
Abstract
It is known that water purified by conventional TiO2 photocatalysts may not be safe enough for drinking, due to the toxicity by tiny existence of TiO2 nanoparticles after water treatment. We herein demonstrate a facile design of a three-dimensional (3D) TiO2 photocatalyst structure with which both the efficiency of purification and the safety level of the final purified water can be improved and ensured, respectively. The structure, consisting of 3D sulfur-doped TiO2 microtubes in nanotubes (eco-TiO2), is suitable for both environmental and bio-medical applications. Investigation of its formation mechanism reveals that anodic aluminum oxide (AAO), owing to a spatial constraint, causes a simple, nanoparticles-to-nanotubes structural rearrangement as a template for nanotube growth. It is found that eco-TiO2 can be activated under visible-light irradiation by non-metal (sulfur; S) doping, after which it shows visible-light photocatalytic activities over a range of solar energy. Importantly, an in vitro cytotoxicity test of well-purified water by eco-TiO2 confirms that eco-TiO2 satisfies the key human safety conditions.
Similar content being viewed by others
Introduction
One of the most significant environmental issues today is the potential of photocatalytic technology to provide solutions to various environmental problems faced by our society1,2. TiO2 particularly, a typical photocatalyst, has various environmental applications3,4,5,6,7,8. For example, it is known to exhibit excellent photocatalytic behavior for decomposition of volatile organic compounds in air/water and bacterial treatments9,10,11,12,13. Practical environmental applications for TiO2 photocatalysts require the synthesis of immobilized TiO2 films on suitable substrates3. However, immobilized TiO2 film occasionally shows lower photocatalytic activity due to surface-area diminution, limited mass diffusion, and/or the application only of UV-light photoactivity1,2,3. It is therefore necessary to improve the photocatalytic activity of TiO2 film by optimization of several factors such as thickness, size, porosity, crystal structure and (non-) metal doping.
Porous TiO2 nanotubes are among the most attractive of the various TiO2 structures, owing to their excellent and highly promising performance in a variety of fields including photocatalysis, photoelectronics, semiconducting and others14,15,16,17,18. Generally, TiO2 nanotubes are prepared using a hard template, for example, an anodic alumina oxide (AAO) nanochannel array architecture15,19,20,21,22,23. Recent publications indicate that highly ordered and vertically oriented nanotube arrays, such as tube-in-tube24, tube-in-tube with a porous wall25 and tube with a mesoporous wall26, can be utilized in electronics, optics, sensing, advanced photocatalysis and energy storage/conversion applications. For fabrication of those TiO2 tubes, anodization of metallic Ti is regarded as a suitable method.
The photocatalyst should retain high activity under visible-light irradiation (λ > 400 nm) in order to effectively utilize natural sunlight and maximize photocatalysis1. In order to utilize the wide solar energy spectrum, many efforts have been made to narrow the band-gap of TiO2 by doping it with nonmetals such as carbon, sulfur, nitrogen and fluorine1,2,3,4. Recently, numerous impressive TiO2-nonmetal doping processes have been devised, especially involving the use of sulfur10,27,28,29. For example, we demonstrated that sulfur-doped TiO2 (S-TiO2) exhibits superior visible-light photocatalytic and antibacterial activities, as well as good recycling properties10. Such technology using sulfur for modification of TiO2 can actually be used in highly efficient air/water treatment, dye-sensitized solar cells, as bactericidal agents and for self-antifouling action27,28,29,30,31.
In the present study, we created three-dimensional (3D) sulfur-doped TiO2 microtubes in nanotubes (eco-TiO2) by a structurally simple rearrangement whereby the tube structure is maintained via a nanoparticles-to-nanotubes structural change within a limited space such as the nanochannels of an AAO template. Since the first implementation of TiO2–based photocatalysts, there has been no report on eco-TiO2 synthesis by the 3D-structural approach. Also, the previous research on photocatalytic TiO2 focused only on final air/water purification or treatment1,2,3,4,5, which were not be directly relevant to the reuse of purified water by photocatalysts, indicating no available data whether the treated water is safe while contacting human interface, although an important factor involved in water purification systems is the reuse of treated water by photocatalytic applications. In the present study, we modeled global eco-TiO2 materials and monitored by-products of treated water in an in vitro cytotoxicity assay measuring the level of water purification after photocatalytic treatment. Further, we studied the detailed eco-TiO2 formation mechanism and its fundamental photochemical properties. Our findings indicated that eco-TiO2 shows an efficient water-treatment performance together with an economic dividend.
Results
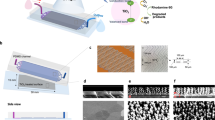
Fig. 1 and S1 show that the 3D TiO2 microtubes in nanotubes (3D-TiO2) covered the entire substrate surface. The enlarged field-emission scanning electron microscopy (FE-SEM) images clearly show the numerous 3D-TiO2 arrays to be both uniform and compact, with lengths of at least several micrometers (~10 μm) (Fig. 1a) and outer tube diameters of a few micrometers (~1 μm) (Figs. 1a and 1b). The inter-nanotubes showed a coarse lateral profile (Fig. 1b, inset) and had a larger contact surface area than conventional structures, which structural morphologies make them good candidates for photocatalytic applications. On high-resolution transmission electron microscopy (HR-TEM) and scanning transmission electron microscopy (STEM) images of the as-grown and eco-TiO2 samples (Figs. 1c, 1d and S1), the eco-TiO2 structures showed uniform diameters throughout their lengths and the TiO2 microtubes revealed their nanotube-like 3D-structural composition (Fig. 1c). In a detailed chemical analysis of the arrays by energy-dispersive X-ray spectroscopy (EDS) (Fig. S2, overlay), the eco-TiO2 nanostructures were composed of Ti, S and O (the Cu and Al signals were caused by the TEM grid and AAO template).

