
Abstract
We propose a series of icosahedral matryoshka clusters of A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd), which possess large HOMO-LUMO gaps (1.29 to 1.54 eV) and low formation energies (0.06 to 0.21 eV/atom). A global minimum search using a genetic algorithm and density functional theory calculations confirms that such onion-like three-shell structures are the ground states for these A21B12 binary clusters. All of these icosahedral matryoshka clusters, including two previously found ones, i.e., [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12−, follow the 108-electron rule, which originates from the high Ih symmetry and consequently the splitting of superatom orbitals of high angular momentum. More interestingly, two magnetic matryoshka clusters, i.e., Sn@Mn12@Sn20 and Pb@Mn12@Pb20, are designed, which combine a large magnetic moment of 28 µB, a moderate HOMO-LUMO gap and weak inter-cluster interaction energy, making them ideal building blocks in novel magnetic materials and devices.
Similar content being viewed by others
Introduction
The first evidence for the formation of bare anionic clusters of the post-transition elements, particularly those of groups 14 and 15, was obtained by Zintl and co-workers1,2,3,4 in the 1930s from the potentiometric titrations of post-transition elements with alkali metals in liquid ammonia. However, definitive structure determinations of these clusters became possible only in the 1970s, when Kummer and Diehl reported the first structurally-authenticated Zintl anion cluster of Na4Sn95. The original post-transition element clusters were empty, i. e., they contained no interstitial atoms in the center of the cluster polyhedron. However, subsequent synthetic studies led to the discovery of clusters containing interstitial transition metals. Examples of structurally characterized such clusters based on 10-vertex outer polyhedra include the anionic indium clusters M@In1010− (M = Ni, Pd, Pt)6 in the intermetallics K10In10M, the anionic lead clusters M@Pb102− in [K(2,2,2-crypt)]2[M@Pb10] (M = Ni, Pd, Pt)7, Fe@Sn103− in [K(2,2,2-crypt)]3[Fe@Sn10]8, the cationic pentagonal antiprismatic bismuth cluster Pd@Bi104+ in Bi14PdBr16 ( = [Pd@Bi10][BiBr4]4)9 and the centered pentagonal prismatic clusters M@Ge103− (M = Co10, Fe11). Interstitial metal atoms are also found in the icosahedral M@Pb122− clusters (M = Ni, Pd, Pt)7,12. Furthermore, Al3+ has been encapsulated in Pb122−, Pb102− and Sn102− clusters, which have been characterized by mass spectroscopy and density functional theory (DFT) calculations13,14.
Attempts to expand the knowledge of transition metal derivatives of post-transition element clusters led to the discovery of even more complicated structures. Of particular interest is the cluster [As@Ni12@As20]3−, isolated as its n-Bu4P+ salt and structurally characterized by X-ray diffraction15. This cluster anion has a perfect icosahedral (Ih) symmetrical three-shell matryoshka doll structure consisting of an outer As20 regular dodecahedron encapsulating an Ni12 icosahedron, which in turn encapsulates an As3− trianion (Figure 1). Beyond the filled d10 shells of the arsenic and nickel atoms, this system has a total of 108 valence electrons, among which 40 skeletal electrons16 correspond to a “magic” number of a jellium sphere17 with 1S21P61D102S21F142P6 molecular orbital configuration.

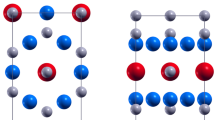
Atomic structure of an icosahedral matryoshka cluster of C@B12@A20 (left) and its schematic plot (right).
Initially the icosahedral matryoshka [As@Ni12@As20]3− structure with 108 valence electrons was considered as an anomaly, unique with an unusual combination of specific electronic and steric properties of its arsenic and nickel components. However, in 2011 Stegmaier and Fässler18 reported a completely analogous [Sn@Cu12@Sn20]12− icosahedral matryoshka doll anion in the M12Cu12Sn21 intermetallics (M = Na, K). This [Sn@Cu12@Sn20]12− anion is exactly isoelectronic with the [As@Ni12@As20]3− anion with 108 valence electrons beyond the filled d10 shells of the tin and copper components. Similar bonding models appear to be applicable to both the [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12− systems. Furthermore, the observation of the icosahedral matryoshka doll structure in both the soluble [As@Ni12@As20]3− anion and the [Sn@Cu12@Sn20]12− anion in the A12Cu12Sn21 intermetallics suggests that such structures are particularly favorable in clusters where 108 total valence electrons are available from the elements composing the three layers of the matryoshka structure.
Parallel to the condensed phased clusters19 such as those discussed above, gas-phase clusters have been intensively studied during the past three decades since they exhibit many fascinating physical and chemical properties that depend on their size, geometry and composition20. Interestingly, some specific clusters with appreciable stability mimic the chemical behavior of elemental atoms in the periodic table and thus can be regarded as “superatoms”21,22,23,24. In these superatom clusters, the electronic states are delocalized over the entire cluster with spatial shapes resembling the atomic orbitals. The corresponding energy eigenvalues can be grouped into atomic-like shells (1S, 1P, 1D, 2S, 1F, 2P, …), depending on the degeneracy and spatial symmetry of the molecular orbitals. Unlike the atoms, the physical and chemical properties of superclusters can be tailored through selection of size and composition, making them very promising building blocks for new materials25,26.
