
Abstract
Contact hypersensitivity (CHS) is a form of delayed-type hypersensitivity triggered by the response to reactive haptens (sensitization) and subsequent challenge (elicitation). Here, we show that ASK1 promotes CHS and that suppression of ASK1 during the elicitation phase is sufficient to attenuate CHS. ASK1 knockout (KO) mice exhibited impaired 2,4-dinitrofluorobenzene (DNFB)-induced CHS. The suppression of ASK1 activity during the elicitation phase through a chemical genetic approach or a specific inhibitory compound significantly reduced the CHS response to a level similar to that observed in ASK1 KO mice. The reduced response was concomitant with the strong inhibition of production of IL-17, a cytokine that plays an important role in CHS and other inflammatory diseases, from sensitized lymph node cells. These results suggest that ASK1 is relevant to the overall CHS response during the elicitation phase and that ASK1 may be a promising therapeutic target for allergic contact dermatitis and other IL-17-related inflammatory diseases.
Similar content being viewed by others


Introduction
Contact hypersensitivity (CHS) is a form of delayed-type hypersensitivity triggered by the response to reactive haptens (sensitization) and subsequent challenge (elicitation)1. Clinicians have focused primarily on the latter because it is the phase when allergic contact dermatitis (ACD) is clinically manifested2. ACD is one of the most common skin diseases caused by complex immune mechanisms in response to a variety of reactive contact sensitizers, such as metals, preservatives and hair dyes and is well-modelled by CHS in mice. In the sensitization phase of CHS, epidermal proteins cross-linked with applied haptens are acquired mainly by two populations of dendritic cells (DCs), epidermal Langerhans cells (LCs) and Langerin-positive dermal DCs (dDCs), which have been shown to function redundantly3,4,5,6,7. These DCs migrate to the draining lymph nodes (LNs), where they prime hapten-specific effector T cells. The elicitation phase is initiated by the subsequent challenge of the sensitized animal with the same hapten, which induces infiltration of hapten-primed T cells to the challenge site. The infiltrated T cells subsequently elicit cutaneous inflammation through the production of cytokines such as IL-17 and IFN-γ8,9, the former of which indeed has recently been shown to be critically involved in the regulation of allergic skin diseases, as well as other various inflammatory diseases10,11. Large-scale efforts have been devoted to elucidating the regulatory mechanisms of CHS in the hope of discovering therapeutic targets for ACD.
Stress-activated p38 mitogen-activated protein kinase (MAPK) signaling has been identified as a mechanism promoting CHS because the pharmacological inhibition of p38 or the heterozygous deletion of the MAPK14 gene that encodes p38α, one of the isoforms of mammalian p38 MAPKs, attenuated the CHS induced by DNFB12,13,14. The roles of p38 MAPKs in innate immunity are well established and apoptosis signal-regulating kinase 1 (ASK1), a member of the MAPK kinase kinase (MAP3K) family, is a critical upstream activator of p38 in DCs15,16,17. However, the mechanism by which p38 promotes CHS and whether or not ASK1 is involved in p38-mediated CHS are not elucidated.
In this study, we found that ASK1 was, as expected, involved in 2,4-dinitrofluorobenzene (DNFB)-induced CHS and that the suppression of ASK1 activity during the elicitation phase through a chemical genetic approach or a specific inhibitory compound significantly attenuated the CHS response, concomitant with the strong inhibition of IL-17 production from sensitized lymph node cells. These results suggest that ASK1 is relevant to the overall CHS response during the elicitation phase.
Results
CHS response is attenuated in ASK1 KO mice
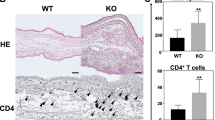
To investigate the involvement of ASK1 in CHS, we examined DNFB-induced CHS in ASK1 KO mice and found that it was attenuated compared with that of wild type (WT) mice (Fig. 1A, B). CHS induced by fluorescein isothiocyanate (FITC) was also attenuated in ASK1 KO mice (Fig. 1C).

