737 SNPs were also identified using this approach, with an average of one SNP was detected per 144.48 bp. Among the 316,276 AFSM markers, 49,831 were distributed in the CDS region, 113,907 in the gene region and 48,058 in the promoter region (Supplementary Table 5).
To further evaluate the AFSM approach, we developed a high-density genetic map. This study is the first attempt to develop a cassava genetic linkage map using only AFSM markers. Presumably, due to the large genome of cassava and the low sequence coverage of the AFSM tags in the cassava KS population, 3,032 out of 15,176 filtered AFSM markers (Supplementary Table 5) were assigned to 20 linkage groups (LGs) and 1,141 markers were located within gene regions (Figure 4). Using this newly developed AFSM approach, the genetic map consisted of 2,331 SNPs, 537 indels and 164 methylation polymorphism markers and it was named the KS genetic map. The markers were not evenly distributed into the different LGs; for example, the largest LG had 625 markers and the two smallest were only comprised of 66 markers. The lengths of the LGs varied from 47.07 cM (LG 20) to 151.55 cM (LG 8) and the KS genetic map spanned a total of 1,970.40 cM. For the KS genetic map, the intervals between adjacent markers varied greatly among the different LGs; the average marker interval was 0.65 cM, with intervals between markers ranging from 0.18 cM (LG 1) to 2.59 cM (LG 4).

Distribution of AFSM markers in the cassava genetic linkage map.
AFSM markers mapped to gene regions are shown by the red dots. Additional markers that mapped to non-gene regions are depicted by the blue dots.
Discussion
The AFSM approach represents a significant improvement compared with other related high-throughput polymorphism discovery and genotyping methods because it is the only method that can detect genome-wide methylation sites in addition to SNPs and indels within a population without the need for increased sequencing efforts. Moreover, fully methylated and hemi-methylated sites can be distinguished using this approach (Supplementary Table 7). The AFSM method provides a streamlined and cost-effective alternative to existing reduced-representation methods. 2b-RAD22, RAD19, GBS20, restriction site tiling analysis (RSTA)32 and single-feature polymorphisms (SFP)33 are able to identify a large number of markers (11,829,959 reads, 41,622 reads, 102,505,713 reads, 50,935 reads and 92,924 reads, respectively) similar to AFSM (99,446,059 reads) (Supplementary Table 7). However, the short oligonucleotide sizes that are utilised for the RSTA (50 bp), SFP (25 bp), 2b-RAD (33 ~ 36 bp) and RAD (26 bp) approaches could add noise to data through non-specific binding, particularly in species with large complex genomes and could fail to detect more subtle differences between genotypes at each polymorphic locus. This noise would necessitate large sample sizes to improve the signal-to-noise ratios for quantitative RSTA, SFP and 2b-RAD genotype data (informative marker rates of 24%, 4% and 42.7%, respectively, have been reported in model organisms with small, simple genomes). The AFSM method may be better suited for species with large genomes or high heterozygosity and may yield cleaner data for heterozygotes because of the longer oligonucleotides used (such as the 2 × 132 bp oligonucleotides used in this study). The informative marker rate that was measured in this study using the AFSM approach was 48.52% in the non-model organism cassava, which possesses high levels of heterozygosity and a large genome. This rate was higher than that which has been reported for other methods (24% for RSTA, 31% for RAD and 4% for SFP with a 5% false discovery rate cut-off). The library that was constructed using the AFSM approach was as uniform as that which has been constructed with GBS (the coefficient of variation was 45.33% for AFSM and 43% for GBS).
For the AFSM approach, the DNA processing and data analysis steps are simpler and more economical compared with those required by other approaches. For RSTA, the DNA processing step necessitates shearing by sonication, restriction digest with specific enzymes and fluorescent labelling, making it costly for genotyping data. For RAD, the DNA processing step also requires shearing by sonication and restriction digest with specific enzymes. For SFP, the microarrays present a high upfront cost and require mask development and there is little chance that such gene chips will become available for many species. In addition, RAD requires EST or genome sequences for mapping, while RSTA and SFP utilise cDNA library databases or 454 transcriptome sequences. The AFSM approach can distinguish among SNPs, indels and methylation sites and it is suitable for the parallel genotyping of large numbers of non-model species without the necessity for the shearing of the DNA by sonication or its fluorescent labelling, thus reducing the cost. This method also does not necessitate an upfront microarray design cost.