(a and b) FE-SEM in which dotted box shows a partially enlarged image of (b) and (c and d) TEM images of the 3D sulfur (S)-doped TiO2 microtubes in nanotubes (eco-TiO2) samples.
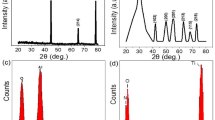
The corresponding X-ray diffraction (XRD) patterns, as recorded from the as-grown and annealed eco-TiO2 samples, are shown in Fig. 2. They evidence the fact that whereas the titanate phase of the 3D-TiO2 samples (JCPDS card No. 00-047-0124) was dominant before annealing, after annealing at 350°C for 1 h, it was transformed into anatase TiO2 (JCPDS card No. 02–0406) with higher photocatalytic activity. The XRD patterns also clearly indicate that the diffraction peaks of the eco-TiO2 samples were shifted to higher diffraction angles (~0.2°) relative to those of the other samples. This implies that, after our method's high-energy treatment, an oxygen atom in TiO2 in the eco-TiO2 lattice was replaced by a sulfur atom10.

XRD patterns of commercial TiO2 (Com.-TiO2), as-grown 3D-TiO2 (3D-aTiO2), 3D-TiO2 and eco-TiO2 samples.
The anatase and titanate peaks are identified as A and T respectively.
For further investigation of the chemical states of the eco-TiO2 samples, high-resolution X-ray photoelectron spectroscopy (HR-XPS) measurements were carried out. In Fig. 3's HR-XPS spectra of the S2p region of the eco-TiO2 samples, it is apparent that the S2p peaks around 167.4 eV and 168.5 eV were shifted negatively by 0.5 eV relative to other research group's reported standard binding energy of sulfur in pure SO32− (S4+, 167.7 eV) and SO42− (S6+, 168.7 eV)32,33,34. All of the sulfur atoms were in the S2− state, with a peak at approximately 160–163 eV33,34. This finding corresponds to the Ti–S bond formed when some of the oxygen atoms in the TiO2 lattice were replaced by sulfur atoms. This leads to the large formation energy required for Ti–S bonds instead of Ti–O bonds. Thus, the replacement of Ti4+ by S4+/S6+ is chemically more favorable than that of O2− by S2− and consequently, Ti–O–S (or Ti-S-O) bonds are formed10,32,33,34. Although anionic sulfur (S2−) doping was reported as a difficulty to be performed because the ionic radius of S2− (1.7 Å) is significantly larger than that of O2− (1.22 Å)10,32 but it was revealed by XPS spectra (Fig. 3b) where the inset exhibited both S4+/S6+ at ~168.0 eV and S2− species at ~162.0 eV in eco-TiO2 lattices10,34,35, confirming that the Ti-S bonds were formed. Note that that as-prepared eco-TiO2 sample was washed with deionized water and 1 M HCl solution several times to remove the surface-adsorbed SO42− or SO32− species.

High-resolution X-ray photoelectron spectra (HR-XPS) of (a) 3D-TiO2 and (b) eco-TiO2 where the inset in (b) indicates the deconvolution fitting of the S 2p of eco-TiO2.
Our simple structural rearrangement is required for the formation of eco-TiO2 photocatalysts, owing to the special 3D nanostructure (Fig. 4). First, using our method, TiO2 nanoparticles, which will be changed into TiO2 nanotubes within the nanochannel arrays of the AAO membrane (anodisc 47, Whatman®), are generated. To achieve 3D-TiO2 of high surface area, surfactant solutions (25 wt% solutions) are first coated onto the internal wall surfaces of the AAO template. This allows the formation of TiO2 nanoparticles (the corresponding synthetic results are available in previously published research15,19,20,21,22,23, specifically by the filling of the titanium isopropoxide (TIP) precursor in the AAO nanochannel arrays with a sol-gel reaction. The TiO2 nanoparticle/AAO composites are then dried for 24 h at 100°C in air. Special attention should be paid to the filling of the AAO nanochannels, as, if the surfactant solution is fully filled within the nanochannel arrays, there would be no space for the precursor. The Ti precursor, therefore, should be filled only after removing the surfactant to some extent by vacuum filtration. Via a simple alkaline hydrothermal reaction, the formed nanoparticles are transformed into titanate nanotubes within the limited space of the nanochannels, AAO providing the template for nanotube growth14,15,16,17,18,19,20. The general mechanism of titanate nanotube formation is as follows: a nanosheet-type structure of sodium titanate (Na2Ti3O7) is formed16,17,18,19,20,21,22,23,24 and then, after exfoliation, is folded into a nanotube; it grows in the axis direction and is protonated via HCl rinsing (H2Ti3O7)14,15,16,17,18,19,20; 3D-TiO2 is formed by the effect of the nanochannel array pattern of AAO; pure anatase 3D-TiO2 and eco-TiO2 are formed by annealing and repetitive ultrasound irradiation/annealing processes10,12. Notably, we found that the as-grown 3D-TiO2 at 300–450°C prevented the formation of brookite and rutile titania. This implies that by controlling the annealing temperature, the titanate of the as-grown eco-TiO2 was successfully crystallized to an anatase phase.

