
Abstract
Saccharomyces cerevisiae strains vary in their ability to develop and enhance sensory attributes of alcoholic beverages and are often found growing in mixed strain fermentations; however, quantifying individual strains is challenging due to quantification inaccuracies, low marker longevity and compromised kinetics. We developed a fluorescent probe, consisting of glutathione molecules conjugated to a quantum dot (QD). Two S. cerevisiae strains were incubated with different coloured probes (QD attached to glutathione molecules, QD-GSH), fermented at multiple ratios and quantified using confocal microscopy. The QD method was compared with a culture method using microsatellite DNA analysis (MS method). Probes were taken up by an ADP1 encoded transporter, transferred from mother cell to daughter cell, detectable in strains throughout fermentation and were non-toxic. This resulted in a new quantification method that was more accurate and efficient than the MS method.
Similar content being viewed by others

Introduction
Outside the laboratory, microbial monocultures are rare. Rather, most food and beverage fermentations contain a mixed variety of functionally ambiguous yeasts. Some of which strongly influence the sensorial attributes of the final product. Selected for their superior fermentation performance and reduced production of undesirable metabolites, commercially available strains of Saccharomyces cerevisiae are commonly used as fermentation inoculants. Nevertheless, other S. cerevisiae strains can still persist, resulting in co-fermentations between two or more strains of this species1,2. These mixed fermentations are considered quite beneficial in winemaking. For example, they produce more complex sensorial attributes than wines fermented from pure culture3,4. Because of these associated benefits, interactions between multiple S. cerevisiae strains throughout mixed culture fermentations are of considerable interest.
Tracking and quantifying individual strains within a mixed culture is not trivial. Current molecular methods are non-visual and generally require samples to first be grown in culture media before analysis4. Additionally, traditional dyes are not ideal for quantifying cells throughout an extended fermentation (e.g. 5–14 days)5 such as those in winemaking. Most staining techniques require the integrity of the cellular membrane to be compromised before fluorescent tags can be absorbed by the yeast. This may ultimately affect yeast growth and fermentation kinetics. Also, because only a select few organelles are transferred from mother cells to daughter cells during cell division6,7, stains that are not directly taken up or absorbed by the yeast cannot be transferred to newly formed cells8.
In this way, fluorescent tags bound to glutathione have distinctive advantages over traditional fluorescent stains. Glutathione (GSH) is frequently taken up by S. cerevisiae via several transport mechanisms6,9. Yeast are able to metabolize GSH as a sole source of sulfur, or otherwise transfer a portion to storage vacuoles to be used at a later time. Therefore, fluorescent tags bound to GSH can be taken up by yeast and transferred within the cytosol and vacuoles—both of which are transferred from the mother cell to daughter cells during cell division6,7. Commercial kits (i.e., CellTracker™ Invitrogen™, Burlington, Ontario) successfully exploit some of these inherent properties of glutathione. However, these fluorescent tracers are only stable for ~3 days; a timeframe well below the 5–14 days necessary for the completion of a typical wine fermentation5.
In lieu of other tags, highly fluorescent nanoparticles, called quantum dots (QDs), remain photochemically stable for orders of magnitude longer than traditional dyes10. Quantum dots are typically composed of a metal core, generally a chalcogenide (e.g., selenide or sulfide) sheathed within a functional organic polymer. This polymer protects the QD from degradation and provides a chemical surface to which targeting probes can be attached11. Recent studies have documented the uptake of QD probes in bacteria, yeast and multicellular fungi12,13,14,15. Here we describe a novel QD-based method used to visually and quantitatively track multiple S. cerevisiae strains simultaneously throughout mixed culture fermentations in vivo.
Our QD probe consisted of GSH bound to QDs (QD-GSH) (Fig. 1a). Carboxyl groups of GSH were covalently bound to amino terminated blue (495 nm emission) or red (631 nm) QDs (Crystalplex, Pittsburgh, USA). Red QD-GSH was incubated with the S. cerevisiae strain Lalvin® (RC212) and blue QD-GSH with Lalvin® ICV-(D254). These QDs were selected because of their commercial availability, small size (< 5.5 nm diameter) and composition-tuned CdSeS cores wrapped in ZnS shells. Because emission colors were tuned based on core compositions rather than overall QD size, each of our QD-conjugates were equivalent in size and mass regardless of emission color16.

