
Abstract
Aniridia is a congenital panocular disorder caused by the mutations of the paired box gene-6 (PAX6). To investigate the clinical characterization and the underlying genetic defect in a Chinese family with aniridia and other ocular abnormalities, we recruited the family members who underwent ophthalmic examination. Two patients in this family, the proband and his affected son, both have bilateral aniridia, foveal hypoplasia and nystagmus. Moreover, the proband also had presenile cataracts, but his affected son did not show cataracts at the time of examination. Sequencing PAX6 revealed that a heterozygous duplication mutation c.95_105dup11, predicted to generate non-functional truncated protein at position Gly36 (p.G36X), was found in the affected individuals but not in any of the unaffected family members including the parents of the proband. Haplotype analysis showed that the proband and his affected son shared a common disease-related haplotype, which was arisen from the proband's unaffected father through crossing-over. In conclusion, we identified a novel de novo duplication mutation of PAX6 in the aniridia and other ocular abnormalities family. This mutation has occurred de novo on a paternal chromosome by direct duplication, which presumably results from replication slippage or unequal non-sister chromatids exchange during spermatogenesis.
Similar content being viewed by others

Introduction
Aniridia (OMIM#106210) is a rare congenital, autosomal dominant hereditary, bilateral, panocular disorder affecting not only the iris but also the cornea, anterior chamber, lens, retina and optic nerve. About two-thirds of cases are familial with dominant inheritance, high penetrance and variable expressivity. The remaining one-third of cases is sporadic and expected to be transmitted to the next generation in an autosomal dominant fashion1,2,3.
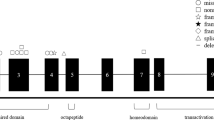
The paired box gene-6 (PAX6) (OMIM#607108) on chromosome 11p13 was described as a candidate for human aniridia by positional cloning4. The PAX6 gene encodes a highly conserved transcriptional regulator involved in oculogenesis and other developmental processes5,6. The PAX6 protein has two DNA binding domains, a paired domain (PD) and a homeodomain (HD), which separated by a glycine rich linker segment (LNK). The C-terminus, a domain rich in proline, serine and threonine (PST), acts as transactivator7. PAX6 mutations cause small eyes in mice8 and eyeless phenotype in Drosophila9. In humans, heterozygous mutations of the PAX6 gene cause aniridia as well as other various congenital abnormalities such as Peters' anomaly, foveal hyperplasia, corneal opacification, congenital cataracts, keratitis and microphthalmia3,7. So far, more than three hundred mutations of PAX6 have been identified in patients with ocular malformations, which were archived in Human PAX6 Allelic Variant Database10.
Here, we reported the clinical characterization of a Chinese family with aniridia and other ocular abnormalities, where a novel de novo duplication mutation of PAX6 was identified in the patients of this family. This duplication mutation was presumably derived from paternal chromosome by replication slippage or unequal non-sister chromatids exchange during spermatogenesis.
Results
Two individuals were affected with aniridia and other ocular abnormalities in Family AN-11.The proband (II:1) was a 40-year-old man with complete absence of the iris and congenital nystagmus in both eyes. He also suffered from bilateral progressive cataracts at the age of 32 years (Fig. 1-A, B). His visual acuity was very poor (0.15 in left eye and 0.12 in right eye). Using the direct ophthalmoscope, his central fovea of macula area was not observed. The 18-year-old son (III:1) of the proband had bilaterally no iris (Fig. 1-C, D) accompanied with congenital nystagmus, foveal hypoplasia(Fig. 1-E, F, G, H) and poor visual acuity (0.4 in left eye and 0.3 in right eye), but without cataracts. The proband's parents (I:1 and I:2) were clinically normal in both eyes.

Clinical features of the affected patients.