Possible mechanism for formation of eco-TiO2 with anatase crystalline while retaining large surface area.
Additionally, the 3D-TiO2 sample showed a broad photoluminescence (PL) emission peak located around 528.9 nm (Fig. 5a). However, for the eco-TiO2 sample, broad visible PL bands centered at 602.1 nm were observed. It can be seen from Figure 5a that the PL intensity of the 3D-TiO2 was much higher than in the spectra for the eco-TiO2 sample, indicating that non-metal (S) doping can effectively inhibit the recombination of excited electrons and holes10. This is the reason for the excited electron transference from the valence band to the new, S-doping-induced defect levels existing near the conduction band minimum, which also decreased the PL intensity. Closer inspection of Fig. 5a reveals a 73.2 nm (eco-TiO2) shift of the emission peaks toward longer wavelengths when nonmetals were doped into the TiO2 nanotubes. The emission signal at 602.1 nm might have originated from the charge-transfer transition from the oxygen vacancies to the new energy levels caused by non-metal (S) doping. Fig. 5b shows the reflectance spectra of the eco-TiO2. Clearly, compared with the TiO2 nanotubes, the action region of the eco-TiO2 extended significantly into the visible-light region. The corresponding action spectra strongly suggest that eco-TiO2 can be activated by visible-light irradiation. In addition, the photocurrent density was increased in the order of eco-TiO2 > S-TiO2 > 3D-TiO2 at all λ < 400 nm, λ > 400 nm and λ > 600 nm. Particularly, the photocurrent density (μA/cm2) with 16–20 of eco-TiO2 was higher than that with 12–18 of S-TiO2 (Fig. S3). It is associated with three-dimensional (3D) structure, corresponding to the PL quenching data (Fig. 3a). Based on the shift of absorbance peak to visible light regions in sulfur doped TiO2 (eco-TiO2), it is indicated that sulfur ions (S2− and S6+) are incorporated and subsequently changed into crystal and electronic structures in eco-TiO2 lattices, corresponding to the rearrangement of eco-TiO2 surfaces in XPS spectra, suggesting that eco-TiO2 photocatalysts are strongly activated on wide visible light ranges10,34,35.

(a) Photoluminescence (PL) and (b) UV-Vis-NIR reflectance spectra of commercial TiO2 (Com.-TiO2), as-grown 3D-TiO2 (3D-aTiO2), 3D-TiO2 and eco-TiO2 samples.
In addition to the confirmation of efficient charge separation in electron-hole (e−/h+) pairs, evidenced by photocurrent and PL quenching data, the delay in charge recombination in eco-TiO2 can be explained with the enhanced photocurrent. Harsh condition, i.e., H2SO4 treated with sonication, for sulfur (S) doping might increase surface oxygen vacancies and defects, leading to the suppression of the recombination of the photoinduced electrons and holes by capturing the photoinduced electrons36,37. Furthermore, in case of sulfur (S)-doped TiO2 nanoparticles, the abundant sulfur (S)-functional groups on the surface of TiO2 nanoparticles can facilely trap electrons (e-) in the charge recombination (e−/h+) pairs, resulting in improvement of the photocatalytic efficiencies of eco-TiO238. Such efforts to sink electrons neighboring microenvironment from the photogenerated charge carriers promoted to decrease in the charge recombination of TiO2 photocatlysts39.
For provision of information on free radicals in environmental and biological systems, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) is frequently used in electron spin resonance (ESR) spin-trapping experiments. In the present study, detection of ·OH radicals by DMPO spin trapping for eco-TiO2 samples under UV light (λ = 365 nm, 5 min irradiation) in a PBS buffer solution was investigated30,31 (Fig. 6). The characteristic 1:2:2:1 quartet signal of a DMPO-·OH adduct appeared in both TiO2 samples31. Significantly, the ESR signal intensity of the eco-TiO2 was strongly enhanced, growing to approximately 3.7 times that of 3D-TiO2. This is attributable to the facilitation of electron transference from the valence band to the conduction band, which results in the reaction of valence band holes (h+vb) with OH− ions, producing ·OH radicals30,31. Importantly, the visible-light-induced ESR signal patterns in the eco-TiO2 in the PBS buffer solution (λ > 400 nm, 5 min irradiation) showed intrinsic signals corresponding to S-doped TiO2, while no signals were detected for those in the 3D-TiO2.

ESR spectra of (a and b) commercial TiO2 (Com.-TiO2), (c and d) 3D-TiO2 and (e and f) eco-TiO2 samples.