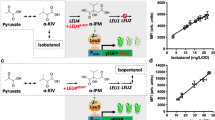
A quantum dot (QD) method to track multiple S. cerevisiae strains in mixed culture.
Conceptual diagram of quantum dot-glutathione (QD-GSH) conjugates (a) and their corresponding reference spectra after being taken up by yeast cells during 24 h of incubation (b). The blue line references QD-GSH (495 nm emission) within D254 and the red line references QD-GSH (631 nm emission) within RC212. (c) Spectral scans were collected from each fermentation, sorbel filtered and analyzed as a two-channel image; where color corresponds to the emission fingerprint associated with each QD-GSH-labelled strain. Spectral scans were collected from 470–670 nm at 5 nm intervals using an Olympus FV1000 confocal microscope. Scale bar is 25 µm. (d) Confocal laser scans of the QD-GSH probe being transferred from mother cell to daughter cell via the cytosol and vacuole. From left to right: bright field image of RC212; QD-GSH fluorescence within the yeast cell; and a superimposed image of the two. Each strain was able to transfer the QD-GSH probe from mother cell to daughter cell throughout the entire course of fermentation. Scale bars are 5 µm. (e) Uptake of QD-GSH by different S. cerevisiae knockout strains. There were five QD-GSH treatments comprised of four knockouts (opt1, ycf1, gex2, adp1) and their analogous wild type BY4743 (WT); which served as a positive control. Knockout strains lacking ADP1 transporters displayed significantly less QD-GSH uptake in comparison to every other strain, including the wild type control (p < 0.0001; n = 4). Consequently, QD-GSH was most likely acquired by yeast cells via an ADP1-mediated transport mechanism. All non-spectral confocal laser scans (535 ± 10 nm emission fluorescence) were collected at 40x using an Olympus FV1000 confocal microscope. Values are average means ± S.E.
Results and Discussion
On average, each of our QD-GSH conjugates were ~5.6 nm in diameter based on Stokes-Einstein equation and raster image correlation spectroscopy14,17. Both yeast strains took up the QD-GSH probes within 8 hours of incubation. After uptake, we identified each strain based on its unique emission spectra using a confocal microscope (Fig. 1b and c). We did not observe any cells that contained both QD-GSH colours. This suggests that there was no plasmogamy or QD-GSH loss and reabsorption occurring between the two strains during the course of fermentation. We determined that the QD-GSH probes were being transferred from mother cell to daughter cell via a portion of cytosol or vacuole (Fig. 1d). These results are consistent with previous studies that have documented the transfer of cytosol, vacuole and GSH from mother cell to daughter cell in S. cerevisiae6,7.
To determine the mechanism for QD-GSH uptake, we focused on genetic knockouts that lacked membrane transporters associated with glutathione movement (based on gene and protein sequences collected from the GenBank® online database). The homozygous diploid deletants in the BY4743 background were obtained from EUROSCARF (Frankfurt, Germany) and included knockouts in one of the following: YJL212C (OPT1), an oligopeptide transporter found in plasma membranes; YKR106W (GEX2), a glutathione antiporter located in vacuolar and plasma membranes; YDR135C (YCF1), a cadmium-glutathione transporter found in vacuolar membranes; and YCR011C (ADP1), a putative ABC permease found in vacuolar and plasma membranes. Our data indicates that QD-GSH uptake mainly occurred via ADP1-associated pathways (Fig. 1e). Although, this transporter was discovered nearly two decades ago, very little is known about its function. This is the first study to document GSH uptake by the ADP1 transporter. Previously, the only link between ADP1 and GSH uptake by S. cerevisiae was based on putative gene sequences found in other organisms (GenBank accession no. NC_001135).
