
Abstract
Essential genes, those critical for the survival of an organism under certain conditions, play a significant role in pharmaceutics and synthetic biology. Knowledge of protein localization is invaluable for understanding their function as well as the interaction of different proteins. However, systematical examination of essential genes from the aspect of the localizations of proteins they encode has not been explored before. Here, a comprehensive protein localization analysis of essential genes in 27 prokaryotes including 24 bacteria, 2 mycoplasmas and 1 archaeon has been performed. Both statistical analysis of localization information in these genomes and GO (Gene Ontology) terms enriched in the essential genes show that proteins encoded by essential genes are enriched in internal location sites, while exist in cell envelope with a lower proportion compared with non-essential ones. Meanwhile, there are few essential proteins in the external subcellular location sites such as flagellum and fimbrium and proteins encoded by non-essential genes tend to have diverse localizations. These results would provide further insights into the understanding of fundamental functions needed to support a cellular life and improve gene essentiality prediction by taking the protein localization and enriched GO terms into consideration.
Similar content being viewed by others
Introduction
Regardless of the immense differences between bacterial genomes in their size and gene repertoires, all the genomes must contain enough information giving the cell the ability to maintain metabolic homeostasis, reproduction and evolvement, the three basic properties of cellular life1. Among all the genes in an organism, what genes are indispensable to fulfill these functions? To address this problem, a concept of essential gene was proposed. Essential genes are those indispensable for the survival of an organism under certain conditions and the functions they encode are therefore considered a foundation of life2,3,4. Investigation of essential genes is becoming an increasingly appealing issue not only because it will shed new light on the understanding of life at its simplest level, but also because it has much significance in practical use such as pharmaceutics and synthetic biology5,6,7.
An intuitive way to identify an essential gene is to detect whether the inactivation of this gene is lethal. Previous approaches used to identify essential genes include global transposon mutagenesis strategies, inhibition of gene expression using antisense RNA and systematic gene inactivation of each individual gene present in a genome2,8. More recently, high-throughput sequencing has been applied together with high-density transposon-mediated mutagenesis, which has increased the number of prokaryotic species involved in gene essentiality research dramatically9. In the last few years, great progresses not only in vivo but also in silico have been made. For example, bacterial essential genes have been showed more evolutionarily conserved than non-essential ones and tend to reside in the leading strand10,11. Based on these progresses, gene essentiality prediction models and tools have also been developed12,13,14,15.
Our study is focused on the protein location of essential genes. In general case, proteins must be transported to the appropriate location to perform their designated function. The location sites in prokaryotic cells can be reduced to three groups: internal structures, cell envelope and external structures. The uppermost internal structure is cytoplasm, a jelly-like substance where all proteins are synthesized and most of them remain16,17. The main structures found in the cytoplasm are the ribosomes and one (or a few) chromosome (s) which are essential to the functions of all prokaryotic cells. The cell envelope is composed of cytoplasmic membrane and cell wall in Gram-positive bacteria. While in Gram-negative bacteria, the cell envelope location sites include the cytoplasmic membrane, the outer membrane and the periplasm, which is the space between the two membranes. Most external structures such as flagella, fimbriae, capsule and slime layer are specific structures that are found in some, but not all bacteria18.
Knowledge of protein localization is invaluable for understanding their function as well as the interaction of different proteins19. When other information is not available, the subcellular localization will also be helpful in the annotation for new proteins. In the medical microbiology, subcellular location knowledge can help identify therapeutic intervention points rapidly during the drug discovery progress. For example, because of their localization, secreted proteins and membrane proteins are easily accessible by drug molecules20. Because of the critical functions of essential genes, it was hypothesized that proteins encoded by essential genes are enriched in internal location sites, while exist in cell envelope with a lower proportion compared with non-essential ones. In the current study, some analyses were performed to test this hypothesis.