Next, the photocatalytic activities of the eco-TiO2 samples were evaluated by measuring the decomposition rate of Rhodamine B (Rho B) solutions under UV- and visible-light irradiation (Fig. 7). Without a photocatalyst, Rho B was hardly degraded under UV- and visible-light irradiation: the dye-color removal was only 1% under 70 min UV-light irradiation and a paltry 0.4% under 70 min visible-light irradiation. Contrastingly, with the eco-TiO2 photocatalysts, 100% color removal was achieved under the identical irradiation conditions. The superior UV-light activity of the eco-TiO2 samples can be ascribed to their higher surface areas and lower electron-hole pair numbers (Fig. 5 and Table S1) because the measured BET surface area (m2/g), pore diameter (nm) and pore volume (cm3/g) of eco-TiO2 were 276.2, 1.85 and 0.13 whose physicochemical properties are enhanced values, compared to 276.2, 1.85 and 0.13 of equivalent 3D-TiO2. As a reference, although pore diameter (nm) of S-TiO2, which was prepared sulfur doped commercial TiO2 using same method of eco-TiO2, showed highest 5.01 nm but BET surface area of that was significantly low value (51.4 m2/g) with approximately 5-fold for that of eco-TiO2. Even so, the TiO2 nanotubes showed very poor photocatalytic activities under visible-light irradiation. The highest degradation rate for eco-TiO2 ([k] = 3.3814 h−1) was 10 times faster than that of 3D-TiO2 ([k] = 0.3278 h−1). In Fig. S4, other dyes such as MB and RB 5 were degraded by eco-TiO2 with its relating by-products measurement as total organic carbon (TOC) results, showing a similar degradation pattern. In order to avoid the self-photosentization of dyes, spectra scans of phenol (Ph) degradation by Com.-TiO2, 3D-aTiO2, 3D-TiO2, S-TiO2 and eco-TiO2 were observed (Fig. S5a–e) and it resulted in the apparent rate constants ([k] = 0.009, 0.014, 0.048, 0.298 and 1.643 h−1) (Fig. S5f), showing eco-TiO2 possesses an efficient photocatalytic activity. As noted above, S doping might possibly have generated an intermediate energy level above the valence band of TiO2, thereby narrowing the band-gap for induction of visible-light absorption. The TiO2 nanotubes exhibited almost no visible-light activity, owing to the approximately 2.8–3.0 eV band-gap energy of the general TiO2 photocatalyst1,2,3,4,5. After S atoms were doped into the TiO2 nanotubes, the visible-light photocatalytic activities of the as-prepared samples increased greatly (Fig. S6). As increase in sulfur doing concentration in 3D-TiO2 (i.e., 0.4%, 0.8% and 1.2%) (Fig. S7a), the bandgap energy (eV) was decreased to 2.26, 2.21 and 2.02, respectively. Proportionally, the Rho B degradation efficiencies were slightly enhanced (Fig. S7b), although BET surface area of 0.8% S 3D-TiO2 was higher (271.4 m2/g) than 1.2% S 3D-TiO2 (i.e., eco-TiO2), showing similar values of pore diameter (nm) and pore volume (cm3/g) in respective 0.8% S 3D-TiO2 and 1.2% S 3D-TiO2 (i.e., eco-TiO2) (Table S2). The quantities of evolved CO2 during the photocatalytic reaction of Rho B with the TiO2 photocatalysts under visible-light irradiation are plotted in Fig. 8a. As is apparent, the evolved CO2 increased gradually as the photocatalytic reaction progressed. After 70 min visible-light irradiation, the quantity of the evolved CO2 for eco-TiO2 (0.115 mmol) was four times higher than that for 3D-TiO2 (0.031 mmol). These results reflect the high degree of mineralization achieved for Rho B. The length of one cycle was 70 min (visible-light irradiation time); after each interval, the decolorization was measured (Fig. 8b). After 15 repetitions, the photocatalytic conversion ratio of Rho B by eco-TiO2 remained at about 92.6%. The decrease in the photocatalytic conversion ratio after every cycle was probably caused by the tiny loss of the eco-TiO2 photocatalyst. However, it is clear that the removal efficiency of organic contaminants using eco-TiO2 remained approximately constant even after 15 cycles. Undoubtedly, in this case, eco-TiO2 exhibited excellent photocatalytic stability.

Decomposition results of Rho B by commercial TiO2 (Com.-TiO2), as-grown 3D-TiO2 (3D-aTiO2), S-TiO2, 3D-TiO2 and eco-TiO2 samples under (a and b) UV and (c and d) visible irradiation.

(a) Evolved CO2 of commercial TiO2 (Com.-TiO2), as-grown 3D-TiO2 (3D-aTiO2), 3D-TiO2 and eco-TiO2 samples under visible irradiation and (b) recycling results of the eco-TiO2 in decolorization for Rho B solution.
The previous research on the photocatalytic activities of metal-oxide materials focused only on final water purification under UV- and visible-light irradiation, which is not directly related to the reuse of ventilated water with a product for dye-contaminated water1,2,3,4,5. Therefore, in the present research, we carried out an in vitro cytotoxicity test for monitoring of by-products in Rho B treated water and measurement of the available level of water. In the results, we found that the purified water by eco-TiO2 was safe enough for human cells. In an evaluation of the cytotoxicity of purified water by eco-TiO2 under visible-light irradiation, the relative viabilities of CHO-K1 (ovary, Chinese hamster), COS-7 (kidney, African green monkey) and HepG2 (hepatocellular carcinoma, human) cells exposed to purified water were measured using the 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay (Fig. 9). Fig. 9 shows the cell viability after incubation for 12, 24 and 72 h with the purified-water solution at purification degrees ranging from 0 to 100%. Below the purification degree of 98%, the cell viabilities were decreased or reduced to zero by azo dye contaminants or other chemicals noxious to organs in the water solution. After 24 h of incubation at the purification degree of 98%, the cell viability increased less than 16–18% (cellular viability: 96.2% ± 5.1% for CHO-K1, 95.5% ± 4.5% for HepG2, 94.8% ± 6.2% for COS-7; mean ± SEM, N = 5; probability value p < 0.005). These observations clearly show that over 12, 24 and 72 h periods, the purified water (at purification degrees of up to 98%) did not exert any cytotoxic effects on the cells, which is one of the key safety criteria. Importantly, at purification degrees typically used in nontoxic cell cultivation (>98%), the cytotoxicity of purified water was found to be marginal. Additionally, it was confirmed for the similar cytotoxicity results, after 24 h incubation, for other dyes treatment by eco-TiO2, resulting in (cellular viability: 89.0% ± 9.0%/92.0% ± 8.0% for CHO-K1, 91.0% ± 8.0%/90.0% ± 8.0% for COS-7; mean ± SEM, N = 5; probability value p < 0.005) for 98% of MB- and RB 5 purified water, respectively (Fig. S8). Therefore, treatment of dye-contaminated water by eco-TiO2, capable of greater-than-98% removal efficiency, can be considered to be an efficient non-cytotoxic clean-up method.