To test the performance of the QD method in comparison to a microsatellite approach (MS method), two separate sets of experimental fermentations were conducted in parallel. One set was analyzed using a novel QD method (where the initial starting cultures were QD-labelled) and the other using a standard microsatellite technique3,18 (where polymorphisms of microsatellite DNA were used to distinguish between strains). For each fermentation set, three initial inoculation treatments (1:1, 3:1 and 9:1 of RC212:D254) were replicated in triplicate. We found that the QD probe was detectable for at least 120 hours during fermentation; the amount of time it took for > 98% of sugars to be depleted (Fig. 2a). Previous studies have documented the photochemical stability of QDs in other organisms over extended periods10,13. For example, after taking up QD-labelled adenine, Bacillus subtilis cells are able to retain their fluorescence for more than a year13. In addition, both cell growth and sugar kinetics were statistically identical in fermentations composed of QD-labelled yeast in comparison to those lacking QDs. This suggests that the QD-GSH probes were likely non-toxic to the yeast, having no significant effect on fermentation kinetics (Fig. 2a). Furthermore, there was a significant correlation between the results of the two methods (R = 0.87; Fig. 2b). Cell counts (RC212:D254) produced by the QD method were statistically identical to those produced by the MS method for the individual inoculation ratios at every stage of fermentation (Fig. 2c, d). However, the QD data was observably less variable than the MS data (Fig. 2c, d). In the QD method, cell counts were significantly different across inoculation ratios at every stage of fermentation; increasing in order from 1:1, 3:1 and 9:1, respectively (Fig. 2d). While similar trends were observed using the MS method, only 2 out of the 3 fermentation stages were significant (Fig. 2c). This variation was likely due to the collective number of cells that were practically assessed by each tracking method. In the QD method, more than 1,600 cells were analyzed per replicate, two orders of magnitude greater than the 20 cells analyzed in the MS method. Even though both methods are able to successfully identify different S. cerevisiae strains from mixed cultures, the QD method is capable of analyzing a much larger cell pool, thereby producing data that is statistically superior, but at a fraction of the time and cost. Generally, 16–24 cell colonies are identified per sample replicate with MS-based methods19,20. Nevertheless, when liberated from time demands and associated costs, it may be possible to improve the quality of data generated by the MS method by merely increasing the number of cells analyzed per sample replicate.

Comparison of the quantum dot (QD) method versus a more traditional microsatellite (MS) method when quantifying multiple yeast strains in mixed culture fermentations.
(a) There were no significant differences between cell growth (p = 0.70) or sugar utilization kinetics (p = 0.78) when comparing fermentations incubated with QD-GSH versus those without (based on ANCOVAs). These data suggest that the QD-GSH was non-toxic to the yeast throughout the entire course of fermentation. (b) Moreover, there was a positive and strong correlation between the QD and the MS method across all ratios and time points (R = 0.87). To visually display these results, this data is presented as a linear regression (r2 = 0.76 and p = 0.003). Individual data points are RC212:D254 ratios of cell numbers. Although similar trends were exhibited in data sets from both the (c) MS and (d) QD method, there was significantly greater variability in the MS results (based on average standard error); albeit only a marginal significance across all fermentation stages (p = 0.07). Significant differences between ratio treatments were determined using a one-way ANOVA (p < 0.05). Values with the same letters at a given fermentation stage are not significantly different according to a Spearman rank test. Values are means ± S.E; dn = 3, 20 yeast cells analyzed per flask per time point; en = 3, >1600 yeast cells analyzed per flask per time point.
More than 95% of the cells analyzed using the QD method contained detectable amounts of QD-GSH. The cells that lacked QD-GSH fluorescence appeared flaccid. Although, we did not directly test cell viability in this study, it is possible that the cells lacking QD-GSH were non-viable. When yeast cells become inactive they start autolysis, which ruptures the vacuoles, discharges the intracellular contents and eventually breaks down the rest of the cell5. Because QD-GSH is located in vacuoles and cytosol within yeast, cells lacking QD-GSH may represent inactive cells that have already started the autolytic process.