The concept of “superatoms” was originally proposed by Khanna and Jena in their pioneering paper21. For example, an Al13− cluster with filled 1S2, 1P6, 1D10, 2S2, 1F14 and 2P6 shells exhibits strong resistance to oxidation27. Meanwhile, with an electron affinity (EA = 3.57 eV)22 only slightly lower than that of the Cl atom (3.62 eV), a neutral Al13 cluster was considered as a superhalogen by Bergeron and co-workers27, though this nomenclature is not consistent with the original definition of superhallogen by Bolydyrev and Gutsev28, which defined superhalogen as a cluster/molecule whose electron affinity is larger than that of any halogen atom. Moreover, a neutral Al14 cluster behaves like an alkaline earth atom29.
Usually, a superatom cluster is stable because it has filled electron shells with paired electrons, corresponding to a nonmagnetic state. The concept of magnetic superatom was initially introduced by Kumar and Kawazoe30, who predicted Mn@X12 clusters (X = Ge, Sn) as icosahedral magnetic superatoms with a high magnetic moment of 5 μB and a large highest occupied molecular orbital-lowest unoccupied molecular orbital (HOMO-LUMO) gap of ~1.1 eV. By appropriately combining the localized d states with magnetism and the delocalized superatom orbitals that stabilize the entire cluster, Khanna and co-workers recently designed a series of magnetic superatoms31, such as VCs832, VNa8−33, Mg8Fe34, Ca8Fe35, MnCa936, MnSr937, ScK12 and ScCs1238. All of them possess enhanced stabilities, substantial HOMO-LUMO gaps (usually about 0.4 ~ 0.7 eV) and large magnetic moments on the transition metal atom (up to 5 μB).
Stimulated by the above-mentioned progresses, here we propose a series of icosahedral matryoshka clusters, i.e., A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd, Mn). Analogous to [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12−, all of these clusters show high stability and alignment of superatom orbitals. In particular, the Sn@Mn12@Sn20 and Pb@Mn12@Pb20 clusters possess a giant magnetic moment of 28 μB, which can be retained in cluster assemblies. Our results thus extend the scope of icosahedral matryoshka clusters and provide novel building blocks for cluster-based materials and devices exhibiting fascinating novel magnetic and optical properties.
Results and Discussion
The binding energies, formation energies, HOMO-LUMO gaps and optical absorption gaps for icosahedral matryoshka A@B12@A20 clusters (A = Sn, Pb; B = Mg, Zn, Cd) are summarized in Table 1. For a given cluster of AxBy, its formation energy is defined by: Ef (AxBy) = E(AxBy) – xE(A) – yE(B), where E(AxBy) is the energy of AxBy cluster, E(A) or E(B) is the energy per atom of the constituent element A or B in the standard states, x or y is the number of A or B atoms in the cluster. Detailed information on geometry parameters and on-site charges for these clusters are given in Table S1 of Supplementary Information.
All the clusters explored here show reasonably low formation energy (typically between 0.1 and 0.2 eV/atom), indicating that they are easy to form thermodynamically. For comparison, we consider several 38-atom elementary metal clusters with a fcc-like truncated octahedron structure39. Higher formation energies are obtained, i.e., 0.225 eV/atom for Pb38, 0.281 eV/atom for Sn38, 0.576 eV/atom for Mg38, 0.497 eV/atom for Zn38 and 0.385 eV/atom for Cd38 clusters, respectively. Using global optimization combined with DFT calculations, Ferrando's group40 has recently achieved a series of three-shell high-symmetry matryoshka clusters, including a Ni13@Mg20 that is isostructural to the clusters considered here. However, the formation energy of this Ni13@Mg20 cluster is as high as 0.468 eV/atom from our DFT calculation. The more favorable formation energies of the present icosahedral matryoshka superatoms clearly demonstrate the stabilization effect of electronic structure, which will be discussed in detail later.
Note that the lengths of Pb-Pb (3.317 to 3.578 Å) and Sn-Sn (3.176 to 3.463 Å) bonds listed in Table S1 are longer than the equilibrium bond lengths of the hollow Pb20 (3.073 Å) and Sn20 (2.891 Å) cages from our DFT optimization. Therefore, the enlarged Pb20 and Sn20 cages are stabilized by the encapsulated C@B12 icosahedron (C = Sn, Pb; B = Mg, Zn, Cd), as previously found41 for [As@Ni12@As20]3−. It is also interesting to compare the structural parameters of Sn@Zn12@Sn20 with its isoelectronic counterpart, i.e., [Sn@Cu12@Sn20]12−. The theoretical Sn-Sn bond lengths in Sn@Zn12@Sn20 of 3.278 Å are comparable to that of 3.290 Å for [Sn@Cu12@Sn20]12− computed with the same level of theory. Note that the experimental Sn-Sn bond lengths of [Sn@Cu12@Sn20]12− in the condensed phase obtained by X-ray crystallography range between 3.076 to 3.133 Å18.