Distinct advantages of this approach compared with other methylation scanning methods are that it facilitates the rapid and high-throughput identification of methylated sites, requires less cost and labour and allows for the construction of methylated maps for populations. The basic principle of the AFSM approach, similar to the methylation-sensitive amplified polymorphism (MSAP) assay, is the discovery of methylation patterns using a pair of isoschizomers that have different sensitivities to methylation of inner or outer cytosines. However, the MSAP assay may have three major constraints that are associated with the methylation-resolving power of the gel. First, only a small proportion of cytosine methylation sites can be investigated by MSAP; 195 methylated sites have been previously detected in rice24, 49 in Acacia mangium25, 369 in barley26, 394 in Jatropha curcas27, 283 in Salvadora persica, 482 in Commiphora wightii, 533 in the male genotype of Simmondsia chinensis, 511 in the female genotype of Simmondsia chinensis, 630 in Withania coagulans28 and 376 in perennial ryegrass29. This limitation occurs because detection is restricted to fragments that are amplified from the gel by primer pairs with 3 ~ 4 selective bases. Second, MSAP produces different bands, each of which represents a recognition site that is cleaved by one or both isoschizomers. Methylation patterns are deduced based on polymorphisms in the amplified band patterns between the two digestions of each sample but not the precise patterns of methylation, e.g., there may be a situation in which only one of the isoschizomers cleaves the recognition site, producing three different bands. This result would lead to the mistaken identification of three methylated cytosines. Third, full methylation of external cytosines (mCCGG) cannot be detected by MSAP. The findings from this study are encouraging because the AFSM approach allowed for the detection of precise cytosine methylation sites, including those that were fully methylated at the external cytosine (mCCGG), fully methylated at the internal cytosine (CmCGG) and hemi-methylated. Moreover, 11,526 polymorphic methylated sites were detected by this method, which is more than 250 times the levels detected in rice24 (46 polymorphic methylated sites), more than 50 times those observed in barley26 (230 polymorphic methylated sites), more than 50 times those reported in Jatropha curcas27 (277 polymorphic methylated sites), more than 370 times those detected in Salvadora persica (31 polymorphic methylated sites), more than 147 times those reported in Commiphora wightii (78 polymorphic methylated sites), more than 162 times those observed in the male genotype of Simmondsia chinensis (71 polymorphic methylated sites), more than 188 times those detected in the female genotype of Simmondsia chinensis (61 polymorphic methylated sites) and more than 160 times those reported in Withania coagulans28 (72 polymorphic methylated sites) by MSAP.
Heritable epigenetic variations could directly or indirectly influence the course of evolution in crop species because they are able to affect the processes of adaptation and divergence through the selection of stable epigenetic variants without the involvement of genetic variations or through evolutionary change, for which epigenetic modifications would guide the selection of correlated genetic variations. DNA methylation is the most well-described epigenetic mechanism and it is involved in nearly all well-documented instances of transgenerational epigenetic inheritance in crops. DNA methylation protects against transposon amplification and the inhibition of genomic imprinting and it plays an important role in regulating gene expression. DNA methylation markers of epigenetic changes tend to be heritable and may affect the process of evolution. Many examples of phenotypic variations in association with the methylation of alleles have been identified. The molecular mechanisms and functions that are associated with DNA methylation are reasonably well understood for model crops and a number of studies have also documented intraspecific epigenetic variations in model species34,35. Heritable methylation patterns have not been fully elucidated for non-model plant populations lacking well-developed genomic resources. This study represents the first report of the distribution of genome-wide methylation at CCGG sites in a non-model plant population, in which an average of one methylation site was detected per 51 bp. This study is also the first report of the full and hemi-methylation patterns in an entire genome of a non-model population (Supplementary Table 4). Moreover, these findings are encouraging because out of all of the genes that were methylated, a substantial fraction possessed known functions (Figure 3c, d and Supplementary Figure 2). All of the methylated sites that were distributed on the KS genetic linkage map were fully methylated, indicating that full methylation may be hereditary. Conversely, hemi-methylation may be non-hereditary and accumulate due to environmental stimuli. Full and hemi-methylation may indeed play different roles in biological systems and possess contrasting modes of heredity. Thus, the AFSM method, which is a simple and efficient tool for the detection of SNPs, indels and DNA methylation sites, should be useful in the study of evolution, genotyping, genetic mapping and a number of other important methylation-associated biological phenomena.