Results and discussion
We selected 27 prokaryotic organisms to analyze the protein location of the essential and non-essential genes. The data used in the current study are obtained from DEG (a database of essential genes, available at http://www.essentialgene.org/)21 and are displayed in Table 1. To elucidate the evolutionary relationship among the organisms, the phylogenetic tree was constructed. The lines at the top of Figure 1 are the phylogenetic tree of the organisms used in the current study. The tree was constructed using the MEGA6 program (Statistical Method: Maximum Likelihood, Test of Phylogeny: Bootstrap method, No. of Bootstrap Replications: 1000)22 with the sequences of 16S ribosomal RNA of the 27 organisms downloaded from the NCBI FTP site (ftp://ftp.ncbi.nih.gov/genomes/Bacteria). Based on the clades of the tree, the organisms can be divided into 4 groups: 20 Gram-negative bacteria (pink clades in Figure 1), 4 Gram-positive bacteria (purple clades), 2 mycoplasmas (blue clades) and 1 archaeon (green clades). To which group Mycobacterium tuberculosis H37Rv should be classified is disputable. We treated it as Gram-negative bacterium due to its cell structure.

The plot of statistically significant GO terms in the category of cellular component incorporating the phylogenetic information.
Every GO term with p value less than 0.05 in over two organisms according to the results of Fisher's exact tests is listed in the vertical axis. If the GO term is over-represented in the organism listed in the horizontal axis, the cell at the crossing of the row and column is red. Blue boxes represent that the GO term is under-represented in the organism of the column. If the GO term is not statistically significant in the organism, the box is white. The lines at the top of the figure are the phylogenetic tree of the organisms used in the current study.
Protein localizations are different between essential and non-essential genes
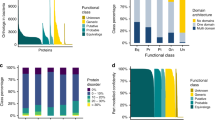
We first submitted the amino acid sequences of both essential and non-essential genes in the 27 organisms to PSORTb and obtained the protein localization records. With precision values >97% for both archaea and bacteria, PSORTb 3.0 is the most precise prokaryotic localization prediction tool available. Compared with other localization prediction tools, PSORTb is able to discriminate between Gram-positive and Gram-negative bacteria, which makes it a more suitable tool for the current study23. The final prediction includes five Gram-negative localization sites (cytoplasm, cytoplasmic membrane, periplasm, outer membrane and extracellular space) and four Gram-positive localization sites (cytoplasm, cytoplasmic membrane, cell wall and extracellular space)24. When a protein might have multiple localization sites, PSORTb will output the most possible localization site. Then we calculated the proportions of proteins located in the location sites for essential and non-essential genes respectively. The results are displayed in Figure 2. The average percentages of proteins located in cytoplasm are 64.40% and 43.88% for essential and non-essential genes, respectively. The Student's t test shows that the difference is statistically significant (p = 1.57 × 10−10). For all the organisms except Vibrio cholerae N16961, the percentages of proteins located in cytoplasm for essential genes are higher than those for non-essential genes (Figure 2a). The reason for the anomalous outcome in Vibrio cholerae N16961 may be the higher proportion of ‘unknown’ predicted results (43.13%) compared with the average percentage (16.43%). To test this hypothesis, we used another tool CELLO25,26 to predict the protein localization in Vibrio cholerae N16961 again. Then the percentages become 71.08% and 54.31% for essential and non-essential genes respectively, which is in accordance with the results of the other 26 organisms. These results suggest that proteins encoded by essential genes are enriched in cytoplasm. The average percentages of proteins located in cytoplasm membrane are 16.73% and 23.35% for essential and non-essential genes, respectively. The Student's t test shows that the difference is statistically significant (p = 1.33 × 10−5). The bars in Figure 2a shows that in 23 (85.19%) of the 27 groups of data, the percentages of proteins located in cytoplasm membrane for non-essential genes are higher than those for essential genes. For both essential and non-essential proteins, the proportions of secreted proteins are quite low, just 0.50% essential proteins and 1.54% non-essential proteins are located in extracellular space. With Student's t test p = 1.95 × 10−4, it's credible that the proportion of non-essential proteins located in extracellular is significantly higher than that of essential ones (Figure 2a).

(a) Percentages of proteins located in cytoplasm, cytoplasm membrane and extracellular for essential and non-essential genes in the 27 genomes. (b) Average percentages of proteins located in periplasm, outer membrane and cell wall for essential and non-essential genes in the related genomes.