Cytotoxicity studies of Rho B purified water treatment (degree of %) by eco-TiO2 photocatalysts under visible-light irradiation after 12 h (a), 24 h (b) and 72 h (c) cell incubation.
To evaluate the cytotoxicity of purified water by eco-TiO2 photocatalysts under visible-light irradiation, the relative viabilities of CHO-K1 (ovary, Chinese hamster), COS-7 (kidney, African green monkey) and HepG2 (hepatocellular carcinoma, human) cells exposed to purified water were measured using the MTT assay. The data is presented as mean ± SE of five replicates.
Discussion
We developed a photocatalyst, eco-TiO2, for purification of organic- pollutant-contaminated water that is suitable for human consumption. The eco-TiO2 was synthesized via a structural rearrangement entailing the changing of nanoparticles to nanotubes within an AAO template that provided a limited space (nanochannels) for TiO2 nanotube growth. Eco-TiO2 was designed to achieve higher surface areas and lowered electron-hole pairs with uniform diameters in a 3D architecture. The as-prepared eco-TiO2 exhibited broad visible PL bands centered at 634.59 nm after S atoms were doped into the TiO2 nanotubes. These results clearly indicated that the photocatalytic activities of the eco-TiO2 under visible light were markedly superior to those of TiO2. Using eco-TiO2, the degradation rate of the dye solution under 70 min visible-light irradiation was 100%. And as for the practical findings, the cellular viabilities of CHO-K1, HepG2 and COS-7 after 12, 24 and 72 h incubation with water solution 98% purified by eco-TiO2 were as high as 96%, indicating no cytotoxicity. In terms of mitigating the impacts of environmental pollution, eco-TiO2 offers a great potential for reduction of water-contamination levels by acting as an effective photocatalyst in practical bio-medical applications.
Methods
Synthesis of 3D TiO2 microtubes in nanotubes
All of the reagents were of analytical grade (Sigma Aldrich, MO, USA) and were used without further purification. Ultrapure water (18.2 MΩ/cm, DI water) from a Milli-Q ultrapure system was used in this study. In the typical synthesis, an AAO (Whatman® anodic 47, Germany) template was filled with 20 mg of a hexadecyltrimethylammonium chloride surfactant solution (C19H42ClN, molecular weight: 320) and 30 mg of a titanium isopropoxide precursor solution at 100°C before drying. Thereafter, the template was subjected to an alkaline hydrothermal reaction for 24 h at 120°C in a 10 M sodium hydroxide solution and washed several times with DI water. It was then rinsed with a 0.1 M hydrochloric acid solution and dried in vacuum for 12 h at 60°C.
Synthesis of eco-TiO2
Eco-TiO2 was prepared as follows. The obtained 3D-TiO2 microtubes with nanotubes were dispersed in 900 mL of 0.1–1.0 M sulfuric acid (H2SO4) solution, to which 100 mL of a hydrogen peroxide (H2O2) solution was added dropwise with vigorous stirring for 5 min at room temperature (RT). Ultrasound irradiation was just applied for 40 min, with the electrical energy input maintained at 100 W/cm2. The reddish-orange-colored TiO2 powder was filtered, washed three times with DI water and finally dried under air at RT. An eco-TiO2 was then calcined up to 623 K in air.
