
Abstract
We have recently developed a scalable algorithm for ordering the instantaneous observations of a dynamical system evolving continuously in time. Here, we apply the method to long molecular dynamics trajectories. The procedure requires only a pairwise, geometrical distance as input. Suitable annotations of both structural and kinetic nature reveal the free energy basins visited by biomolecules. The profile is supplemented by a trace of the temporal evolution of the system highlighting the sequence of events. We demonstrate that the resultant SAPPHIRE (States And Pathways Projected with HIgh REsolution) plots provide a comprehensive picture of the thermodynamics and kinetics of complex, molecular systems exhibiting dynamics covering a range of time and length scales. Information on pathways connecting states and the level of recurrence are quickly inferred from the visualisation. The considerable advantages of our approach are speed and resolution: the SAPPHIRE plot is scalable to very large data sets and represents every single snapshot. This minimizes the risk of missing states because of overlap or prior coarse-graining of the data.
Similar content being viewed by others
Introduction
Present day science or more broadly society record observations as a function of time in diverse contexts1. Data on meteorological phenomena, communication tracking, or financial markets, to name a few, are all mined for the generation of predictive models. The raw data are usually unfit for human consumption due to their high dimensionality and sheer size2,3. Both aspects limit the types of analyses performed on these “big data” to those algorithms with satisfactory scaling properties4. In biophysics, long computer simulations of the trajectories of complex macromolecules with high-dimensional representations have become commonplace5 and this is where our particular interest lies6.
Molecular dynamics (MD) simulations of proteins and other biomolecules7 record stochastic trajectories, in which the macromolecule visits a number of different, metastable states (free energy basins) connected by an ensemble of pathways of interconversion. The latter report on the barriers of the underlying free energy landscape8. Because millions of snapshots are now routinely recorded for thousands of coupled degrees of freedom3, MD trajectories call for scalable algorithms that are able to provide information-preserving projections for this specific class of complex systems. We have recently introduced such an algorithm9 and provide a brief description next.
Given a definition of distance between trajectory snapshots, the entire data set is considered as a complete graph with vertices corresponding to snapshots and edge weights given by the pairwise distances between snapshots. Either the exact or an approximation to the minimum spanning tree are computed. From a generally arbitrary starting point, the available edge with smallest weight is followed to define a sequence of snapshots, the so-called progress index. The available edges at each point are those connecting any snapshot not considered yet (we refer to this set of snapshots as A) with any snapshot already included (the set S). The resulting sequence has the crucial property of stepping through high density regions one by one. It can therefore be expected that all free energy basins will appear as groups of nearby points along the progress index. Importantly, the progress index does not reflect the temporal nature of the input data in any way, i.e., it is generally independent of input order. Because every snapshot is considered, the limiting resolution is optimal given the time resolution of the input trajectory.
The progress index can be annotated both kinetically and structurally to provide an informative and compact representation of all major states visited by the input trajectory. The procedure has several advantages over projections using geometric or kinetic distances from a reference state to order snapshots. First, it maximizes resolution as mentioned above. Second, it avoids overlap precisely because the ordering is not with respect to a particular state. Third, it is easy to use, scalable and requires a notion of distance between snapshots as the only “parameter”. Like most data mining methods exploiting pairwise similarity as a guide, e.g., clustering10, it requires sufficient sampling density. The sampling weights of individual basins can in general be resolved quantitatively.
In the present, short contribution, we apply the method of Blöchliger et al.9 to two molecular dynamics trajectories of proteins, which were produced using dedicated hardware11. We annotate the plot threefold, viz., structurally, kinetically and with times of occurrence in the original trajectories (called dynamical trace hereafter). We demonstrate that the information summarized in the resultant SAPPHIRE (States And Pathways Projected with HIgh REsolution) plot provides an efficient means of identifying the statistically reliable states visited by a complex, dynamical system while enabling a rapid assessment of state interconversion and recurrence, which provide information on kinetic pathways and simulation convergence, respectively.