According to the on-site population analysis (see Supplementary Table S1), the B atoms in the intermediate shell donate some charge to the A atoms in the exterior shell, while the central A atom gains more charge, ranging from 0.245 to 1.362 electrons (by Mulliken definition). The trend of charge transfer remains the same for different systems, suggesting that all these clusters share the same pattern of chemical bonding. The direction of charge transfer can be easily understood by the electronegativity differences, that is, the Pauling electronegativities of Pb (2.33) and Sn (1.96) are higher than those of Mg (1.31), Zn (1.65), Cd (1.69) and Mn (1.55).
As shown in Table 1, the HOMO-LUMO gaps of the A21B12 clusters with B = Mg, Zn, Cd range from 1.27 to 1.54 eV, comparable to those of [As@Ni12@As20]3− (1.44 eV) and [Sn@Cu12@Sn20]12− (1.39 eV) using the same computational scheme. Previous DFT calculations yielded similar band gaps for [As@Ni12@As20]3− (1.44 eV41) and [Sn@Cu12@Sn20]12− (1.34 eV18). The high symmetries of these clusters brings about multiple degeneracy of the frontier molecular orbitals and severe selection rules in optical transitions. Our TD-DFT calculations further reveal that the lowest singlet excitation (HOMO-to-LUMO) is optically forbidden due to the LUMO and HOMO symmetries. The first optically-allowed excitation (i.e., optical absorption gap Eog) and the associated energy levels are summarized in Table 1. The computed Eog by TD-DFT ranges from 1.72 to 1.94 eV (i.e., the visible light region), making them potentially useful as nanoscale building blocks for optoelectronic devices25,41.
As aforementioned, the experimentally found icosahedral matryoshka clusters, i.e., [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12−, are exactly isoelectronic with 108 valence electrons beyond the filled d10 shells (for As, Ni, Sn, Cu). The same 108-electron rule applies to all binary A@B12@A20 clusters considered here, noting that Mg has no d electrons. However, 108 does not explicitly belong to any existing magic numbers for various models, e.g., the shell model within the jellium approximation23, the Wade-Mingos rules42, the spherical aromatic model43, the octet or eighteen-electron rule23. Therefore, we explore the origin of the “magic number” of 108 for these spherical multi-shell clusters as a guide to design other matryoshka clusters exhibiting high stabilities and sizeable electronic gaps.
For simplicity, we start with Sn@Mg12@Sn20 that does not involve any d electrons in the valence shells of the component atoms. The energy levels and spatial distribution for the Kohn-Sham molecular orbitals of Sn@Mg12@Sn20 are presented in Figure 2 and Figure 3, respectively. According to the energetic sequence and the nodal shape of the orbitals, we can identify a series of superatom orbitals in the sequence 1S, 1P, 2S, 1D, 1F, 1G, 2P, 3S, 2D, 1H, 2F, 3P, 1I corresponding to the 54 lowest-lying valence molecular orbitals (108 valence electrons) up to and including HOMO. Among them, 1S, 1P, 2S, 1D, 1F, 1G, 2P, 3S, 2D, 3P orbitals are completely filled. In the icosahedral ligand field44, the 2F, 1H and 1I superatom orbitals are split into two, three and four components, respectively. The lowest 1I orbital (gg) is occupied as the HOMO of superatom. The lowest two (t2u and hu) of 1H orbitals are occupied, while the triply degenerate middle 1H t1u orbital is the LUMO of superatom. The lower gu component of 2F orbitials is occupied and the higher t2u one is empty as LUMO+1. Even without closure of any specific superatom orbital, a large HOMO-LUMO gap of 1.383 eV separates the occupied gg component of the 1I orbital and the empty t1u component of the 1H orbital of Sn@Mg12@Sn20. Therefore, the “magic number” of 108 electrons of the icosahedral matryoshka clusters originates from the high Ih symmetry and consequently the splitting of molecular orbitals of high angular momentum (2F, 1H and 1I)44. Previously, Martin and co-workers45 had shown that both electronic and geometric effects can lead to extraordinary stability for a cluster (i.e., a magic cluster).

Energy levels (in eV) for the molecular orbitals of the Sn@Mg12@Sn20 cluster, which can be assigned to a series of superatom orbitals corresponding to the indicated spherical harmonics.
A few unoccupied levels are shown and highlighted by green.

Isosurfaces for the spatial distribution of the occupied superatom orbitals of the Sn@Mg12@Sn20 cluster.