Characterization
The crystalline structures of the eco-TiO2 samples were investigated by reference to the XRD (Rigaku RDA-cA X-ray diffractometer, Japan) patterns obtained using Cu Kα radiation passed through a nickel filter. The morphology and size distribution of the eco-TiO2 particles were recorded by FE-SEM (Hitachi, S-4700, Japan) and HRTEM (JEOL, JEM 2200, Japan). Preparatory to the analyses, the samples were placed onto the surfaces of copper grids and dried under ambient conditions. The Brunauer–Emmett–Teller (BET) surface areas, pore volumes and pore diameters of the eco-TiO2 particles were determined using a BET analyzer (Micromeritics ASAP 2020, USA). HR-XPS carried out using monochromatic Al Kα X-ray radiation (hν = 1486.6 eV) with a power of 120 W (Kratos Analytical, AXIS Nova, UK) was utilized to investigate the samples' surface properties. The shift in the binding energy due to relative surface charging was corrected with reference to an internal standard, namely the C1s level at 284.6 eV. A He-Cd laser (Kimmon, 1 K, Japan) of 325 nm wavelength and a power of 50 mW was utilized as an excitation source for PL measurements carried out using a spectrograph (f = 0.5 m, Acton Research Co., Spectrograph 500i, USA) with an intensified charge-coupled device (CCD; PI-MAX3, Princeton Instruments, IRY1024, USA). For free radical detection by 5,5-dimethyl-1-pyrroline N-oxide (DMPO, 0.3 M in PBS buffer at pH 7.2, Sigma-Aldrich, USA) spin trap agent, an aliquot of as-prepared samples (100 μL of 5 mg TiO2 sample mixed with 300 μL DMPO solution) was filled into a capillary tube and directly irradiated with a UV (λ = 365 nm) or LED light (>400 nm) source for 5 min, after which the spectra were recorded by ESR spectrometry (JES-FA200, JEOL, Japan; center field: 327 mT; powder: 1 mW; amplitude: 5.0 × 100; modulation width: 0.4 × 1; sweep width: 1 × 10; sweep time: 30 s)40,41. For the photocurrent density (μA/cm2) measurement, it was prepared with photo-electrodes by screen printing of each TiO2 photocatalysts onto fluoride doped tin oxide-coated (FTO) glass substrate (1 × 1 inch2)43,44. KI3 solution (0.5 M) was used as an electrolyte. It was assembled the two glass (working electrode acting as photoanode-TiO2 on FTO and reference electrode-carbon black on FTO) plates with coated sides together, but offset so that uncoated glass extends beyond the sandwich42,43,44. Add a drop of electrolyte solution to opposite edges of the plate. Capillary action will cause the electrolyte solution to travel between the two plates. After, it was connected a multimeter (Agilent 34420A, Agilent Technologies, USA) using an alligator clip to each plate. The photocurrent was observed for each switch-on/off event by using a Xenon lamp (450 W) with a 400–600 nm cut-off filter.
Measurement of photocatalytic and antibacterial activities
The photocatalytic degradations of phenol (Ph; 3.0 mg/L, Aldrich, USA), Reactive black 5 (RB 5; 3 mg/L, Aldrich, USA), Rhodamine B (Rho B; 3 mg/L, Sigma-Aldrich, USA) and Methylene blue (MB; RB 5; 3 mg/L, Aldrich, USA) solutions by catalyst samples (0.5 g/L) were carried out under UV (source: 4 W, 365 nm, VSLAB VL-4CL, Korea) and visible-light (source: 150 W Xe lamp, λ > 420 nm, SCHOTT, USA) irradiation and the absorbance of the solutions was measured using a UV-Vis-NIR spectrophotometer (Varian, Cary 5000, Australia) in the wavelength region 200–800 nm. The concentrations of Ph, RB 5, Rho B and MB in the solutions after photoirradiation were measured from the absorbance peak intensities of the solutions at 270, 598, 555 and 664 nm, respectively10,12,45. Total organic carbon (TOC) of the solution was also determined by a Shimadzu TOC-V analyzer. Light source was positioned at 15 cm height from the experimental desk. The changes in the concentration of the dye solution [ln(C0/C) = kt, where k is the apparent reaction rate constant and C0 and C are the initial and reaction concentrations, respectively, of Rho B] with reaction time also were investigated for all of the samples10. To demonstrate the stability of the photocatalysts, eco-TiO2 was recycled for testing of other photocatalytic activities. The recycling tests evaluating the photocatalytic activity of eco-TiO2 were performed after washing the samples three times with DI water and drying them in an oven for 6 h after each cycle10,12. Evolved CO2 was analyzed with a TM IGC120-MB gas chromatograph equipped with a Q column. Additionally, an action-spectrum analysis of the photocatalytic reactions was carried out under monochromatic light irradiation from a JASCO CRM-FD diffraction-grating-type illuminator.
In vitro cytotoxicity test of purified water using eco-TiO2
The cytotoxicities of the samples were evaluated by a MTT assay46. Briefly, CHO-K1 (ovary, Chinese hamster), HepG2 (hepatocellular liver carcinoma, human) and COS-7 (kidney, African green monkey) cells were seeded in a 96-well plate at a density of 8 × 103 cells per well and cultured in a humidified incubator at 37°C for 24 and 72 h under a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium (DMEM) and/or Roswell Park Memorial Institute (RPMI)-1640 supplemented with 10% FBS and 1% penicillin antibiotics. The DMEM and/or RPMI-1640 media were used to purify water samples (to 0, 25, 50, 75, 90, 95, 98 and 100% degrees) using eco-TiO2 photocatalysts, after which they were incubated for 24 and 72 h. Then, 20 μL of 0.2 mg/mL MTT solution in medium was added to each well and incubated at 37°C for 2 h. Finally, the optical density (OD) was measured at 490 nm with an absorbance microplate reader (EMax microplate reader, Bucher Biotec AG, Basel, Switzerland).
In the measurements of the photocatalytic activities for the antimicrobial and following cytotoxicity tests, the data were averaged and expressed as mean ± standard deviations (SE). Each test was repeated up to four times. A statistical analysis was performed by analysis of variance (ANOVA); p-values less than 0.05 were considered significant.
References
Kubacka, A., Fernandez-García, M. & Colon, G. Advanced nanoarchitectures for solar photocatalytic applications. Chem. Rev. 112, 1555–1614 (2012).
Chen, H., Nanayakkara, C. E. & Grassian, V. H. Titanium dioxide photocatalysis in atmospheric chemistry. Chem. Rev. 112, 5919–5948 (2012).