The QD method is able to visually and quantitatively track multiple S. cerevisiae strains throughout an entire fermentation, providing precise in vivo data at a fraction of time and cost associated with current MS techniques. Because this method can distinguish between multiple emission spectra, even those that closely overlap, a multitude of different strains can be tracked simultaneously using different QD colours. Nonetheless, given the extensive use of S.cerevisiae as a model organism, the pragmatic benefits of the QD method extend well beyond the wine-based questions addressed in this study. For example, defining how yeast behave in mixed cultures could help improve our ability to predict how genetically different cells will react in close proximity to one another. Also, since the QD method enables the tagging of individual cells and consequently their progeny, cell development can be examined while in close proximity to other cells or strains, such as those found on solid media or in biofilms. The impact of these interactions on gene expression could be determined by separating the QD-labelled cells using a fluorescence activated cell sorter (FACS) before molecular analysis. Moreover, the novel ADP1 transport mechanism described in this study may provide a pathway to deliver pharmaceutically important molecules to eukaryotic cells. For example, nanoparticle drug delivery has become increasingly focused on GSH-mediated release mechanisms21,22. By weakly associating (e.g., hydrostatic associations) an outer GSH monolayer to a drug-carrying nanoparticle, the conjugate may function as a Trojan horse; first being taken up by natural GSH pathways and then unpackaged within the cell via GSH-mediated release., Furthermore, recent studies have documented elevated GSH levels in cancerous cells23. Thus, the ability to quantify QD-GSH uptake rates in vivo could parenthetically serve as a proxy to identify, sort and possibly even treat diseased cells.
Methods
Quantum dot conjugation
Quantum dot-glutathione (QD-GSH) conjugates were composed of glutathione molecules (Sigma-Aldrich, Oakville, Canada) covalently bound to amino-terminated blue (495 nm emission), or red (631 nm) quantum dots (Crystalplex, Pittsburgh, USA). Two separate binding reactions, one for each QD color, were performed simultaneously, in the dark, in 2 ml amber glass vials. Each reaction contained 3.17 nmol QDs and 4.52 nmol GSH at a 1 to 33 ratio (QD:GSH). Reactions were performed in 1 ml of 10 mM borate buffer at pH 6.8 with an excess amount (~200 mg) of the binding reagent 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (Sigma-Aldrich, Oakville, Canada). Each reaction vial was vortexed for 20 minutes. Reaction vials were then individually dialyzed against 1 L of sterile water for 30 minutes, after which, 1.05 mM of each QD-GSH solution was transferred to an amber vial, wrapped in foil and stored at 4°C in the dark. The QDs used in this study were selected because of their commercial availability, small size (< 5.5 nm diameter) and composition-tuned CdSeS cores wrapped in ZnS shells. Because emission colors were tuned based on core compositions rather than overall QD size, each of our QD-conjugates were equivalent in size and mass regardless of emission color16. On average, each of our QD-GSH conjugates were 5.6 nm. Quantum dot size (i.e., average hydrodynamic diameter) was estimated before and after conjugation using Stokes-Einstein equation and raster image correlation spectroscopy14,17.
Experimental fermentations
To test the performance of the QD method in comparison to a more standard molecular approach, two separate sets of experimental fermentations were conducted in parallel. One set of fermentations was analyzed using the QD method and the other using standard microsatellite (MS) techniques. Commercial wine strains of S. cerevisiae, Lalvin® RC212 (RC212) and Lalvin® ICV-D254 (D254), were sourced directly from commercially available active dried yeast preparations (Lallemand Montreal Quebec, Canada). Each strain was rehydrated separately in 30 ml of YEPD media. After rehydration, cell numbers were determined via direct microscopic count using a hemocytometer. Starter cultures were initiated by adding each strain to 1 × 106 cells per ml in separate aliquots of 0.22 μm filter-sterilized Pinot Noir juice diluted 1:1 with sterile water (yielding ~ 10.5 °Brix). The two starter cultures were grown aerobically on an incubator shaker (120 rpm) at 28°C. After 20 h, each starter culture was checked for purity and cells were added at 5 × 106 cells per ml to individual 250 ml fermentation flasks containing 100 ml of 0.22 μm filter-sterilized full strength (21 °Brix) Pinot Noir juice. Each fermentation flask was fitted with an airlock to maintain an anaerobic environment. There were three initial inoculation treatments for each fermentation (1:1, 3:1 and 9:1 of RC212:D254 respectively) and three replicate flasks for each treatment. There were separate fermentations and controls for each cell-count method, which are described in the corresponding sections below.