Meanwhile, the suitable matching of atomic sizes of the component elements ensures the geometry stability of such unique three-layer matryoshka with high Ih symmetry. As summarized in the Supplementary Table S2, the covalent radii46 of the constituent elements in an icosahedral matryoshka cluster have to match with each other (in the range of 1.22 ~ 1.46 Å). Indeed, we have also investigated other possible icosahedral matryoshka A@B12@A20 clusters with B = Be, Ca (and A = Sn, Pb). However, all of them exhibited several large imaginary vibrational frequencies due to mismatch of atomic radius and thus were not further discussed here.
The energy levels for the four icosahedral matryoshka clusters, including [As@Ni12@As20]3−, [Sn@Cu12@Sn20]12−, Sn@Mg12@Sn20 and Sn@Zn12@Sn20, are compared in Supplementary Figure S1. The arrangement of molecular orbitals in each species is generally similar. The differences for those clusters are related to the specific energy of each orbital as well as the relative location of the d orbitals in the Ni, Cu, or Zn derivatives. However, the d orbitals, which locate only in a narrow energy range, have no noticeable influence on the overall superatom orbitals. On the other hand, the very deep d levels for As, Sn and Pb do not overlap with the superatom orbitals. As discussed above, the universal arrangement of molecular orbitals must originate from the unique icosahedral nesting doll structure. This demonstrates further the existence of a class of matryoshka clusters with similar geometries and electronic properties.
Analogous to Cu, Ni, Zn, Cd elements with filled d10 shells, we further design two new icosahedral matryoshka clusters by using Mn with a half-filled d5 shell, that is, Sn@Mn12@Sn20 and Pb@Mn12@Pb20. According to Hund's rule, the Mn atoms are expected to carry certain magnetic moments. Indeed, our DFT calculations show that both Sn@Mn12@Sn20 and Pb@Mn12@Pb20 possess a giant magnetic moment of 28 μB. Mulliken population analysis reveals that the magnetism mainly resides on the Mn atom, i.e., 2.880 (3.013) μB on each Mn atom for Sn21Mn12 (Pb21Mn12), whereas there are certain induced antiferromagnetic moments (about −0.3 ~ 0.8 μB) on the central and exterior Sn or Pb atoms (see Table 2). Even with the half-filled d5 shell and large spin polarization, these two magnetic superatoms exhibit moderate HOMO-LUMO gaps of 0.382 eV (Sn21Mn12) and 0.614 eV (Pb21Mn12), respectively, which are comparable to the gap magnitude (0.4 ~ 0.7 eV) for previously proposed magnetic superatoms, i.e., VNa8−33, Mg8Fe34, Ca8Fe35, MnCa936, MnSr937, TcMg847, ScK12 and ScCs1238.
The spin-polarized density of states for Sn@Mn12@Sn20 and Pb@Mn12@Pb20 clusters are presented in Figure 4. Even with spin polarization, the alignments of superatom orbitals in Sn@Mn12@Sn20 and Pb@Mn12@Pb20 still resemble those of their non-magnetic counterparts shown in Supplementary Figure S1. This suggests that the 3d orbitals of Mn in such species are effectively non-bonding and contribute only to the overall magnetic moment of the clusters. As demonstrated by the spin density distribution in Figure 4(c) and (d), the magnetism mainly originates from the unpaired 3d electrons from the Mn atoms, while some superatom orbitals (like 2P, 2D, 3S) also exhibit certain exchange splitting between the majority and minority spin states. We also observe some hybridization between the 3d states of Mn and the superatom orbitals (2F, 1G, 1H, 1I). Previously, Khanna and co-workers32,33,34,35 suggested that the hybridization between transition metal d states and superatom states as well as the combined action of the crystal field splitting and the exchange splitting are the key factors to stabilize a magnetic superatom and to ensure a large magnetic moment. Although the size and geometry of the current clusters are entirely different from the previous ones, this design principle for magnetic superatoms remains valid.

Spin-polarized density of states for (a) Sn@Mn12@Sn20 and (c) Pb@Mn12@Pb20 clusters.The occupied superatom orbitals are labelled. The d states from Mn atoms are highlighted by shadow peaks. The HOMO levels are shown by dashed lines. Atomic structure along with isosurface of spin density distribution (green areas) of (b) Sn@Mn12@Sn20 and (d) Pb@Mn12@Pb20 clusters are also presented (color code: dark gray for Sn and Pb, light purple for Mn).
Previously, an [Mn@Mn12@Au20]− cluster, iso-structural to the current icosahedral matryoshka, was also predicted to possess a giant magnetic moment of 44 μB48, but with a smaller HOMO-LUMO gap (0.25 eV). Using current theoretical scheme, the calculated formation energy for Mn@Mn12@Au20 is 0.795 eV/atom, about twice of Sn21Mn12. Again, this indicates that design of highly stable superatoms requires the consideration of electronic structures, i.e., closure of electronic shell or subshell. Also note that Sun et al.49 designed an onion-like caged clusters Fe@Au12@Au42 in which the magnetic moment on Fe atom (3 μB) is enhanced over bulk value, while Wu and Jena50 recently proposed a series of stable ferromagnetic hollow cages with large magnetic moments, i.e., Co12C6 (14 μB), Mn12C6 (38 μB) and Mn24C18 (70 μB).