Methods
Plant Materials and DNA Extraction
Samples included parents and 83 F1 offspring from the KS mapping population, descending from a cross between two non-inbred lines, including KU50 as the female parent and SC124 as the male parent. KU50 possessed many elite economic traits, such as a high proportion of starch and the ability to produce high yields, but it was intolerant of cold temperatures. In contrast, SC124 was high yielding and cold-resistant but possessed a low proportion of starch. Stem cuttings from the 83 F1 progenies and parents were planted using a randomised block design and two replicates. From each plant, one offspring was grown under a common, controlled environment. Total genomic DNA was extracted from the leaves of 5-month-old crops at the same phenological stage (bearing fully expanded leaves) using a Plant DNeasy Maxi kit (Qiagen) and the two biological replicates were pooled for each individual. We constructed an 85-plex library (combined from EcoRI-MspI and EcoRI-HpaII 85-plex libraries) from the cassava KS population using two sets of samples. Each sample set consisted of a single sample from each F1 individual and one replicated sample from each parent. DNA was quantified using a NanoDrop 2000C spectrophotometer and DNA concentrations were normalised to 100 ng/μL. DNA quality and integrity were assessed by electrophoresis on 1.0% agarose gels.
Choosing Restriction enzymes and Restriction Digest
We chose a combination of two restriction enzyme pairs, EcoRI-MspI and EcoRI-HpaII. The isoschizomers HpaII and MspI were used as frequent cutters, while EcoRI was used as a rare cutter. Methylation-susceptible, anonymous 5′-CCGG sequences and their methylation statuses can be assessed by the AFSM method. To detect the AFSM sites, two digestion reactions were set up at the same time. In the first reaction, genomic DNA (200 ng) was digested in a 20 μl reaction volume of NEB Buffer 4 with 10 U of EcoRI (New England BioLabs Inc., Ipswich, MA, R0101) and 10 U of MspI (New England BioLabs Inc., Ipswich, MA, R0106). The second digestion reaction was carried in exactly the same manner except that HpaII and NEB Buffer 1 were used in place of MspI and NEB Buffer 4. The two sets of DNA samples were digested for 8 h at 37°C. Then, incubation was carried out at 65°C for 30 min to inactivate the enzymes.
Adapter Design
A compatible set of 96 developed barcode sequences were developed for multiplex sequencing is provided as supporting information (Supplementary Table 1) and 85 of them (No.1-85) were used in this study. To minimise the possibility of misidentifying samples as a result of sequencing or adapter synthesis error, all pairwise combinations of barcodes differed by a minimum of 2 bp and barcodes containing runs of 3 or more bp of the same nucleotide were excluded. Hence, it should be possible to correctly assign samples with single-base barcode sequencing errors.
Ligation
The ligation reaction was completed using NEB 1x T4 DNA Ligase Reaction Buffer with 50 mM Tris-HCl, 10 mM MgCl2, 10 mM DTT and 1 mM ATP and 100 U of T4 DNA Ligase (New England BioLabs Inc., Ipswich, MA, M0202) for each sample. For cassava, a 1.5-μM barcode adapter and 1.5-μM HpaII-methylation adapter/MspI-methylation adapter were added to the two sets of samples. The ligation reaction was incubated over night at 16°C and then the ligase was inactivated prior to the pooling of the samples by incubation at 65°C for 20 min.