To test and compare each method during the three main stages of fermentation (early, mid and end), each fermentation was monitored every 12 h by refractive index of the supernatant; starting with an initial reading of 21 °Brix (~200 g l−1 of sugar). When the fermentations dropped below 10 °Brix, Benedict's solution was used on fermentation supernatant samples to determine the residual sugar concentration. Fermentations were considered complete when residual sugar was less than 2 g l−1. Fermentation stages were defined as early (20 h, ~150 g l−1 sugars), mid (45 h, ~90 g l−1) and end (120 h < 2 g l−1).
Cell growth and kinetics
To determine cell growth and kinetics, samples from each fermentation flask were analyzed every 12 h throughout the entire course of fermentation. Culture growth (cell count ml−1) was determined from duplicate counts using a hemocytometer on a light microscope at 40x. Fermentation kinetics was estimated based on the metabolism of D-fructose and D-glucose throughout fermentation. Culture supernatants were prepared from each ferment flask by centrifuging 1 ml samples for 1 min at 21,000 × g. Residual sugar concentrations were determined using a D-fructose and D-glucose assay kit (Megazyme, Bray, Ireland).
Quantum dot quantification of yeast strains
To quantify the abundance of each yeast strain using QD-GSH, we labelled the starter cultures of RC212 and D254 with either blue or red QD-GSH before the start of fermentation. Each starter culture was inoculated with 3 ml (1.05 mM) of QD-GSH. Cultures were incubated at 28°C anaerobically; where red QD-GSH conjugates were mixed with RC212 and blue conjugates with D254. After 23 h of incubation, QD-tagged yeasts (5 × 106 cells mL−1) were used to start a set of QD-GSH-labelled fermentations. In addition, there were two accompanying sets of control fermentations; QD-control fermentations to test for non-specific binding of unbound QDs and no-treatment control fermentations to serve as a baseline for cellular growth, kinetics and autofluorescence. Each fermentation set was started at three initial RC212 to D2654 ratios (1:1, 3:1 and 9:1, respectively). There were three replicates per treatment for a total of 27 individual fermentations. Samples (1 ml) were collected from each flask at early, mid and end stages of fermentation. Each sample was washed by centrifugation with 10 mM borate buffer at pH 6.8. After resuspension in fresh buffer, 75 µl was sandwiched between two coverslips for confocal analysis.
Spectral scans were observed from each coverslide at 20x using emission fingerprinting confocal microscopy. Emission fingerprinting is a spectral-based technique used to analyze multiple fluorescent fluorophores despite overlapping emission profiles. Because emission fingerprinting is used to identify the unique spectral profile of individual particles, rather than the fluorescence intensity across a short wavelength of color, this technique is ideal in systems subject to autofluorescence and spectral overlap. For example, since most yeast autofluoresce24, this technique can be used to discriminate between different yeast strains tagged with different fluorophores despite the amount of spectral overlap in the system. We created reference spectra for RC212 and D254 after 24 h of QD-GSH incubation while in pure culture (Fig. 1b). These fingerprints were used as standards to convert the data from each spectral scan into two-channel images (red RC212 cells versus blue D254). To help standardize the size and shape of each fluorescently-labelled yeast, each two-channel image was filtered using sorbel operators (Olympus FluoView™ FV1000 spectral unmixing) (Fig. 1c). Filtered images were imported into Fiji ImageJ25 and converted to binary files. Each image was individually analyzed for cell counts using an automated particle analysis approach following Schindelin et al25. Each channel was analyzed independently of one another. A ratio of RC212:D254 (cell number) was calculated from the total number RC212 and D254 observed per spectral scan. Roughly 400 cells were analyzed per coverslide. Four coverslides were averaged per flask; for a collective total of nearly 1600 cells analyzed per flask replicate (n = 3). Spectral scans were taken from 470–670 nm at 5 nm intervals using an Olympus FluoView™ FV1000 confocal microscope (Markham, Ontario, Canada).