In addition, a [Mn12] single molecular magnet, i.e., Mn12O12(CH3COO)16(H2O)4, which possesses a magnetic moment of 20 μB51, has been intensively studied52. To some extent, the present magnetic superatom clusters of Sn21Mn12 and Pb21Mn12 can be regarded as a kind of ligand-free single molecular magnet of [Mn12] with slightly enhanced magnetic moment (28 μB vs. 20 μB). The high symmetries, appreciable HOMO-LUMO gaps and giant magnetic moments makes Sn21Mn12 and Pb21Mn12 clusters promising building units for novel cluster-assembled magnetic materials and devices, which have potential applications in high-density information storage53, molecular spintronics54, quantum computing55 and magnetic resonance imaging (MRI)56, etc.
In the cluster-assembled materials, clusters are either encapsulated into a zeolite matrix57 or deposited on a substrate58. It is thus crucial to examine whether every clusters in the assembly can keep their identity without coalescing into larger clusters, like the previous case of VCs832. More importantly, the fantastic magnetic characteristics of the individual clusters must be well retained. As a simplest prototype of cluster assemblies, here we investigate the cluster-cluster interaction by considering the dimers of Sn@Mn12@Sn20 and Pb@Mn12@Pb20. The atomic structure and binding curve for [Pb@Mn12@Pb20]2 dimer are shown in Figure 5 as a representative. These two clusters are stacked along a 5-fold symmetry axis with parallel pentagonal faces in a staggered orientation, which is more stable than the direct orientation by 0.180 eV. Upon formation of the dimer, there is no noticeable deformation of cluster configuration. Accordingly, the interaction strength between the two spherical clusters is rather weak, as demonstrated by the large inter-cluster distances (d = 3.787 Å for [Sn@Mn12@Sn20]2 and d = 4.153 Å for [Pb@Mn12@Pb20]2, see Figure 5) and small inter-cluster binding energies (0.359 eV for [Sn@Mn12@Sn20]2 and 0.291 eV for [Pb@Mn12@Pb20]2. Such weak cluster-cluster interaction might be attributed to the moderate HOMO-LUMO gap as well as the sphere-like icosahedral geometry of the clusters. Most excitingly, the total magnetic moments of both cluster dimers are exactly twice of the individual ones, i.e., 56 μB. Indeed, the on-site local moments in the cluster dimers are barely affected by the neighboring cluster (see Table 2). To consider the possible antiferromagnetic state, we initially set different spin directions (spin up vs. spin down) for the Mn atoms in each of the two clusters in [Sn@Mn12@Sn20]2 or [Pb@Mn12@Pb20]2 dimer. However, upon relaxation of spin-polarized electron wavefunction, the spin state of the cluster dimer spontaneously transforms from antiferromagnetic to ferromagnetic. Moreover, inclusion of dispersion correction for the inter-cluster interaction only moderately increases the magnitude of binding energy Eint but has no effect on the magnetism of the cluster dimers. These results clearly indicate that the Sn@Mn12@Sn20 or Pb@Mn12@Pb20 clusters may keep their identities and retain their superior magnetism in cluster-assembled materials and devices. This is a vital prerequisite for their future applications.

Atomic structure (upper) and binding curve (lower) for [Pb@Mn12@Pb20]2 dimer.
The equilibrium distance (d) and optimal inter-cluster binding energy (Eint) are labelled.
Conclusion
A series of three-shell icosahedral matryoshka clusters related to the experimentally known [As@Ni12@As20]3− and [Sn@Cu12@Sn20]12− are proposed. A DFT-based global minimum search confirms that such icosahedral matryoshka structures are ground state configurations for these A21B12 (A = Sn, Pb; B = Mg, Zn, Cd) binary clusters. In addition, the high stabilities of these spherical matryoshka clusters are demonstrated by their large HOMO-LUMO gaps and low formation energies. The molecular orbitals for different icosahedral matryoshka clusters share the same pattern of superatom orbitals. The “magic number” of 108 electrons can be attributed to the high Ih symmetry and consequently the splitting of superatom orbitals of high angular momentum. Such “108-electron rule” should be extendable to other icosahedral matryoshka superatoms with different charge states, which might lead to future experimental discoveries of new metal clusters in solution as well as novel solid-state intermetallic compounds. Two related magnetic icosahedral matryoshka clusters, namely Sn@Mn12@Sn20 and Pb@Mn12@Pb20, are predicted to exhibit large magnetic moments (28 μB) and moderate HOMO-LUMO gaps. More impressively, after formation of cluster assemblies, these two clusters are able to keep their identities and retain their magnetic moments due to weak inter-cluster interaction. These novel binary clusters with unique icosahedral nesting doll geometry, high thermodynamic stabilities and interesting physical properties (such as optical gaps in the visible region and giant magnetic moments), are expected to be useful building blocks in future nanoscale materials and devices.