Cytoplasm, cytoplasm membrane and extracellular are protein location sites involved in all the 4 groups of organisms. Figure 2a shows percentages of proteins located in the three location sites of all the genomes. In Figure 2b, the 3 location sites mentioned are not ubiquitous. Just the 4 Gram-positive bacteria and 1 archaeon have the cell wall structure. Outer membrane and periplasm are the unique structure of Gram-negative bacteria. Figure 2b presents the average percentages of proteins located in the three location sites for essential and non-essential genes in the related genomes. The proportions of non-essential proteins located in periplasm, outer membrane and cell wall are higher than those of essential proteins. The corresponding p values are 4.06 × 10−6, 3.06 × 10−3 and 4.68 × 10−2. All the values are less than 0.05, which means that the differences are statistically significant. Since cytoplasm membrane, cell wall, periplasm and outer membrane together form cell envelope, we could reach the conclusion that proteins encoded by essential genes exist in cell envelope with a lower proportion compared with non-essential ones. We found that protein localization differences between essential and non-essential genes are more significant in Gram-positive bacteria than those in Gram-negative bacteria, which may be due to the simple cell structures in Gram-positive bacteria.
Other factors that may influence the protein localization differences, such as the multiple localization of a protein, the reliability of protein localization prediction and the source of non-essential genes, are also discussed here. On average, 2.47% of the essential proteins and 2.70% of the non-essential proteins in the prediction of PSORTb have been annotated with multiple localization sites (the percentages of multiple localization proteins in each dataset are listed in Table 1). Therefore, the issue of multiple localization of a protein only bring a very slight impact on the accuracy of the statistical results due to the low percentages. Since the prediction result might not be perfectly precise, some experimental data were also employed. The protein localization information was obtained from the Universal Protein Resource (UniProt; http://www.uniprot.org)27. Captured from literatures, the data in UniProt is credible. We selected Bacillus subtilis 168, Escherichia coli MG1655 and Mycoplasma genitalium G37 as model genomes for Gram-positive bacteria, Gram-negative bacteria and mycoplasmas respectively, due to their higher percentages of the proteins with localization information. On average, 47.03% of the essential genes and 44.91% of the non-essential genes in these genomes have annotated localization information. We defined “unknown” as subcellular location for the proteins without annotated localization information. Among the proteins with localization information, 3.54% of the essential proteins and 3.35% of the non-essential proteins have multiple localization sites, which is close to the statistical result obtained from the prediction of PSORTb. Since multiple localization protein can locate in any site mentioned in its annotation, all the related site groups counted the protein in the calculation here. Figure 3 shows the distribution of essential proteins (the inner ring of the doughnut chart) and non-essential proteins (the outer ring of the doughnut chart) in B. subtilis 168, E. coli MG1655 and M. genitalium G37. In all the three doughnut charts, the percentages of the essential proteins located in cytoplasm are higher than those of non-essential proteins and the proteins encoded by essential genes exist in cell envelope with a lower proportion compared with non-essential ones. These conclusions are consistent with the prediction results of PSORTb. Comparisons were also made between groups classified according to the source of non-essential genes presented in Table 1. We found the differences are more significant in the organisms whose non-essential genes are obtained based on the original literatures. The reason may be that non-essential from the original literatures are more reliable than those from the complementary set of essential genes.

Distribution of essential proteins (the inner ring of the doughnut chart) and non-essential proteins (the outer ring of the doughnut chart) in (a) Bacillus subtilis 168, (b) Escherichia coli MG1655 and (c) Mycoplasma genitalium G37.
Protein localization analysis of essential genes based on GO terms
The Gene Ontology (GO) is one of the most useful terms and controlled vocabularies for describing the roles of genes and gene product characteristics. The ontology covers three domains: cellular component, molecular function and biological process. Cellular component refers to the place in the cell where a gene product is active. The molecular function is the elemental activities of a gene product at the molecular level. Biological process is defined as a biological objective to which the gene or gene product contributes28.