Results
We present results on two different proteins. The data on Fip3512, a small WW domain, come from two long MD trajectories and describe reversible transitions of this peptide between the folded state, a three-stranded β-sheet and a coil-like unfolded state. The single MD trajectory obtained for the 58-residue bovine pancreatic trypsin inhibitor (BPTI), a protein with a mixed α/β fold13, exhibits few transitions between distinct folded states that differ prominently in the isomerization states of disulphide bridges. We analyse both of these data sets with SAPPHIRE plots. The general annotation functions we use are as follows:
-
1
The sets A and S allow us to stipulate a two-state Markov state model and we can derive the mean first passage times in either direction9. A cut function as used elsewhere14 allows an analytical evaluation. We define the average of the two values as τMFP. This kinetic annotation is expected to highlight barriers reliably with the caveat that it cannot be interpreted quantitatively due to the simplicity of the two-state model. For data sets obtained by concatenating many short MD trajectories, the cut function is adjusted to ignore the spurious transitions at the break points between two trajectories.
-
2
The actual time of occurrence in the input data (dynamical trace) is plotted for each snapshot as an annotation highlighting direct transitions between states (pathways). Because the progress index is expected to be free of overlap, this allows a straightforward assessment of recurrence. This annotation is less informative if the data set is a concatenation of short trajectories where each continuous segment visits only one or few basins.
-
3
States themselves are characterized by a structural annotation. This is necessarily system-specific and requires prior knowledge of the system and data. An informative, geometric annotation can be exceptionally helpful in connecting the states identified by the kinetic annotation with a structural interpretation fit for human consumption. Structural annotations do not depend on input order, i.e., they are useful even for unordered input data.
Reversible folding of a 35-residue protein domain
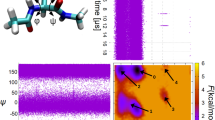
FiP3512 exhibits reversible folding at a simulation temperature of 395 K in explicit solvent molecular dynamics runs of a total length of 200 μs. Specifically, the trajectories show that FiP35 converts 10–15 times between an unfolded state that is very low in secondary structure content and the native topology, viz., a twisted, three-stranded β-sheet11,15,16. All following results refer to a specific computational model and sampling protocol11 underlying the trajectories being analysed. Due to the protein's small size, it is possible to provide a comprehensive, structural representation at the backbone level using a DSSP annotation17 resolved by residue. Fig. 1(a) shows the SAPPHIRE plot for the composite trajectory using this annotation and it is immediately apparent that the native topology is observed more than 50% of the time. The native basin is delineated by the kinetic annotation (black line) as expected. The unfolded state shows no consistent secondary structure and is kinetically homogeneous, suggesting that FiP35 should be described well as a two-state folder.

SAPPHIRE plot for FiP35.
(a) The progress index, of 106 snapshots from 200 μs of MD data, is annotated with kinetic information (τMFP, black curve), dynamical trace (red dots), DSSP assignment17 by residue (legend on top) and the state partitioning of Berezovska et al.20 These annotations are only shown for every 1000th, 100th, 1000th and 500th snapshots, respectively, in order to maintain readability at fixed figure resolution. The limits of possible definitions of the folded and unfolded states for the computation of transition path times are indicated by the blue, horizontal lines. Cartoons31 of a snapshot in the native state and an unfolded conformation are shown. (b) Zoom-in on the transition region of the SAPPHIRE plot shown in (a). The various annotations are shown for every 100th, 10th, 50th and 250th snapshots, respectively. Representative conformations of I1 and I2 are shown as cartoons. The box highlights a particular state (see text).
Fig. 1(b) highlights one of the major advantages of our approach, i.e., its high resolution. Here, we zoom in on the transition region. Previous analyses of the same data suggested the existence of at least two intermediates18,19,20 and we have additionally annotated the SAPPHIRE plot with the state partitioning proposed by Berezovska et al.20 Referring to their Fig. 3, we denote the larger and smaller of the two unlabelled states as L and S, respectively. By also taking into account the dynamical trace, Fig. 1 allows us to quickly extract the following results:
-
I2 is identified with a very homogeneous state sampled extensively only once during the 200 μs and characterized by a three-stranded β-sheet topology with shifted registry for the N-terminal hairpin (see DSSP annotation in Fig. 1(b)). It is referred to as a kinetic trap elsewhere16 and its sampling weight is 0.5–1.0%.