Methods
Ab initio calculations were performed using the spin-polarized density functional theory as implemented in the DMol3 program59. The core electrons were treated by an all-electron relativistic method including scalar relativistic effects. The generalized gradient approximation (GGA) with PW91 parameterization60 was adopted to describe the exchange-correlation interaction and the double numerical basis set including d-polarization functions (DND) were employed. Vibrational analyses were performed for each cluster to ensure that the optimized structures are the true minima on the potential energy surface. In addition, time-dependent density functional theory (TD-DFT) calculations with PW91 functional were carried out for those A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd) clusters to obtain the optical adsorption spectra and optical gaps.
To confirm that the three-shell icosahedral configuration in Figure 1 is the ground state for the binary A21B12 clusters (A = Sn, Pb; B = Mg, Zn, Cd, Mn), we performed an unbiased global minimum search of two representative clusters (Sn21Mg12 and Sn21Zn12) using a genetic algorithm (GA) incorporated with DFT calculations implemented in the DMol3 package, which was described in our previous publication61. In the GA search, sixteen initial configurations were generated from scratch. Any two individuals in this population were then chosen as parents to produce a child cluster via a “cut and splice” crossover operation, followed by an optional mutation operation (either a small random displacement on each atom or exchange of a pair of different types of atoms) of 35% probability. The diversity of the populations was filtered by the inertia of each cluster. After about 3000 GA iterations, the icosahedral matryoshka structure was obtained for both Sn@Mg12@Sn20 and Sn@Zn12@Sn20 clusters as the ground state. In addition, some metastable isomers (Supplementary Figure S2) are found from GA-DFT search, but are less energetically favorable and will not be further discussed.
References
Zintl, E., Goubeau, J. & Dullenkopf, W. Metals and alloys. I. Salt-like compounds and intermetallic phases of sodium in liquid ammonia. Z. Phys. Chem., Abt. A 154, 1–46 (1931).
Zintl, E. & Harder, A. Metals and alloys. II. Polyplumbides, polystannides and their transition into metal phases. Z. Phys. Chem., Abt. A 154, 47–91 (1931).
Zintl, E. & Dullenkopf, W. Metals and alloys. III. Polyantimonides, polybismuthides and their transformation into alloys. Z. Phys. Chem., Abt. B 16, 183–194 (1932).
Zintl, E. & Kaiser, H. Metals and alloys. VI. Ability of elements to form negative ions. Z. Anorg. Allgem. Chem. 211, 113–131 (1933).
Kummer, D. & Diehl, L. Preparation and Properties of a Crystalline Compound Na4Sn9 · 6–8 Ethylenediamine. Angew. Chem., Int. Ed. 9, 895–895 (1970).
Henning, R. W. & Corbett, J. D. Formation of Isolated Nickel-Centered Gallium Clusters in Na10Ga10Ni and a 2-D Network of Gallium Octahedra in K2Ga3 . Inorg. Chem. 38, 3883–3888 (1999).
Esenturk, E. N., Fettinger, J. & Eichhorn, B. The Pb122− and Pb102− Zintl Ions and the M@Pb122− and M@Pb102− Cluster Series Where M = Ni, Pd, Pt J. Am. Chem. Soc. 128, 9178–9186 (2006).
Krämer, T. et al. Structural trends in ten-vertex endohedral clusters, M@E10 and the synthesis of a new member of the family, [Fe@Sn10]3−. Dalton Trans. 42, 12120–12129 (2013).
Ruck, M., Dubenskyy, V. & Söhnel, T. Structure and Bonding of Pd@[Bi10]4+ in the Subbromide Bi14PdBr16 . Angew. Chem. 115, 3086–3090 (2003).
Wang, J. Q., Stegmaier, S. & Fässler, T. F. [Co@Ge10]3−: An Intermetalloid Cluster with Archimedean Pentagonal Prismatic Structure. Angew. Chem. 121, 2032–2036 (2009).
Zhou, B., Denning, M. S., Kays, D. L. & Goicoechea, J. M. Synthesis and isolation of [Fe@Ge10]3−: A pentagonal prismatic Zintl ion cage encapsulating an interstitial iron atom. J. Am. Chem. Soc. 131, 2802–2803 (2009).
Esenturk, E. N., Fettinger, J., Lam, Y.-F. & Eichhorn, B. [Pt@Pb12]2−. Angew. Chem. Int. Ed. 116, 2184–2186 (2004).
Neukermans, S. et al. Extremely Stable Metal-Encapsulated AlPb10+ and AlPb12+ Clusters: Mass-Spectrometric Discovery and Density Functional Theory Study. Phys. Rev. Lett. 92, 163401 (2004).