Multiplexing, Amplification and Sequencing
One set of 85 EcoRI-HpaII ligated samples were pooled and purified in one tube and another set of 85 EcoRI-MspI ligated samples were pooled and purified in another tube using an E.Z.N.A. Cycle-Pure Kit (Omega Bio-tek, Inc., US, D6492). Restriction fragments from each library were then amplified in 50 μL volumes containing 10 μL of pooled DNA fragments, NEB 2x Taq Master Mix (New England Biolabs) and 20 μM each of the primer pairs. The EcoRI-HpaII primer pair sequences were 5′-TAGCTCGTAGACACCGTCAG-3′ and 5′-GTCATGCCTCATCTCACCGG-3′ and the EcoRI-MspI primer pair sequences were 5′-TAGCTCGTAGACACCGTCAG-3′ and 5′-GTCATGCCTCATTAGTCCGG-3′. PCR was performed at 95°C (30s), followed by 23 cycles consisting of 95°C (30 sec), 55°C (30 sec) and 68°C (30 sec) and a final step at 72°C (5 min). The samples were then run on a 2% agarose gel (Sigma). PCR products ranging from 250 bp to 500 bp in size were isolated using an E.Z.N.A. Gel Extraction Kit (Omega Bio-tek Inc., US, D2500-1). After EcoRI-MspI 85-plex library and EcoRI-HpaII 85-plex library had been constructed, equal amount of the two libraries were combined into one 85-plex library. Then paired-end sequencing was performed in one lane using an Illumina HiSeq 2500 (Illumina Inc., San Diego, CA) by Shanghai South Gene Technology Co., Ltd.
Processing of Raw Illumina Data
Raw Illumina sequence reads were processed using custom Perl scripts (http://afsmseq.sourceforge.net/) to optimise read numbers and reduce artefacts within the data. Sequences were assigned to individual samples using barcodes and trimmed to 2 × 132 bp. Those sequences with ambiguous ‘N’ nucleotides were eliminated. Only the sequences that were exact matches to a barcode followed by the expected 5-bp sequence remaining from the EcoRI cleavage site were retained. The full set of reads was then examined for unique tags that were present in more than five different lines.
DNA Sequence Alignments and SNP Calling
The filtered sequence reads were aligned to the AM560 cassava reference genome (obtained from JGI) using Bowtie230, allowing for a maximum of four mismatches and one gap of up to 3 bp. SNPs were identified using the SAMtools36 and VCFtools_v0.1.9 (http://vcftools.sourceforge.net/).
Methylation Analyses
Analyses of the AFSM methylation results were based on comparisons of the EcoRI-HpaII- and EcoRI-MspI-assembled sequences with methylated cytosines at the 5′-CCGG sites using custom Perl scripts (http://afsmseq.sourceforge.net/) for individual plants. HpaII and MspI were a pair of isoschizomers that recognised the same restriction site (5′-CCGG) but had different sensitivities to cytosine methylation. Specifically, HpaII did not cut if either of the cytosines was fully (double-strand) methylated, but it did cut if the external C was hemi-methylated (single-stranded); in contrast, MspI only failed to cut external cytosines that were fully or hemi-methylated. Thus, for a given DNA sample, three methylation states at the CCGG sites, i.e., hemi-methylation of the external cytosine, full methylation of the internal cytosine, or full methylation of the external cytosine, would be revealed as different presentations of 5′-CCGG in the EcoRI + HpaII versus EcoRI + MspI digests. According to previous studies, internal hemi-methylation at CCGG sites occurs only infrequently; therefore, this state was excluded24. We henceforth refer to the hemi-methylation state as the hemi-methylation of the external cytosine and to the full methylation state as the full methylation of the internal cytosine and the full methylation of the external cytosine. The total methylation state describes both the hemi-methylation and full methylation states mentioned above. For each individual assembled sequence, it was first determined whether those with CCGG sites were: (1) present only in the HpaII cleavage sites of the EcoRI-HpaII products and the body sequences of the EcoRI-MspI products but not in the MspI-cleaved sites; (2) present only in the MspI cleavage sites of the EcoRI-MspI products and the body sequences of the EcoRI-HpaII products but not in the HpaII cleavage sites; (3) present in the body sequences of both the EcoRI-HpaII and EcoRI-MspI products but not in the HpaII or MspI cleavage sites. Condition (1) denotes a hemi-methylated state and conditions (2) and (3) correspond to fully methylated states. Methylated sites that were similarly methylated in less than two samples were excluded.