The Fisher's exact test was employed to obtain the GO terms enriched in the essential genes of 27 prokaryotes. P values less than 0.05 were considered statistically significant. Figure 1 represents the statistically significant GO terms in the category of cell component. As can be seen from this figure, in this category, GO:0005737 (cytoplasm), GO:0005840 (ribosome) and GO:0015935 (small ribosomal subunit) are the over-represented essential GO terms in all the 4 groups of organisms under analysis. Essential genes are more enriched in cytoplasm, ribosome, small ribosomal subunit and large ribosomal subunit, which are the major cell components for cell functions such as energy metabolism, nucleic acid translation and transcription, whereas GO:0016021 (integral component of membrane), GO:0016020 (membrane), GO:0005886 (plasma membrane), GO:0005622 (intracellular) and GO:0009279 (cell outer membrane) are under-represented in over 6 organisms, which means that proteins in cell components such as membrane have no much relationship with essential genes and are more likely to be encoded by non-essential genes. In addition, the membrane related GO terms are species-specific due to the different structure of cell envelope. For example, besides GO:0009279 (cell outer membrane), GO:0030288 (outer membrane-bounded periplasmic space) and GO:0042597 (periplasmic space) are particularly under-represented in E. coli MG1655, which is caused by the complicated cell structure of Gram-negative bacteria. This result can be construed as another evidence to the conclusion that proteins encoded by essential genes tend to locate in cytoplasm.
The Fisher's exact test was also employed to obtain enriched GO terms in the category of biological process and molecular function. GO:0007049 (cell cycle), GO:0006260 (DNA replication), GO:0009252 (peptidoglycan biosynthetic progress), GO:0051301 (cell division), GO:0065002 (intracellular protein transmembrane transport), GO:0006265 (DNA topological change) and GO:0006184 (GTP catabolic process) are the most significantly over-represented biological process GO terms. These progress are all indispensable for a cell and take place in cytoplasm or ribosome. The GO terms under-represented in over 6 organisms in this category are GO:0006355 (regulation of transcription, DNA-templated), GO:0035556 (intracellular signal transduction), GO:0006200 (ATP catabolic process), GO:0005975 (carbohydrate metabolic process) and GO:0055114 (oxidation-reduction process). For the GO terms relating to molecular function, the most significantly over-represented molecular functions are GO:0003735 (structural constituent of ribosome), GO:0019843 (rRNA binding), GO:0005524 (ATP binding), GO:0000049 (tRNA binding), GO:0000287 (magnesium ion binding) and GO:0005525 (GTP binding). While GO:0003700 (sequence-specific DNA binding transcription factor activity), GO:0003677 (DNA binding), GO:0003824 (catalytic activity), GO:0051539 (4 iron, 4 sulfur cluster binding), GO:0000155 (phosphorelay sensor kinase activity), GO:0043565 (sequence-specific DNA binding), GO:0000156 (phosphorelay response regulator activity) and GO:0004872 (receptor activity) are significantly under-represented in more than 6 organisms.
Conclusion
Our results show the protein localization difference between essential and non-essential genes in prokaryotes. Essential proteins are enriched in cytoplasm. The proportions of non-essential genes locating in cytoplasm membrane, periplasm, outer membrane, cell wall and extracellular are significantly higher than those of essential genes. The Fisher's exact test of GO terms reached a coincident conclusion. Taking the protein localization and protein function into consideration comprehensively, we can know more about essential genes. These results would provide further insights into the understanding of fundamental functions needed to support a cellular life and improve gene essentiality prediction by taking the protein localization and enriched GO terms into consideration.
Methods
Bioinformatics Databases
DEG is a database of essential genes (http://www.essentialgene.org/). The newly released DEG 10 has been developed to accommodate the quantitative and qualitative advancements brought by the progressive identification methods. Currently available records of both essential and non-essential genes among a wide range of organisms can be downloaded from DEG 10, making it possible to compare the two different types of genes in many aspects21.