Chen, Z. et al. To Achieve Stable Spherical Clusters: General Principles and Experimental Confirmations. J. Am. Chem. Soc. 128, 12829–12834 (2006).
Moses, M. J., Fettinger, J. C. & Eichhorn, B. W. Interpenetrating As20 Fullerene and Ni12 Icosahedra in the Onion-Skin [As@Ni12@As20]3− Ion. Science 300, 778–780 (2003).
King, R. B. & Zhao, J. The isolable matryoshka nesting doll icosahedral cluster [As@Ni12@As20]3− as a “superatom”: analogy with the jellium cluster Al13− generated in the gas phase by laser vaporization. Chem. Commun., 4204–4205 (2006).
King, R. B. & Silaghi-Dumitrescu, I. The role of “external” lone pairs in the chemical bonding of bare post-transition element clusters: the Wade-Mingos rules versus the jellium model. Dalton Trans., 6083–6088 (2008).
Stegmaier, S. & Fässler, T. F. A Bronze Matryoshka: The Discrete Intermetalloid Cluster [Sn@Cu12@Sn20]12− in the Ternary Phases A12Cu12Sn21 (A = Na, K). J. Am. Chem. Soc. 133, 19758–19768 (2011).
Scharfe, S., Kraus, F., Stegmaier, S., Schier, A. & Fässler, T. F. Zintl Ions, Cage Compounds and Intermetalloid Clusters of Group 14 and Group 15 Elements. Angew. Chem. Int. Ed. 50, 3630–3670 (2011).
Jena, P. & Castleman, J. A. W. Naoclusters: a bridge across disciplines. (Elsevier, UK, 2010).
Khanna, S. N. & Jena, P. Assembling crystals from clusters. Phys. Rev. Lett. 69, 1664–1667 (1992).
Castleman, A. W. & Khanna, S. N. Clusters, Superatoms and Building Blocks of New Materials. J. Phys. Chem. C 113, 2664–2675 (2009).
Jena, P. Beyond the Periodic Table of Elements: The Role of Superatoms. J. Phys. Chem. Lett. 4, 1432–1442 (2013).
Castleman, A. W. From Elements to Clusters: The Periodic Table Revisited. J. Phys. Chem. Lett. 2, 1062–1069 (2011).
Claridge, S. A. et al. Cluster-Assembled Materials. ACS Nano 3, 244–255 (2009).
Castleman, A. W. et al. From Designer Clusters to Synthetic Crystalline Nanoassemblies. Nano Lett. 7, 2734–2741 (2007).
Bergeron, D. E., Castleman, A. W., Morisato, T. & Khanna, S. N. Formation of Al13I−: Evidence for the superhalogen character of Al13 . Science 304, 84–87 (2004).
Gutsev, G. L. & Boldyrev, A. I. DVM-Xα calculations on the ionization potentials of MXk+1− complex anions and the electron affinities of MXk+1 “superhalogens”. Chem. Phys. 56, 277–283 (1981).
Bergeron, D. E., Roach, P. J., Castleman, A. W., Jones, N. O. & Khanna, S. N. Al Cluster Superatoms as Halogens in Polyhalides and as Alkaline Earths in Iodide Salts. Science 307, 231–235 (2005).
Kumar, V. & Kawazoe, Y. Metal-doped magic clusters of Si, Ge and Sn: The finding of a magnetic superatom. Appl. Phys. Lett. 83, 2677–2679 (2003).
King, R. B. Theoretical chemistry: Magnetic superatoms. Nat. Chem. 1, 260–261 (2009).
Reveles, J. U. et al. Designer magnetic superatoms. Nat. Chem. 1, 310–315 (2009).
Zhang, X. et al. On the Existence of Designer Magnetic Superatoms. J. Am. Chem. Soc. 135, 4856–4861 (2013).
Medel, V. M. et al. Hund's rule in superatoms with transition metal impurities. Proc. Nat. Acad. Sci. 108, 10062–10066 (2011).
Chauhan, V., Medel, V. M., Ulises Reveles, J., Khanna, S. N. & Sen, P. Shell magnetism in transition metal doped calcium superatom. Chem. Phys. Lett. 528, 39–43 (2012).
Medel, V. M., Reveles, J. U., Islam, M. F. & Khanna, S. N. Robust Magnetic Moments on Impurities in Metallic Clusters: Localized Magnetic States in Superatoms. J. Phys. Chem. A 117, 4297–4303 (2013).
Medel, V., Reveles, J. U. & Khanna, S. N. Magnetism of electrons in atoms and superatoms. J. Appl. Phys. 112, 064313 (2012).
Pradhan, K., Reveles, J. U., Sen, P. & Khanna, S. Enhanced magnetic moments of alkali metal coated Sc clusters: New magnetic superatoms. J. Chem. Phys. 132, 124302 (2010).
Parks, E. K., Nieman, G. C., Kerns, K. P. & Riley, S. J. Reactions of Ni38 with N2,H2 and CO: Cluster structure and adsorbate binding sites. J. Chem. Phys. 107, 1861–1871 (1997).