Go Classifications
GO annotations of the cassava genes were downloaded from the JGI Phytozome (http://www.phytozome.org/cassava.php). GO comparative analysis between the methylated gene groups of interest were performed using BGI WEGO (http://wego.genomics.org.cn/cgi-bin/wego/index.pl)31.
Genetic Linkage Map Construction
To identify the SNPs, indels and methylation polymorphism sites, all pairs of tags were evaluated for the presence of at least two reads. JoinMap 3.037 was used to construct the cassava linkage map for the KS population. Bi-allelic SNPs, indels and methylation polymorphism sites were identified by querying the filtered tags for pairs of sequences with the following characteristics: 1) identical in at least two reads; 2) present in >50% of the individuals; 3) passed a Fisher's exact test for independence; 4) fit to the expected Mendelian segregation ratio as demonstrated by a chi-squared test at a P<0.01; 5) possessed a threshold of ≤5.0 with a LOD score of >1.0; 6) possessed a recombination frequency of <0.4; and 7) had AFSM markers specific to the female or male parent that fit to a 1:1 segregation ratio in addition to shared AFSM markers that fit to a 3:1 segregation ratio in the KS population. If a SNP, indel or methylation polymorphism site call was heterozygous, presumably due to sequencing errors, then the call was classified as missing data. According to JoinMap 3.0, the KS population could be considered as a CP population according to the genetic background if the two parents were heterozygous. The parent-specific AFSM markers, which segregated at a 1:1 ratio in the population, were recorded as lm×ll (marker in the female parent) and nn×np (marker in the male parent). The AFSM markers that were present in both parents and segregated at a 3:1 ratio in the population were recorded as hk×hk (marker present in both parents).
Additional Information
Accession codes: Sequence Read Archive (SRA) database SRR1282409 and SRR1282413.
References
Lei, M. et al. Arabidopsis EDM2 promotes IBM1 distal polyadenylation and regulates genome DNA methylation patterns. Proc Natl Acad Sci U S A 111, 527–532 (2014).
Lippman, Z. et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 430, 471–476 (2004).
Singer, T., Yordan, C. & Martienssen, R. A. Robertson's Mutator transposons in A. thaliana are regulated by the chromatin-remodeling gene Decrease in DNA Methylation (DDM1). Genes Dev 15, 591–602 (2001).
Cubas, P., Vincent, C. & Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161 (1999).
Marfil, C. F., Camadro, E. L. & Masuelli, R. W. Phenotypic instability and epigenetic variability in diploid potato of hybrid origin, Solanum ruiz-lealii. BMC Plant Biol 9, 21 (2009).
Ronemus, M. J. et al. Demethylation-induced developmental pleiotropy in Arabidopsis. Science 273, 654–657 (1996).
Sherman, J. D. & Talbert, L. E. Vernalization-induced changes of the DNA methylation patter in winer wheat. Genome 45, 253–260 (2002).
Bibikova, M. et al. Human embryonic stem cells have a unique epigenetic signature. Genome Res 16, 1075–1083 (2006).
Down, T. A. et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol 26, 779–785 (2008).
Hatada, I. et al. A genomic scanning method for higher organisms using restriction sites as landmarks. Proc Natl Acad Sci U S A 88, 9523–9527 (1991).
Kim, K. D., Baidouri, M. E. & Jackson, S. A. Accessing epigenetic variation in the plant methylome. Brief Funct Genomics 10.1093/bfgp/elu003. (2014).
Oda, M. et al. High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res 37, 3829–3839 (2009).
Zhang, X. et al. Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 126, 1189–1201 (2006).
Cao, J. et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nature Genet 43, 956–963 (2011).
Fournier-Level, A. et al. A Map of Local Adaptation in Arabidopsis thaliana. Science 334, 86–89 (2011).
Huang, X. et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497–501 (2012).
Kump, K. L. et al. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nature Genet 43, 163–168 (2011).
Hyten, D. L. et al. High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics 11, 38 (2010).
Baird, N. A. et al. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS One 3, e3376 (2008).
Elshire, R. J. et al. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. Plos One 6, 319379 (2011).
Miller, M. R. et al. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17, 240–248 (2007).
Wang, S. et al. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Meth 9, 808–810 (2012).