Microsatellite quantification of yeast strains
To quantify the abundance of each strain throughout fermentation using a molecular based approach, standard microsatellite (MS) techniques were used following Legras et al18 and Howell et al.3. For MS analysis, 1 ml samples were plated onto YEPD agar (yeast extract 10 g l−1, peptone 20 g l−1, glucose 20 g l−1, agar 20 g l−1) from each sampled fermentation stage from all three ratio treatments. Twenty colonies were randomly selected from each culture plate, DNA was extracted and analyzed using a slight modification of the standard MS operating procedures18. Extraction of genomic DNA from each colony was performed using a Sigma DNA extraction kit (Sigma-Aldrich, Oakville, Canada). Colonies along with 50 μl of the extraction solution were aseptically placed into a well of a 96 well PCR plate. The plates containing extraction solution and colonies were heated to 95°C for 15 min in an Applied Biosystems Veriti Thermal Cycler (Foster City, CA, USA) and then stored frozen prior to further processing. Two microsatellite loci (C11 and SCYOR267c) were used to distinguish the two different S. cerevisiae strains. DNA fragments were amplified using primer pairs described for C11 and SCYOR267c microsatellite loci18. Fragments sizes of the loci were quantified using a 3130xl DNA sequencer (Applied Biosystems, Foster City, CA, USA). RC212:D254 ratios were calculated across the twenty colonies for each treatment at each stage.
Characterization of QD-GSH uptake by yeast strains
To determine the uptake mechanisms used by S. cerevisiae during acquisition of QD-labelled glutathione, we quantified and compared QD-GSH uptake across four knockout strains and their associated wild type. We focused on genetic knockouts that lacked membrane transporters associated with glutathione movement (based on gene and protein sequences collected from the online GenBank® database). Of our four knockout strains, each lacked one of the following: YJL212C (OPT1), a gene encoding for an oligopeptide transporter found in plasma membranes; YKR106W (GEX2), a gene encoding for a glutathione antiporter located in both vacuolar and plasma membranes; YDR135C (YCF1), a gene encoding for cadmium and glutathione transporter found in vacuolar membranes; and YCR011C (ADP1), a gene encoding for a putative ABC permease (GenBank® Database). Each deletion strain was sourced from EUROSCARF having been constructed in the diploid laboratory-based wild type background of BY474326.
In total, there were five QD-GSH treatments comprised of the four knockouts (opt1, ycf1, gex2, adp1) and their analogous wild type (BY4743); which served as a positive control. There were two additional control treatments: a QD-control; where BY4743 was inoculated with unconjugated QDs to test for non-specific QD uptake and a no-treatment control; where BY4743 was analyzed without any QD treatment to determine yeast autofluorescence. There were four replicates per treatment for a total of 28 cultures.
For the QD-GSH treatments, individual strains were supplemented with 1.05 mM of green QD-GSH (535 nm emission) in 25 ml of YEPD (200 g l−1 sugar). The QD-control cultures were composed of 1 × 106 BY4743 cells treated with unconjugated amino-terminated QDs in 25 ml of YEPD and the no-treatment controls consisted of BY4743 (1 × 106 cells) in 25 ml of YEPD. Cultures were incubated at 28°C for 24 h with shaking (120 rpm). After incubation, 50 μl was taken from each culture and immobilized on cover slides with 100 μl polyvinyl alcohol-lactic acid-glycerol (PVLG). Slides were cured overnight at 60°C. As the media hardened, 1 μl of PS-Speck™ calibration beads (1 μm diameter; 520–540 nm emission) (Life Technologies Invitrogen, Burlington Ontario, Canada) was added to each slide. Coverslips were stored in the dark at 4°C. Green QDs (535 nm emission; Crystalplex, Pittsburgh, USA) were used in each of the five QD-GSH treatments.