27 prokaryotic organisms including 24 bacteria, 2 mycoplasmas and Methanococcus maripaludis S2, the only record of the Archaea domain were selected to analyze the protein localization and GO distribution of the essential and non-essential genes. There are 31 bacterial records corresponding to 27 organisms in the database in total and 26 sets of data were selected in the current study. Streptococcus pneumonia was not chosen for the lack of non-essential genes. Since the essential genes were not genome-widely identified, it's not reasonable to regard the complementary set of essential genes as non-essential genes in Streptococcus pneumonia29,30. In the case of multiple records for one organism, the one with the most convincing experimental methods was chosen. The non-essential genes in Methanococcus maripaludis S2 and 13 bacteria such as Escherichia coli MG1655 are obtained based on the original literatures, while non-essential genes in other 12 organisms such as Bacillus subtilis 168 are the complementary set of essential genes. The information of the organisms used in the current study are displayed in Table 1.
The three model genomes' subcellular location information and the Gene Ontology (GO) terms used for the analysis in the current study were downloaded from the Universal Protein Resource (UniProt; http://www.uniprot.org). Maintained by the UniProt Consortium, UniProt is committed to providing biologists with a comprehensive, high-quality and freely accessible resource of protein sequences and functional annotation27. Among the wealth of annotation data, detailed GO annotation statements are included. A comprehensive set of evidenced-based associations between terms from the GO resource and UniProtKB proteins are provided in GO annotation dataset, which is maintained by external collaborating GO Consortium groups31.
Software Tools
PSORTb is the most precise bacterial localization prediction tool available24. PSORTb 3.0 comprises multiple analytical modules, each of them is carried out independently. Every module returns to a prediction either a protein is belonging or not belonging to a particular localization site, or a result of ‘unknown’ by analyzing one biological feature known to influence subcellular location. All the results are then integrated to generate a final prediction. The likelihood of a protein being at a specific localization site is showed by a score. When a protein might locate in more than one sites, PSORTb will output the most possible localization site. Because PSORTb 3.0 added the capability of predicting subcellular localizations of archaeal proteins, we can obtain the localization information of Methanococcus maripaludis S2 with this tool. Compared with other localization prediction tools, PSORTb is able to discriminate between Gram-positive and Gram-negative bacteria, which makes it a more suitable tool for the current study23.
CELLO is another localization prediction tool which uses the support vector machines trained by multiple feature vectors based on n-peptide compositions25,26. We used this tool to predict the protein localizations of Vibrio cholerae N16961, for whom the result presented by PSORTb had a high proportion of ‘unknown’. The phylogenetic tree was constructed using the MEGA6 program22.
Test Method
The Student's t test was performed to test the significance of difference between the proportions of proteins located in the sites for essential and non-essential genes. The Student's t test is a method of testing whether the means of two groups are statistically different from each other. P values less than 0.05 were considered statistically significant.
To obtain the GO terms enriched in the essential genes of 27 prokaryotes, the Fisher's exact test was employed. Fisher's exact test is a statistical significance test used for small sample sizes. The most common use of Fisher's exact test is for 2 × 2 tables, but it is valid for all sample sizes32. P values less than 0.05 were considered statistically significant.
References
Luisi, P. L., Oberholzer, T. & Lazcano, A. The notion of a DNA minimal cell: a general discourse and some guidelines for an experimental approach. Helvetica Chimica Acta 85, 1759–1777 (2002).
Gil, R., Silva, F. J., Peretó, J. & Moya, A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol R 68, 518–537 (2004).
Koonin, E. V. How Many Genes Can Make a Cell: The Minimal-Gene-Set Concept 1. Annu Rev Genom Hum G 1, 99–116 (2000).
Zhang, R., Ou, H. Y. & Zhang, C. T. DEG: a database of essential genes. Nucleic Acids Res 32, D271–D272 (2004).
de S Cameron, N. M. & Caplan, A. Our synthetic future. Nat Biotechnol 27, 1103 (2009).
Juhas, M., Eberl, L. & Church, G. M. Essential genes as antimicrobial targets and cornerstones of synthetic biology. Trends Biotechnol 30, 601–607 (2012).
Re, C. et al. Synthetic genome brings new life to bacterium. Science 18, 965 (2007).
Juhas, M., Eberl, L. & Glass, J. I. Essence of life: essential genes of minimal genomes. Trends Cell Biol 21, 562–568 (2011).