Chen, X. & Mao, S. S. Titanium dioxide nanomaterials: synthesis, properties, modifications and applications. Chem. Rev. 107, 2891–2959 (2007).
Tong, H. et al. Nano-photocatalytic materials: possibilities and challenges. Adv. Mater. 24, 229–251 (2012).
Kisch, H. Semiconductor photocatalysis-mechanistic and synthetic aspects. Angew. Chem.-Int. Edit. 52, 812–847 (2013).
Albu, S. P., Ghicov, A., Macak, J. M., Hahn, R. & Schmuki, P. Self-organized, free-standing TiO2 nanotube membrane for flow-through photocatalytic applications. Nano Lett. 7, 1286–1289 (2007).
Wang, X., Li, Z., Shi, J. & Yu, Y. One-dimensional titanium dioxide nanomaterials: nanowires, nanorods and nanobelts. Chem. Rev. 10.1021/cr400633s (2014).
Xiong, Z. & Zhao, X. S. Nitrogen-doped titanate-anatase core–shell nanobelts with exposed {101} anatase facets and enhanced visible light photocatalytic activity. J. Am. Chem. Soc. 134, 5754–5757 (2012).
Li, Y., Zhang, W., Niu, J. & Chen, Y. Mechanism of photogenerated reactive oxygen species and correlation with the antibacterial properties of engineered metal-oxide nanoparticles. ACS Nano 6, 5164–5173 (2012).
Lee, H. U. et al. Highly visible-light active nanoporous TiO2 photocatalysts for efficient solar photocatalytic applications. Appl. Catal. B-Environ. 129, 106–113 (2013).
Tong, T. et al. Effects of material morphology on the phototoxicity of nano-TiO2 to bacteria. Environ. Sci. Technol. 47, 12486–12495 (2013).
Lee, H. U. et al. Influence of visible-light irradiation on physicochemical and photocatalytic properties of nitrogen-doped three-dimensional (3D) titanium dioxide. J. Hazard. Mater. 258, 10–18 (2013).
Kamegawa, T., Shimizu, Y. & Yamashita, H. Superhydrophobic surfaces with photocatalytic self-cleaning properties by nanocomposite coating of TiO2 and polytetrafluoroethylene. Adv. Mater. 24, 3697–3700 (2012).
Wang, K., Wei, M., Morris, M. A., Zhou, H. & Holmes, J. D. Mesoporous titania nanotubes: their preparation and application as electrode materials for rechargeable lithium batteries. Adv. Mater. 19, 3016–3020 (2007).
Zheng, Q. et al. Self-organized TiO2 nanotube array sensor for the determination of chemical oxygen demand. Adv. Mater. 20, 1044–1049 (2008).
Zhang, Z., Zhang, L., Hedhili, M. N., Zhang, H. & Wang, P. Plasmonic gold nanocrystals coupled with photonic crystal seamlessly on TiO2 nanotube photoelectrodes for efficient visible light photoelectrochemical water splitting. Nano Lett. 13, 14–20 (2013).
Lu, X. et al. Hydrogenated TiO2 nanotube arrays for supercapacitors. Nano Lett. 12, 1690–1696 (2012).
Zhang, J., Bang, J. H., Tang, C. & Kamat, P. V. Tailored TiO2–SrTiO3 heterostructure nanotube arrays for improved photoelectrochemical performance. ACS Nano 4, 387–395 (2010).
Yuan, L., Meng, S., Zhou, Y. & Yue, Z. Controlled synthesis of anatase TiO2 nanotube and nanowire arrays via AAO template-based hydrolysis. J. Mater. Chem. A 1, 2552–2557 (2013).
Yoo, H. et al. Understanding photoluminescence of monodispersed crystalline anatase TiO2 nanotube arrays. J. Phys. Chem. C 118, 9726–9732 (2014).
Li, S., Zhang, G., Guo, D., Yu, L. & Zhang, W. Anodization fabrication of highly ordered TiO2 nanotubes. J. Phys. Chem. C 113, 12759–12765 (2009).
Naito, K., Tachikawa, T., Fujitsuka, M. & Majima, T. Single-molecule observation of photocatalytic reaction in TiO2 nanotube: importance of molecular transport through porous structures. J. Am. Chem. Soc. 131, 934–936 (2009).
Liu, R. & Sen, A. Controlled synthesis of heterogeneous metal-titania nanostructures and their applications. J. Am. Chem. Soc. 134, 17505–17512 (2012).
Bavykin, D. V., Passoni, L. & Walsh, F. C. Hierarchical tube-in-tube structures prepared by electrophoretic deposition of nanostructured titanates into a TiO2 nanotube array. Chem. Commun. 49, 7007–7009 (2013).
Xu, X. et al. Tube-in-tube TiO2 nanotubes with porous walls: fabrication, formation mechanism and photocatalytic properties. Small 7, 445–449 (2011).
Li, G., Liu, J. & Jiang, G. Facile synthesis of spiny mesoporous titania tubes with enhanced photocatalytic activity. Chem. Commun. 47, 7443–7445 (2011).
Pathakoti, K. et al. Photoinactivation of Escherichia coli by sulfur-doped and nitrogen–fluorine-codoped TiO2 nanoparticles under solar simulated light and visible light irradiation. Environ. Sci. Technol. 47, 9988–9996 (2013).
Nam, S. H., Kim, T. K. & Boo, J. H. Physical property and photo-catalytic activity of sulfur doped TiO2 catalysts responding to visible light. Catal. Today 185, 259–262 (2012).