Confocal laser scans (535 ± 10 nm emission fluorescence) were collected at 40x from each coverslide. Fluorescence intensity (A.U.) per µm3 yeast was determined using Fiji ImageJ25 with each yeast being analyzed at a 0.6 µm focal depth. To minimize variability across samples, the observed fluorescence intensity of each strain was divided by the average intensity of ten calibration beads; located within the same focal plane as the observed yeast. These calibrated ratios were then converted to nmol QD-GSH concentration using a normalized QD-GSH (535 nm emission) calibration curve composed of eight QD concentrations: 3.33 × 10−5 nmol ml−1, 3.33 × 10−4 nmol ml−1, 3.33 × 10−3 nmol ml−1, 3.33 × 10−2 nmol ml−1, 0.333 nmol ml−1, 0.666 nmol ml−1, 1.665 nmol ml−1 and 3.33 nmol ml−1. Each concentration was transferred to a welled microscope slide sealed with a pap pen and each well received 1 μl of PS-Speck™ (520–540 nm emission) calibration beads. Ten fluorescence intensities were calculated per calibration well. These values were averaged per concentration and divided by the average intensity of ten randomly chosen calibration beads within the same focal plane. The resulting calibration ratios were regressed against the known QD-GSH concentrations to convert fluorescent intensity to nmol QD-GSH (r2 = 0.998, p < 0.0001; based on linear regression). All confocal spectroscopy was performed using an Olympus FluoView™ FV1000 confocal microscope.
Data analysis
We performed analyses of variance (ANOVAs) to determine QD-GSH uptake mechanisms in S. cerevisiae. Specifically, we used two-tailed ANOVAs to test for differences in QD-GSH uptake by knockout strain; where the dependent variables were (opt1, ycf1, gex2, adp1) or wild type (BY4743). To determine whether each method (QD method versus MS method) provided results that were significantly different across starting inoculation ratios at each time point, a one-way ANOVA was performed; with cell counts (RC212:D254 ratio) as the dependent variable and initial fermentation ratios (1:1, 3:1, or 9:1) as the independent variables. When significance was indicated, a multiple comparison Tukey's honest significant difference (HSD) test was used (α = 0.05 level). Because we were unable to transform the data for this test to meet the assumptions of normality, this analysis was performed on Conover and Iman ranked data. To test whether the MS method and QD method produced comparable data and led to statically similar results, a Pearson correlation was performed on cell count data produced by the QD method versus the MS method. Cell counts were included across all time points and initial starting ratios. To visually display the results of this correlation, the data was graphed as a linear regression with corresponding statistical values (Fig. 2b). Additionally, the average standard error of both methods was compared using a one-way ANOVA. To determine whether QD-GSH had negative effects on yeast growth or kinetics, separate ANCOVAs were performed on either cell growth data or sugar concentrations collected from QD-fermentations versus non-QD fermentations. To meet the linear assumptions of ANCOVA, cell growth data and sugar concentrations were log transformed for this statistical test. Differences were considered significant when p < 0.05. All statistical analyses were performed using JMP version 11.0 (SAS Institute Inc., Cary, North Carolina).
References
Hall, B., Durall, D. M. & Stanley, G. Population dynamics of Saccharomyces cerevisiae during spontaneous fermentation at a British Columbia winery. Am. J. Enol. Viticult. 62, 66–72 (2011).
Pramateftaki, P., Lanaridis, P. & Typas, M. Molecular identification of wine yeasts at species or strain level: a case study with strains from two vine growing areas of Greece. J. Appl. Microbiol. 89, 236–248 (2000).
Howell, K. S. et al. Metabolic profiling as a tool for revealing Saccharomyces interactions during wine fermentation. FEMS Yeast Res. 6, 91–101 (2006).
Saberi, S., Cliff, M. & van Vuuren, H. Impact of mixed S. cerevisiae strains on the production of volatiles and estimated sensory profiles of Chardonnay wines. Food Res. Int. 48, 725–735 (2012).
Boulton, R. B. et al. Principles and Practices of Winemaking. (Springer., 2010).
Zadziński, R., Maszewski, J. & Bartosz, G. Transport of glutathione s-conjugates in the yeast Saccharomyces cerevisiae. Cell Biol. Int. 20, 325–330 (1996).
Tsai, I. T. et al. Interorganelle interactions and inheritance patterns of nuclei and vacuoles in budding yeast meiosis. Autophagy 10, 285–295 (2014).