-
In state I1, only the N-terminal hairpin is formed with the C-terminus largely coil-like. This is the state sampled most often when converting between folded and unfolded states (F and U, respectively) via an intermediate and its weight is ~2.5%.
-
Structurally, L consists largely of a boundary (barrier) region between I1 and U. Our data suggest that the kinetically and geometrically homogeneous state highlighted by the black box in Fig. 1(b) should have been separated out.
-
States U and F are explored for extended periods of time less than 10 times each. Several excursions into intermediate states are unproductive.
-
We cannot assign obvious meaning to state S.
-
Fig. 1(a) suggests the existence of an additional state with a weight of ~5%, in which the N-terminal turn is in an alternative conformation. Similar differences are observed when comparing NMR structures of apo and holo forms of related WW domains21. While kinetically distinct, this state may have been ignored by Berezovska et al. because it is not on-pathway. Indeed, it is likely to correspond to the state labelled holo by Lane et al.19.
The picture emerging from the above is overall congruent with analyses of the same data reported elsewhere16,18,19,20,22,23. This also extends to the pathway information regarding dominant routes of folding. The main advantage of carefully constructed Markov state models is of course that probability flux and time scales are explicit and quantitative. This bears the caveat that the required lag times may be so large that useful information on or below the timescale of this lag time is lost. Conversely, Fig. 1 allows many of the important conclusions arrived at in the literature from a single plot that can be produced in near-linear time with respect to the number of snapshots. Some of the kinetic information such as probability flux and pathways are qualitative in nature only. It may be necessary to rescale the plot to resolve some of the finer details. It is also important to keep in mind that the sequence from left to right does not correspond to real pathways taken by the trajectory even though it may sometimes appear that way. Pathway information is gleaned exclusively from the dynamical trace, which is read vertically from bottom to top.
The SAPPHIRE plot allows a very straightforward grouping of snapshots into states. From these groupings, we can compute further quantities such as the times taken to reach the folded state from the unfolded state and vice versa (transition path times). Fig. 1(a) indicates two extreme definitions of folded and unfolded states and within these limits the computed transition path times range from 20 to 180 ns. Experiments suggest more roughness of the underlying landscape leading to longer transition times (~1 μs)24. We reemphasize that Fig. 1 is conditional upon a specific model, i.e., force field and sampling protocol, which in all likelihood are prone to both systematic and statistical errors. We merely perform the analysis here to demonstrate how the SAPPHIRE plot is also an excellent starting point for further efforts in characterizing and understanding the data and system at hand.
Native state dynamics of a folded protein
Analyses of MD simulations of the folded state ensemble of the 58-residue bovine pancreatic trypsin inhibitor (BPTI) have revealed that a number of states identified by NMR experiments25,26 are populated significantly in the trajectories albeit with inaccurate weights. These metastable states can often be correlated with isomerizations of the disulphide bridges, in particular Cys14-Cys3827,28. Shaw et al.11 used a stochastic algorithm to obtain a coarse, kinetic clustering of their 1.03 ms MD trajectory of BPTI relying on the autocorrelation of interatomic distances. Empirically, they found that five states with significant populations could be identified reliably. These states were annotated structurally. The most important states (the smaller of the two is the one most resembling the crystal structure) are clearly seen in the SAPPHIRE plot as the first two basins from the left (Fig. 2). The structural annotation we select here confirms that the barrier identified by the kinetic analysis (black line) is related to the isomerization of the disulphide bond Cys14-Cys38. The dynamical trace uses the colour scheme of Shaw et al. (distinguishing the red, blue, green, purple and black states). It unmasks that both major states are long-lived and that there is a clear separation of time scales with respect to the mixing time within each basin.

SAPPHIRE plot for BPTI.