Han, C. et al. Innovative visible light-activated sulfur doped TiO2 films for water treatment. Appl. Catal. B-Environ. 107, 77–87 (2011).
Rengifo-Herrera, J. A. et al. Abatement of organics and Escherichia coli by N, S co-doped TiO2 under UV and visible light. implications of the formation of singlet oxygen (1O2) under visible light. Appl. Catal. B-Environ. 88, 398–406 (2009).
Dozzi, M. V., Livraghi, S., Giamello, E. & Selli, E. Photocatalytic activity of S- and F-doped TiO2 in formic acid mineralization. Photochem. Photobiol. Sci. 10, 343–349 (2011).
Dong, F., Zhao, W. & Wu, Z. Characterization and photocatalytic activities of C, N and S co-doped TiO2 with 1D nanostructure prepared by the nano-confinement effect. Nanotechnology 19, 365607 (1–10) (2008).
Xu, J. H. et al. Simple fabrication of twist-like helix N,S-codoped titania photocatalyst with visible-light response. Appl. Catal. B-Environ. 79, 72–80 (2008).
Yu, J. C. et al. Efficient visible-light-induced photocatalytic disinfection on sulfur-doped nanocrystalline titania. Environ. Sci. Technol. 39, 1175–1179 (2005).
Rockafellow, E. M., Stewart, L. K. & Jenks, W. S. Is sulfur-doped TiO2 an effective visible light photocatalyst for remediation? Appl. Catal. B-Environ. 91, 554–562 (2009).
Nam, S.-H., Kim, T. K. & Boo, J.-H. Physical property and photo-catalytic activity of sulfur doped TiO2 catalysts responding to visible light. Catal. Today 185, 259–262 (2012).
Pelaez, M. et al. A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B-Environ. 125, 331–349 (2012).
Asiri, A. M. et al. Enhanced visible light photodegradation of water pollutants over N-, S-doped titanium dioxide and n-titanium dioxide in the presence of inorganic anions. J. Saudi Chem. Soc. 18, 155–163 (2014).
Marschall, R. & Wang, L. Non-metal doping of transition metal oxides for visible-light photocatalysts. Catal. Today 225, 111–135 (2014).
Lee, Y.-C. et al. Self-assembled grapheme oxide with organo-building blocks of Fe-aminoclay for heterogeneous Fenton-like reaction at near-neutral pH: a batch experiment. Appl. Catal. B-Environ. 142–143, 494–503 (2013).
Lee, Y.-C. et al. Oil extraction by aminoparticle-based H2O2 activation via wet microalgae harvesting. RSC Adv. 3, 12802–12809 (2013).
Grätzel, M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg. Chem. 44, 6841–6851 (2005).
Boschloo, G. & Hagfeldt, A. Characteristics of the iodide/triiodide redox mediator in dye-sensitized solar cells. Accounts Chem. Res. 42, 1819–1826 (2009).
Lee, J. S., You, K. H. & Park, C. B. Highly photoactive, low bandgap TiO2 nanoparticles wrapped by grapheme. Adv. Mater. 24, 1084–1088 (2012).
Chowdhury, P., Moreira, J., Gomaa, H. & Ray, A. K. Visible-solar-light-driven photocatalytic degradation of phenol with dye-sensitized TiO2: parametric and kinetic study. Ind. Eng. Chem. Res. 51, 4523–4532 (2012).
Lee, H. U. et al. Photoluminescent carbon nanotags from harmful cyanobacteria for drug delivery and imaging in cancer cells. Sci. Rep. 4, 4665 (1–7) (2014).
Acknowledgements
This research was supported by the KBSI research Grant No. E34800.
Author information
Authors and Affiliations
Contributions
H.U.L., S.C.L., Y.-C.L., Y.-S.L. and J.L. designed the project, organized the entire research. H.U.L., S.C.L., Y.-C.L., S.Y.P., S.C. and J.L. wrote the manuscript. H.U.L., S.C.L., Y.-C.L., J.W.L. and S.Y.P. carried out the sample preparation and characterization. B.S. performed the XPS analysis. Y.K. measured the photocurrents. H.U.L., Y.-C.L., S.Y.P., S.C. and Y.-K.O. performed the photocatalytic activities and cytotoxicity testing of dye purified water by eco-TiO2. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Lee, H., Lee, S., Lee, YC. et al. Innovative three-dimensional (3D) eco-TiO2 photocatalysts for practical environmental and bio-medical applications. Sci Rep 4, 6740 (2014). https://doi.org/10.1038/srep06740
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06740
This article is cited by
-
3D structured nanocomposites by FDM process: a novel approach for large-scale photocatalytic applications
Applied Physics A (2018)
-
Corn-like, recoverable γ-Fe2O3@SiO2@TiO2 photocatalyst induced by magnetic dipole interactions
Scientific Reports (2017)
-
Novel Bio-functional Magnesium Coating on Porous Ti6Al4V Orthopaedic Implants: In vitro and In vivo Study
Scientific Reports (2017)
-
Synthesis, growth mechanism, and photocatalytic activity of Zinc oxide nanostructures: porous microparticles versus nonporous nanoparticles
Journal of Materials Science (2017)
-
Highly hydrophilic poly(vinylidene fluoride)/meso-titania hybrid mesoporous membrane for photocatalytic membrane reactor in water
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