The proband (II:1) with complete aniridia and presenile cataracts (A, B) and his affected son (III:1) with aniridia (C, D) by slit lamp examination; the foveal hypoplasia of the proband's son(III:1) by funduscopy photographs (E, F) and by optical coherence tomography (G, H). OD and OS stand for right eye and left eye, respectively.
Mutation analysis of PAX6 in the proband revealed a heterozygous duplication mutation c.95_105dup11TAGCTCACAGC in exon 5 (Fig. 2), which resulted in the introduction of a premature termination codon (PTC) into the N-terminal subdomain of paired domain of PAX6 (p.G36X). The mutation was also detected in his affected son, but not in his parents, suggesting that it represents a de novo and inheritable mutation. This mutation was not detected in other unaffected members of this family and 103 unrelated normal controls (Fig. 2). In addition, the identified mutation had not been documented in database of single nucleotide polymorphisms (dbSNP) or in the 1000 genomes project dataset (http://browser.1000genomes.org). Since this duplication mutation has not been reported previously, we deposited it in Human PAX6 Allelic Variant Database (ID No. PAX6_00668)10.

DNA sequence chromatograms of the c.95_105dup11 mutation in exon5 of human PAX6 gene.
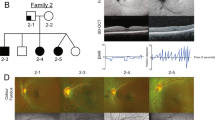
To establish the parental origin of the de novo PAX6 mutation, we performed the genotyping with four selected microsatellite markers (D11S904, D11S914, D11S1751 and D11S935) flanking PAX6 gene in available family members. Both the proband (II:1) and his affected son (III:1) shared the same disease-related haplotype, which was arisen from non-sister chromatids of his unaffected father (I:1) by crossing-over (Fig. 3). To verify paternity, we genotyped additional microsatellite markers located on different autosomes (D1S218, D2S177, D5S2501, D10S1216 and D22S1167) and confirmed that individual I:1 is indeed the biological father of the proband (II:1).

Pedigree and haplotype analysis of Family AN-11 with aniridia and other ocular abnormalities.
Squares and circles symbolize males and females, respectively. Filled symbols denote affected status. The proband is indicated by an arrow. Four selected microsatellite markers (D11S904, D11S914, D11S1751 and D11S935) flanking PAX6 gene listed in descending order from the centromeric end. PAX6 gene is located between D11S914 and D11S1751 on 11q13. The disease-related haplotype is arisen from non-sister chromatids of the proband's father (I:1) by crossing-over. The proband (II:1) transmitted it to his affected son(III:1).
Discussion
We here reported two members in Family AN-11 who were affected with aniridia, foveal hypoplasia and congenital nystagmus. Moreover, the proband was also affected with presenile cataract (onset before age 40 years). Except for aniridia, these clinical features were similar to those described by Thomas et al12. The affected son of the proband has not been found to have cataracts at the time of examination, but the risk of developing cataracts is supposed to take place later in his life. Our patients were caused by a heterozygous duplication insertion (c.95_105dup11), leading to a PTC mutation within the paired domain of PAX6 protein (p.G36X), which consistent with most PTC mutations tend to generate relatively severe phenotypes7,13. The PTC mutant mRNAs are generally detected and degraded by the nonsense mediated decay (NMD)7,14 and therefore we predict that our duplication mutation is probably functionally null.
More than one-third of PAX6 mutations are de novo10, but there are a few reports of the parental origin of them15,16. In this study, we determined that the duplication mutation c.95_105dup11 of PAX6 has occurred de novo on a chromosome inherited from the proband's father and transmitted to his son (Fig. 3). The paternity was unequivocally confirmed by testing with four independent microsatellite markers. The WAGR syndrome (Wilms tumor, aniridia, genital anomalies and mental retardation) is caused by deletion of band 11p13, which involved in WT1 tumor-suppressor gene and PAX6 gene4,17,18. Approximately 90% of these deletions are de novo, most frequently of paternal origin19. Thus we supposed that de novo insertion and/or deletion mutations in PAX6 were preferential susceptibility of paternal origin in aniridia. In fact, all PAX6 de novo mutations reported to date occur exclusively on the paternal allele15,16, which also supported the above inference. However, it needs further verification in more cases.