(Upper panel) The progress index, of 41250 snapshots from 1.03 ms of MD data, is annotated with kinetic information (τMFP, black curve), dynamical trace (dots coloured according to the kinetic clustering of Shaw et al.)11 and selected dihedral angles. These annotations are only shown for every 20th, 2nd and 2nd snapshots, respectively, in order to maintain readability at fixed figure resolution. The annotation with dihedral angles uses binning into up to three bins with boundaries chosen as follows: Cys14 χ1 (−120°, −5°, 120°), Cys14 χ2 (−140°, 0°, 130°), Cys14-Cys38 χ3 (0°, 150°), Cys38 χ2 (−155°, −105°, 120°), Cys38 χ1 (−120°, 0°, 140°), Arg42 ψ (−100°, 75°) and Asp3 φ (0°, 100°). These boundaries were obtained from direct inspection of the individual histograms for each angle. Boxes highlight two brief stretches of the trajectory referred to in the text. (Lower panel) Zoom-in on a thin time slice of the dynamical trace to visualise a particular transition from the red to the black state. End points of this transition are shown as cartoons with Arg1 and Phe4 in a stick-like representation31. The plot is annotated further by the distance between the Cγ atom of Phe4 and the Cδ of Arg1, which is shown for every 5th snapshot.
Fig. 2 indicates that there is a mismatch in assignment between that of Shaw et al. and the positions on the x-axis for several, short excursions into a given state. As an example, we consider the highlighted trajectory segment sampled at ~0.5 ms that is annotated by Shaw et al. to be in the red state but that is placed by the SAPPHIRE plot in the basin corresponding to the blue state. To understand why this may be the case, we first note that the structural annotations generally reveal a small amount of mixing that may be considered erroneous. Indeed, for the segment in question, inspection of instantaneous values yields that the Cys14 side chain angles adopt the values for the blue state, but the χ3 angle and the χ2 angle of Cys38 do not (not shown). The combination of values for the dihedral angles places this segment outside of the list of states characterized previously28. It appears kinetically homogeneous and may correspond to an incomplete or blocked transition. Its sampling weight is so low that neither the SAPPHIRE plot nor the kinetic clustering are sensitive enough to resolve it as an independent state. Due to its intermediate nature, it is lumped into either one of the adjacent states. A very similar effect is observed for a second, highlighted segment (at ~0.75 ms), for which just the two Cys38 side chain angles deviate from the blue state.
The SAPPHIRE plot for BPTI also reveals that over the course of the 1.03 ms trajectory the purple and black states are sampled extensively just once and twice, respectively. This allows us to infer a lack of recurrence, i.e., sampling weights are unlikely to be converged. Poor sampling may also limit the number of states obtainable from Markov state models29 and decrease the accuracy of any extracted passage times. The bottom panel of Fig. 2 zooms into a very thin time slice to illustrate the pathway taken to reach the black state. This is annotated by cartoons and a specific, interatomic distance involving a residue identified by the original authors as being discriminative for this state11. The final result we want to mention in this short note is that the SAPPHIRE plot suggests the green state to be partitioned further. The kinetic annotation is consistent with the dynamical trace in that the two major substates of the green state are homogeneous with respect to the times they were sampled at (no mixing). This is despite the fact that they appear to be directly adjacent to one another in terms of transition pathways.
We conclude the description of the performance of the SAPPHIRE plot with a note of caution. In Fig. 2, toward the right side of the largest basin, there is a region of both temporal and geometric ambiguity most clearly seen by the overlap of blue and red dots in the dynamical trace. Here, the progress index is placing “fringe” regions of both basins. This weakness results from an insufficient sampling density for these lower likelihood regions that immediately surround well-defined states. It is rectified by having better time resolution or, at the risk of a decrease in resolution, by lowering the dimensionality of representation. We show the data on the sparsely sampled trajectory here to illustrate both the general robustness and possible errors encountered with smaller data sets.
Discussion
With growing computing resources and growing data sets, it has become paramount to use tools that quickly and efficiently improve our understanding of a system as complex as a biomolecule. The data required for the SAPPHIRE plot with all three annotations can usually be computed in near-linear time in a single run by the CAMPARI simulation and analysis package (http://campari.sourceforge.net). The plots are ideally generated as fully scalable vector graphics. At fixed resolution, readability may be improved by displaying annotations more sparsely and we have done this for both figures. The required user input is the definition of a suitable measure of pairwise distance and this choice may also help determine which structural annotations to use.