Cabib, E., Roberts, R. & Bowers, B. Synthesis of the yeast cell wall and its regulation. Annu. Rev. Biochem 51, 763–793 (1982).
Penninckx, M. J. An overview on glutathione in Saccharomyces versus non-conventional yeasts. FEMS Yeast Res. 2, 295–305 (2002).
Resch-Genger, U. et al. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 5, 763–775 (2008).
Biju, V., Itoh, T. & Ishikawa, M. Delivering quantum dots to cells: bioconjugated quantum dots for targeted and nonspecific extracellular and intracellular imaging. Chem. Soc. Rev. 39, 3031–3056 (2010).
Coulon, J. et al. Glycosylated quantum dots for the selective labelling of Kluyveromyces bulgaricus and Saccharomyces cerevisiae yeast strains. J. Fluorescence 20, 591–597 (2010).
Kloepfer, J., Mielke, R. & Nadeau, J. Uptake of CdSe and CdSe/ZnS quantum dots into bacteria via purine-dependent mechanisms. Appl. Environ. Microbiol. 71, 2548–2557 (2005).
Whiteside, M. D. et al. Organic nitrogen uptake by arbuscular mycorrhizal fungi in a boreal forest. Soil Biol. Biochem. 55, 7–13 (2012).
Whiteside, M. D., Treseder, K. K. & Atsatt, P. R. The brighter side of soils: Quantum dots track organic nitrogen through fungi and plants. Ecology 90, 100–108 (2009).
Bailey, R. E. & Nie, S. Alloyed semiconductor quantum dots: Tuning the optical properties without changing the particle size. J. Am. Chem. Soc. 125, 7100–7106 (2003).
Digman, M. A. et al. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 89, 1317–1327 (2005).
Legras, J.-L. et al. Selection of hypervariable microsatellite loci for the characterization of Saccharomyces cerevisiae strains. Int. J. Food Microbiol. 102, 73–83 (2005).
Lange, J. N. et al. Implantation and persistence of yeast inoculum in Pinot noir fermentations at three Canadian wineries. Int. J. Food Microbiol. 180, 56–61 (2014).
Capece, A. et al. Diversity of Saccharomyces cerevisiae yeasts associated to spontaneously fermenting grapes from an Italian “heroic vine-growing area”. Food Microbiol. 31, 159–166 (2012).
Cheng, R. et al. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J. Control. Release 152, 2–12 (2011).
Hong, R. et al. Glutathione-mediated delivery and release using monolayer protected nanoparticle carriers J. Am. Chem. Soc. 128, 1078–1079 (2006).
Tan, L., Wan, A. & Li, H. Synthesis of near-infrared quantum dots in cultured cancer cells. ACS Appl. Mater. Interfaces 6, 18–23 (2013).
Billinton, N. & Knight, A. W. Seeing the wood through the trees: A review of techniques for distinguishing green fluorescent protein from endogenous autofluorescence. Anal. Biochem. 291, 175–197 (2001).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Winzeler, E. A. et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 (1999).
Acknowledgements
This work was supported by Quails' Gate Estate Winery and Natural Sciences and Research Engineering Council (NSERC) through a NSERC Collaborative Research Development (CRD) grant CRDPJ 406796-10 as well as a UBC internal wine grant. V,J.'s contribution was supported in part by funding from the Grape and Wine Research and Development Corporation (Project UA 1101). We acknowledge the use of the instruments in the Facility for Environmental and Biological Imaging on the Okanagan Campus at UBC, funded through the CFI, BCKDF and Olympus Canada. We thank Lallemand Inc. for donation of Lalvin® yeasts.
Author information
Authors and Affiliations
Contributions
F.S.G. and M.D.W. developed the concept, designed and performed the experiments and analyzed the data. V.J. and D.M.D. supervised the project. F.S.G., M.D.W., V.J. and D.M.D. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Gustafsson, F., Whiteside, M., Jiranek, V. et al. Development and use of a quantum dot probe to track multiple yeast strains in mixed culture. Sci Rep 4, 6971 (2014). https://doi.org/10.1038/srep06971
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06971
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