Damianos, K., Solokha, P. & Ferrando, R. Core-shell and matryoshka structures in MgNi nanoalloys: a computational study. RSC Advances 3, 9419–9430 (2013).
Zhao, J. & Xie, R.-H. Density functional study of onion-skin-like [As@Ni12As20]3− and [Sb@Pd12Sb20]3− cluster ions. Chem. Phys. Lett. 396, 161–166 (2004).
Mingos, D. M. P. Polyhedral skeletal electron pair approach. Acc. Chem. Res. 17, 311–319 (1984).
Hirsch, A., Chen, Z. & Jiao, H. Spherical aromaticity in Ih symmetrical fullerenes: the 2(N+1)2 rule. Angew. Chem. Int. Ed. 39, 3915–3917 (2000).
King, R. B. Chemical Applications of Topology and Group Theory. 31. Atomic Orbital Graphs and the Shapes of the g and h Orbitals1. J. Phys. Chem. A 101, 4653–4656 (1997).
Martin, T. P., Bergmann, T., Gijhlich, H. & Lange, T. Observation of electronic shells and shells of atoms in large Na clusters. Chem. Phys. Lett. 172, 209–213 (1990).
Cordero, B. et al. Covalent radii revisited. Dalton Trans., 2832–2838 (2008).
Ge, G.-X., Han, Y., Wan, J.-G., Zhao, J.-J. & Wang, G.-H. First-principles prediction of magnetic superatoms in 4d-transition-metal-doped magnesium clusters. J. Chem. Phys. 139, 174309 (2013).
Wang, J., Bai, J., Jellinek, J. & Zeng, X. C. Gold-Coated Transition-Metal Anion [Mn13@Au20]− with Ultrahigh Magnetic Moment. J. Am. Chem. Soc. 129, 4110–4111 (2007).
Sun, Q. et al. Effect of Au coating on the magnetic and structural properties of Fe nanoclusters for use in biomedical applications: A density-functional theory study. Phys. Rev. B 73, 134409 (2006).
Wu, M. & Jena, P. Magnetic hollow cages with colossal moments. J. Chem. Phys. 139, 044301 (2013).
Caneschi, A. et al. Alternating current susceptibility, high field magnetization and millimeter band EPR evidence for a ground S = 10 state in [Mn12O12(Ch3COO)16(H2O)4]·2CH3COOH·4H2O. J. Am. Chem. Soc. 113, 5873–5874 (1991).
Rogez, G. et al. The Quest for Nanoscale Magnets: The example of [Mn12] Single Molecule Magnets. Adv. Mater. 21, 4323–4333 (2009).
Gubin, S. P. Magnetic Nanoparticles. (Wiley-VCH, Weinheim, 2009).
Bogani, L. & Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 7, 179–186 (2008).
Leuenberger, M. N. & Loss, D. Quantum computing in molecular magnets. Nature 410, 789–793 (2001).
Na, H. B., Song, I. C. & Hyeon, T. Inorganic Nanoparticles for MRI Contrast Agents. Adv. Mater. 21, 2133–2148 (2009).
Mikhailov, M. N., Kustov, L. M. & Kazansky, V. B. The State and Reactivity of Pt6 Particles in ZSM-5 Zeolite. Catal. Lett. 120, 8–13 (2008).
Popok, V. N., Barke, I., Campbell, E. E. B. & Meiwes-Broer, K.-H. Cluster–surface interaction: From soft landing to implantation. Surf. Sci. Rep. 66, 347–377 (2011).
Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92, 508–517 (1990).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244–13249 (1992).
Sai, L., Tang, L., Zhao, J., Wang, J. & Kumar, V. Lowest-energy structures and electronic properties of Na-Si binary clusters from ab initio global search. J. Chem. Phys. 135, 184305–184309 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 11134005, 11304030), the Fundamental Research Funds for the Central Universities of China (No. DUT13ZD207, DUT14LK19), the U. S. National Science Foundation (Grants CHE-1057466 and EPS-1010094) and the U. S. Department of Defence (Grant W911NF-12-1-0083).
Author information
Authors and Affiliations
Contributions
J.Z. designed the models and calculations. X.H., J.Z. and Y.S. did the calculations. X.H. and J.Z. prepared all the figures. J.Z., Z.C. and R.B.K. wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Huang, X., Zhao, J., Su, Y. et al. Design of Three-shell Icosahedral Matryoshka Clusters A@B12@A20 (A = Sn, Pb; B = Mg, Zn, Cd, Mn). Sci Rep 4, 6915 (2014). https://doi.org/10.1038/srep06915
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06915
This article is cited by
-
Cu12@Au30Ag12: a magnetic pentakis icosidodecahedron molecule with core–shell configuration
Journal of Molecular Modeling (2022)
-
Iron-based magnetic superhalogens with pseudohalogens as ligands: An unbiased structure search
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