In Figs. 1 and 2, we have shown that SAPPHIRE plots offer an efficient procedure for the analysis and comprehensive pictorial description of complex systems undergoing stochastic evolution, such as proteins. Thermodynamics are resolved quantitatively and the construction of the ordering of snapshots minimizes the risk of state overlap. Major basins are delineated easily by all three annotation functions. Qualitative information about pathways is available at the temporal resolution offered by the trajectory itself. The rapid availability of this information is not only valuable per se but can also be used to guide further simulations and analyses.
Methods
The algorithm underlying the SAPPHIRE plot has been describe qualitatively above (see Introduction and Results). For a complete description we refer the reader to the original publication9. In terms of efficiency, the overall annotation procedure requires linear time with respect to the number of snapshots. The calculation of the required spanning tree is the most expensive step of the algorithm and is aided by heuristics in either variant (exact or approximate). The approximate version can be scaled to very large data sets. When using this version, it will generally be useful to rerun the analysis a few times due to the stochastic nature of the spanning tree. In particular, the kinetic annotation function is sensitive to where a basin appears in the progress index and how well basins to the left have been captured.
The FiP35 trajectory encompasses 106 snapshots saved every 200 ps, while the 41250 snapshots of data on BPTI have a coarser time resolution of 25 ns. Pairwise distances were defined as the coordinate root mean square deviation (RMSD) computed over the backbone oxygen and nitrogen atoms of residues 7–29 for FiP35 and over 695 nonsymmetric atoms for BPTI. These choices reflect the different levels of variance in the two data sets. The approximate algorithm was used for both systems. It requires additional parameters as follows. The number of guesses to find putative nearest neighbours from within a limited space defined by preorganization of the data via clustering30 was set to a value of 1000 throughout. The lower threshold radii for clusters were 3.0 and 2.5Å for FiP35 and BPTI, respectively and the upper threshold radii were 10.0 and 3.0Å. The required input data took ~11 and ~5 hours to compute on a single Intel Xeon core (either E5435 or E5410) for Figs. 1 and 2, respectively.
References
Fu, T.-C. A review on time series data mining. Eng. Appl. Artif. Intel. 24, 164–181 (2011).
Kehrer, J. & Hauser, H. Visualization and visual analysis of multifaceted scientific data: A survey. IEEE Trans. Vis. Comput. Graph. 19, 495–513 (2013).
Rysavy, S. J., Bromley, D. & Daggett, V. DIVE: A graph-based visual-analytics framework for big data. IEEE Comput. Graph. Appl. 34, 26–37 (2014).
Bohlouli, M. et al. in Integration of Practice-Oriented Knowledge Technology: Trends and Prospectives (ed Fathi, M.) Ch. Towards an integrated platform for big data analysis, 47–56 (Springer, 2013).
Dror, R. O., Dirks, R. M., Grossman, J. P., Xu, H. & Shaw, D. E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 41, 429–452 (2012).
Spiliotopoulos, D. & Caflisch, A. Molecular dynamics simulations of bromodomains reveal binding-site flexibility and multiple binding modes of the natural ligand acetyl-lysine. Isr. J. Chem. in press, 10.1002/ijch.201400009 (2014).
Karplus, M. & McCammon, J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9, 646–652 (2002).
Onuchic, J. N., Luthey-Schulten, Z. & Wolynes, P. G. Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem. 48, 545–600 (1997).
Blöchliger, N., Vitalis, A. & Caflisch, A. A scalable algorithm to order and annotate continuous observations reveals the metastable states visited by dynamical systems. Comp. Phys. Comm. 184, 2446–2453 (2013).
Xu, R. & Wunsch II, D. C. Clustering algorithms in biomedical research: A review. IEEE Rev. Biomed. Eng. 3, 120–154 (2010).
Shaw, D. E. et al. Atomic-level characterization of the structural dynamics of proteins. Science 330, 341–346 (2010).
Liu, F. et al. An experimental survey of the transition between two-state and downhill protein folding scenarios. Proc. Natl. Acad. Sci. USA 105, 2369–2374 (2008).
Berndt, K. D., Güntert, P., Orbons, L. P. M. & Wüthrich, K. Determination of a high-quality nuclear magnetic resonance solution structure of the bovine pancreatic trypsin inhibitor and comparison with three crystal structures. J. Mol. Biol. 227, 757–775 (1992).