Barquist, L., Boinett, C. J. & Cain, A. K. Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biol 10, 1–9 (2013).
Jordan, I. K., Rogozin, I. B., Wolf, Y. I. & Koonin, E. V. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res 12, 962–968 (2002).
Rocha, E. P. & Danchin, A. Gene essentiality determines chromosome organisation in bacteria. Nucleic Acids Res 31, 7056–7056 (2003).
Lin, Y. & Zhang, R. R. Putative essential and core-essential genes in Mycoplasma genomes. Sci Rep 1, 53 (2011).
Wei, W., Ning, L.-W., Ye, Y.-N. & Guo, F.-B. Geptop: A Gene Essentiality Prediction Tool for Sequenced Bacterial Genomes Based on Orthology and Phylogeny. PloS ONE 8, e72343 (2013).
Zhong, J., Wang, J., Peng, W., Zhang, Z. & Pan, Y. Prediction of essential proteins based on gene expression programming. Bmc Genomics 14, 1–8 (2013).
Wang, J., Peng, W. & Wu, F. X. Computational approaches to predicting essential proteins: a survey. Proteom Clin Appl 7, 181–192 (2013).
Saleh, M. T., Fillon, M., Brennan, P. J. & Belisle, J. T. Identification of putative exported/secreted proteins in prokaryotic proteomes. Gene 269, 195–204 (2001).
Schatz, G. & Dobberstein, B. Common principles of protein translocation across membranes. Science 271, 1519–1526 (1996).
Silhavy, T. J., Kahne, D. & Walker, S. The bacterial cell envelope. CSH Perspect Biol 2, a000414 (2010).
Nevo-Dinur, K., Govindarajan, S. & Amster-Choder, O. Subcellular localization of RNA and proteins in prokaryotes. Trends Genet 28, 314–322 (2012).
Hung, M.-C. & Link, W. Protein localization in disease and therapy. J Cell Sci 124, 3381–3392 (2011).
Luo, H., Lin, Y., Gao, F., Zhang, C.-T. & Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res 42 (2014).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Bio Evol 30, 2725–2729 (2013).
Gardy, J. L. & Brinkman, F. S. L. Methods for predicting bacterial protein subcellular localization. Nat Rev Microbiol 4, 741–751 (2006).
Nancy, Y. Y. et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 (2010).
Yu, C. S., Lin, C. J. & Hwang, J. K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci 13, 1402–1406 (2004).
Yu, C. S., Chen, Y. C., Lu, C. H. & Hwang, J. K. Prediction of protein subcellular localization. Proteins 64, 643–651 (2006).
Consortium, U. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42, D191–D198 (2014).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat Genet 25, 25–29 (2000).
Song, J.-H. et al. Identification of essential genes in Streptococcus pneumoniae by allelic replacement mutagenesis. Mol Cells 19, 365–374 (2005).
Thanassi, J. A., Hartman-Neumann, S. L., Dougherty, T. J., Dougherty, B. A. & Pucci, M. J. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res 30, 3152–3162 (2002).
Dimmer, E. C. et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res 40, D565–D570 (2012).
Agresti, A. A survey of exact inference for contingency tables. Stat Sci 7, 131–153 (1992).
Acknowledgements
The authors would like to thank Prof. Chun-Ting Zhang for the invaluable assistance and inspiring discussions. They would also like to thank Dr. McGarvey for providing assistance in obtaining the GO IDs of the genes. The present work was supported in part by National Natural Science Foundation of China (Grant Nos. 31171238 and 30800642) and Program for New Century Excellent Talents in University (No. NCET-12-0396).
Author information
Authors and Affiliations
Contributions
F.G. conceived and designed the study. C.P. performed the data analysis and drafted the manuscript. Both authors edited the manuscript and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Peng, C., Gao, F. Protein Localization Analysis of Essential Genes in Prokaryotes. Sci Rep 4, 6001 (2014). https://doi.org/10.1038/srep06001
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06001
This article is cited by
-
Comparative analysis of essential genes in prokaryotic genomic islands
Scientific Reports (2015)
-
Evolutionary conservation analysis between the essential and nonessential genes in bacterial genomes
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