Microdeletions and microinsertions causing inherited disease account for 24% logged mutations in the Human Gene Mutation Database (www.hgmd.org)20. Interestingly, of all the reported mutation types in PAX6, both deletions and insertions were found with a considerably high frequency i.e., 145 of 346(41.9%)8, which reflects a hypermutability state of the PAX6 gene, but the potentially underlying mechanism remains unclear. The present de novo duplication mutation may result from an unequal crossing-over between non-sister chromatids during spermatogenesis, when the breakpoints and junction occurred exactly at the mutation site. However, such coincidence appears to be rare and unequal crossing over should often lead to relatively large duplications or deletions21. Thus, the most likely explanation is that putative mechanism appears to be occur non-sister chromatid exchange and following slipped mispairing mediated by runs of repeat element (AGC) surrounding the mutational position during DNA replication20. However, we are not able to rule out the occurrence of the other mechanisms due to the small number of patients in our recruited family.
In conclusion, we found a novel de novo duplication mutation of PAX6 in a Chinese family with aniridia and other ocular abnormalities. The de novo mutation was of paternal origin, mostly resulting from unequal non-sister chromatids by cross-over during spermatogenesis or slipped-strand mispairing of a direct repeat during replication.
Methods
Subjects and DNA specimens
This study followed the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of Fujian Medical University. The methods were carried out in accordance with the approved guidelines. Written informed consent was obtained from all participants or parents/legal guardians of all the subjects who were studied.
A three-generation family (Family AN-11) with aniridia and other ocular conditions was recruited and all of five individuals (2 affected and 3 unaffected individuals) took part in this study (Fig. 3). Clinical and ophthalmological examinations were performed on the affected individuals, as well as on the unaffected family members. Phenotype was documented by slit lamp photography, funduscopy and optical coherence tomography. Blood samples were obtained from the above subjects and 103 unrelated normal controls from the same ethnic background prior to the study. Genomic DNA was extracted from peripheral blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Beijing, China), according to manufacturer's instructions.
Mutation screening
The entire coding exons and splice junctions of the human PAX6 gene were amplified by PCR using previously reported PCR primers and conditions11, which were listed in Table 1. PCR products were purified using Wizard SV Gel and PCR Clean-Up System (Promega, Beijing, China) according to the manufacturer's instructions and were directly sequenced using M13 forward primer and M13 reverse primer (Table 1). When a suspected mutation is found in the proband, it was further confirmed in all of available other family members as well as in 103 normal unrelated individuals from the same ethnic background. Mutation descriptions follow the nomenclature recommended by the Human Genomic Variation Society.
Haplotyping analysis
To determine the parental origin of the de novo mutation, the genotyping was performed with four selected microsatellite markers (D11S904, D11S914, D11S1751 and D11S935) flanking PAX6 gene in available family members. The additional microsatellite markers located on different autosomes (D1S218, D2S177, D5S2501, D10S1216 and D22S1167) were performed haplotyping analysis for verification of paternity. Briefly, PCR products from each DNA sample were separated by gel electrophoresis with a fluoresence-based on ABI 3730 automated sequencer (Applied Biosystems) using ROX-500 as the internal lane size standard. The amplified DNA fragment lengths were assigned to allelic sizes with GeneMarker Version 2.4.0 software (SoftGenetics, State College, Pennsylvania, USA). Pedigree and haplotype data were managed using Cyrillic (version 2.1) software.
References
Kokotas, H. & Petersen, M. B. Clinical and molecular aspects of aniridia. Clin Genet. 77, 409–420 (2010).
Hingorani, M., Hanson, I. & van Heyningen, V. Aniridia. Eur J Hum Genet. 20, 1011–1017 (2012).