Krivov, S. V. & Karplus, M. One-dimensional free-energy profiles of complex systems: Progress variables that preserve the barriers. J. Phys. Chem. B 110, 12689–12698 (2006).
Lindorff-Larsen, K., Piana, S., Dror, R. O. & Shaw, D. E. How fast-folding proteins fold. Science 334, 517–520 (2011).
Kellogg, E. H., Lange, O. F. & Baker, D. Evaluation and optimization of discrete state models of protein folding. J. Phys. Chem. B 116, 11405–11413 (2012).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Krivov, S. V. The free energy landscape analysis of protein (FIP35) folding dynamics. J. Phys. Chem. B 115, 12315–12324 (2011).
Lane, T. J., Bowman, G. R., Beauchamp, K., Voelz, V. A. & Pande, V. S. Markov state model reveals folding and functional dynamics in ultra-long MD trajectories. J. Am. Chem. Soc. 133, 18413–18419 (2011).
Berezovska, G., Prada-Garcia, D. & Rao, F. Consensus for the Fip35 folding mechanism? J. Chem. Phys. 139, 035102 (2013).
Wintjens, R. et al. 1H NMR study on the binding of Pin1 Trp-Trp domain with phosphothreonine peptides. J. Biol. Chem. 276, 25150–25156 (2001).
a Beccara, S., Škrbić, T., Covino, R. & Faccioli, P. Dominant folding pathways of a WW domain. Proc. Natl. Acad. Sci. USA 109, 2330–2335 (2012).
McGibbon, R. T. & Pande, V. S. Learning kinetic distance metrics for Markov state models of protein conformational dynamics. J. Chem. Theor. Comput. 9, 2900–2906 (2013).
Liu, F., Nakaema, M. & Gruebele, M. The transition state transit time of WW domain folding is controlled by energy landscape roughness. J. Chem. Phys. 131, 195101 (2009).
Otting, G., Liepinsh, E. & Wüthrich, K. Disulfide bond isomerization in BPTI and BPTI(G36S): An NMR study of correlated mobility in proteins. Biochemistry 32, 3571–3582 (1993).
Grey, M. J., Wang, C. & Palmer III, A. G. Disulfide bond isomerization in basic pancreatic trypsin inhibitor: Multisite chemical exchange quantified by CPMG relaxation dispersion and chemical shift modeling. J. Am. Chem. Soc. 125, 14324–14335 (2003).
Long, D. & Brüschweiler, R. Atomistic kinetic model for population shift and allostery in biomolecules. J. Am. Chem. Soc. 133, 18999–19005 (2011).
Xue, Y., Ward, J. M., Yuwen, T., Podkorytov, I. S. & Skrynnikov, N. R. Microsecond time-scale conformational exchange in proteins: Using long molecular dynamics trajectory to simulate NMR relaxation dispersion data. J. Am. Chem. Soc. 134, 2555–2562 (2012).
Noé, F., Wu, H., Prinz, J.-H. & Plattner, N. Projected and hidden Markov models for calculating kinetics and metastable states of complex molecules. J. Chem. Phys. 139, 184114 (2013).
Vitalis, A. & Caflisch, A. Efficient construction of mesostate networks from molecular dynamics trajectories. J. Chem. Theor. Comput. 8, 1108–1120 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Acknowledgements
The authors thank D.E. Shaw Research for sharing the trajectory data and their state annotation for BPTI (colour code in Fig. 2). We thank Dr. Francesco Rao for providing the state partitioning for FiP35 we use in Fig. 1. AV acknowledges financial support from the Holcim foundation. This work was supported in part by a grant from the Swiss National Science Foundation to AC.
Author information
Authors and Affiliations
Contributions
N.B., A.V. and A.C. contributed to study design. N.B. analysed the data and created the figures and captions. A.V. wrote the manuscript, which was reviewed by all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Blöchliger, N., Vitalis, A. & Caflisch, A. High-Resolution Visualisation of the States and Pathways Sampled in Molecular Dynamics Simulations. Sci Rep 4, 6264 (2014). https://doi.org/10.1038/srep06264
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06264
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