Rabbi, I. Y. et al. An EST-derived SNP and SSR genetic linkage map of cassava (Manihot esculenta Crantz). Theor Appl Genet 125, 329–342 (2012).
Xiong, L. Z. et al. Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261, 439–446 (1999).
Baurens, F. C. et al. Genomic DNA methylation of juvenile and mature Acacia mangium micropropagated in vitro with reference to leaf morphology as a phase change marker. Tree Physiol 24, 401–407 (2004).
Li, Y. D. et al. Utility of the methylation-sensitive amplified polymorphism (MSAP) marker for detection of DNA methylation polymorphism and epigenetic population structure in a wild barley species (Hordeum brevisubulatum). Ecol Res 23, 927–930 (2008).
Mastan, S. G. et al. Assessment of changes in DNA methylation by methylation-sensitive amplification polymorphism in Jatrophacurcas L. subjected to salinity stress. Gene 508, 125–129 (2012).
Rathore, M. S. et al. Evaluation of DNA methylation using methylation-sensitive amplification polymorphism in plant tissues grown in vivoand in vitro. Plant Growth Regul 10.1007/s10725-014-9926-8 (2014).
Tang, X. M. et al. Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol Genet Genomics 10.1007/s00438-014-0869-6 (2014).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34, W293–297 (2006).
Pespeni, M. H. et al. Restriction Site Tiling Analysis: accurate discovery and quantitative genotyping of genome-wide polymorphisms using nucleotide array. Genome Biol 11, R44 (2010).
Borevitz, J. O. et al. Large-scale identification of single-feature polymorphisms in complex genomes. Genome Res 13, 513–523 (2003).
Feng, S. H. & Jacobsen, S. E. Epigenetic modifications in plants: an evolutionary perspective. Curr Opin Plant Biol 14, 179–186 (2011).
Osabe, K. et al. Genetic and DNA Methylation Changes in Cotton (Gossypium) Genotypes and Tissues. Plos One 9, e86049 (2014).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Stam, P. Construction of integrated genetic linkage maps by means of a new computer package: JoinMap. Plant J 5, 739–744 (1993).
Acknowledgements
This work was supported by the National Science Foundations of China (31261140363) and the 973 Program: National Basic Research Program of China (2010CB126601) and by grants from the National Science Foundations of China (31301102 and 31171230). We are grateful to C. Lu for growing cassava samples. We also wish to thank Dr. T. Aimee, Dr. T. Afton and Dr. M. Nicole from Nature Publishing Group Language Editing and Dr. C. Laura from American Journal Experts, for editing this paper.
Author information
Authors and Affiliations
Contributions
Z.X. and W.W. conceived and designed the study. Z.X. developed the original protocol for AFSM library preparation and M.Z. developed modifications of the protocol. M.Z., Z.X. and S.Z. prepared AFSM libraries for Illumina HiSeq 2500 sequencing. Z.X. created bioinformatics scripts and conducted sequence analysis and constructed the genetic linkage map. B.F. created part of the bioinformatics scripts. M.Z. wrote the paper. W.W. supervised the whole study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary File
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Xia, Z., Zou, M., Zhang, S. et al. AFSM sequencing approach: a simple and rapid method for genome-wide SNP and methylation site discovery and genetic mapping. Sci Rep 4, 7300 (2014). https://doi.org/10.1038/srep07300
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07300
This article is cited by
-
Genotyping by sequencing-based linkage map construction and identification of quantitative trait loci for yield-related traits and oil content in Jatropha (Jatropha curcas L.)
Molecular Biology Reports (2022)
-
Construction of a high-density linkage map and QTL mapping for important agronomic traits in Stylosanthes guianensis (Aubl.) Sw.
Scientific Reports (2019)
-
Construction of an ultrahigh-density genetic linkage map for Jatropha curcas L. and identification of QTL for fruit yield
Biotechnology for Biofuels (2018)
-
msgbsR: An R package for analysing methylation-sensitive restriction enzyme sequencing data
Scientific Reports (2018)
-
Spatial and temporal expression of cytosine-5 DNA methyltransferase and DNA demethylase gene families of the Ricinus communis during seed development and drought stress
Plant Growth Regulation (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