Lee, H. J. & Colby, K. A. A review of the clinical and genetic aspects of aniridia. Semin Ophthalmol. 28, 306–312 (2013).
Ton, C. C. et al. Positional cloning and characterization of a paired box-and homeobox-containing gene from the aniridia region. Cell. 67, 1059–1074 (1991).
Glaser, T., Walton, D. S. & Maas, R. L. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 2, 232–239 (1992).
Shaham, O., Menuchin, Y., Farhy, C. & Ashery-Padan, R. Pax6: a multi-level regulator of ocular development. Prog Retin Eye Res. 31, 351–376 (2012).
Tzoulaki, I., White, I. M. & Hanson, I. M. PAX6 mutations: genotype-phenotype correlations. BMC Genet. 6, 27 (2005).
Hill, R. E. et al. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 354, 522–525 (1991).
Quiring, R., Walldorf, U., Kloter, U. & Gehring, W. J. Homology of the eyeless gene of Drosophila to the small eye gene in mice and aniridia in humans. Science. 265, 785–789 (1994).
The PAX6 Allelic Variant Database [http://pax6.hgu.mrc.ac.uk/]
Sun, D. G., Yang, J. H., Tong, Y., Zhao, G. J. & Ma, X. A novel PAX6 mutation (c.1286delC) in the patients with hereditary congenital aniridia. Yi Chuan. 30, 1301–1306 (2008).
Thomas, S. et al. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur J Hum Genet. 2013 10.1038/ejhg.2013.162.
Hingorani, M., Williamson, K. A., Moore, A. T. & van Heyningen, V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Invest Ophthalmol Vis Sci. 50, 2581–2590 (2009).
Khajavi, M., Inoue, K. & Lupski, J. R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 14, 1074–1081(2006).
Bandah, D. et al. A novel de novo PAX6 mutation in an Ashkenazi-Jewish family with aniridia. Mol Vis. 14, 142–145 (2008).
Chao, L. Y., Huff, V., Strong, L. C. & Saunders, G. F. Mutation in the PAX6 gene in twenty patients with aniridia. Hum Mutat. 15, 332–339 (2000).
Call, K. M. et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 60, 509–520(1990).
Gessler, M. et al. Homozygous deletions in Wilms' tumours of a zinc-finger gene identified by chromosome jumping. Nature. 343, 774–778 (1990).
Huff, V., Meadows, A., Riccardi, V. M., Strong, L. C. & Saunders, G. F. Parental origin of de novo constitutional deletions of chromosomal band 11p13. Am J Hum Genet. 47, 155–160 (1990).
Ball, E. V. et al. Microdeletions and microinsertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat. 26, 205–213 (2005).
Kotzot, D. et al. Parental origin and mechanisms of formation of cytogenetically recognisable de novo direct and inverted duplications. J Med Genet. 37, 281–286 (2000).
Acknowledgements
The authors thank the families and all subjects for taking part in this study. This work was supported by grants from the Natural Science Foundation of Fujian Province (2010J06010), Program for New Century Excellent Talents in Fujian Province University (JA10127) and Professor Academic Development Fund of Fujian Medical University (JS12003). National Natural science Foundation of China (81270999), the Key Program of Scientific Research of Fujian Medical University (09ZD016).
Author information
Authors and Affiliations
Contributions
Study design: J.H.Y. and K.X.Z. collected the samples and performed the experiments: J.F.Z., X.L.C., Z.H.T. and Y.H.Z. Data interpretation and analysis: J.H.Y., J.F.Z. and X.L.C. Wrote the manuscript: J.H.Y. and X.L.C. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Zhuang, J., Chen, X., Tan, Z. et al. A novel de novo duplication mutation of PAX6 in a Chinese family with aniridia and other ocular abnormalities. Sci Rep 4, 4836 (2014). https://doi.org/10.1038/srep04836
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04836
This article is cited by
-
Impaired DNA-binding affinity of novel PAX6 mutations
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