CHS response is attenuated, but morphology and functions of Langerhans cells are not affected in ASK1 KO mice.
(A, B) DNFB-induced CHS response is attenuated in ASK1 KO mice. Twenty-four hr after treatment of the ears of DNFB-sensitive WT and ASK1 KO mice, the ears were excised for histological analysis (A). Representative histological sections are shown (hematoxylin and eosin staining; scale bar = 300 µm). Twenty-four hours after treatment of the ear of sensitized WT and ASK1 KO mice, ear swelling was determined (B). Data are shown as the mean ± SEM (n = 4). (C) FITC-induced CHS response is attenuated in ASK1 KO mice. Twenty-four hours after treatment of the ears of FITC-sensitized WT and ASK1 KO mice, ear swelling was determined. Data are shown as the mean ± SEM (n = 4) and are representative of four experiments. (D, E) Morphology and density of Langerhans cells are not affected in ASK1 KO mice. Langerhans cells in the epidermal sheets from WT and ASK1 KO mice were stained with PE-conjugated MHC class II antibodies. Representative immunofluorescence images (D) and the calculated density of Langerhans cells in the epidermal sheets (E; n = 5) are shown. (F) FITC-induced migration of Langerhans cells to regional LNs is not affected in ASK1 KO mice. MHC class II expression and FITC fluorescence in regional LN cells isolated from mice at 24 hr after the application of FITC were analyzed by flow cytometry. Data are representative of five experiments.
We further examined the role of ASK1 in the regulation of the nature and function of epidermal and dermal DCs. Staining of epidermal sheets with MHC class II antibodies revealed that the morphology and density of LCs were similar between WT and ASK1 KO mice, suggesting that ASK1 was dispensable for the generation and maintenance of LCs (Fig. 1D, E). FITC was also applied to the skin as a contact sensitizer used to trace DCs, which acquire FITC, migrate to the draining LNs and are detected as FITC-positive cells. At 24 hr after the FITC application to the skin, we isolated the draining LNs and determined the proportion of migrated DCs that was positive for both MHC II and FITC. There was no obvious difference between the WT and ASK1 KO mice, indicating that the FITC-induced migration of DCs to the draining LNs was not affected in the absence of ASK1 (Fig. 1F). Taken together, ASK1 does not appear to play a major role in epidermal and dermal DCs, at least during the sensitization phase of CHS.
IL-17-producing cells are reduced in ASK1 KO mice
To examine the involvement of ASK1 in lymphocytes, another set of cells important for CHS, we carried out adoptive transfer experiments. Naive ASK1 KO mice transferred with LN cells from DNFB-sensitized WT mice did not significantly affect the development of CHS (Fig. 2A). In contrast, naive WT mice transferred with LN cells from sensitized ASK1 KO mice showed a significant reduction in ear swelling, suggesting that ASK1 plays a crucial role in lymphocytes in CHS. In the sensitized LNs, hapten-specific T lymphocytes are primed and differentiated and upon rechallenge with the same hapten, these cells expand and rapidly accumulate in the stimulated site, where they secret proinflammatory cytokines. LN cells from DNFB-sensitized WT and ASK1 KO mice proliferated similarly following stimulation with 2,4-dinitrobenzenesulfonic acid (DNBS), a water-soluble compound with the same antigenicity as DNFB (Fig. 2B); however, the DNBS-dependent production of IL-17, but not of IFN-γ, was attenuated in ASK1 KO cells compared with WT cells, suggesting that ASK1 is selectively required for IL-17 production by sensitized LN cells (Fig. 2C).

IL-17-producing cells are reduced in ASK1 KO mice.
(A) CHS response is attenuated in WT recipient mice adoptively transferred with LN cells from sensitized ASK1 KO mice. Naive WT or ASK1 KO mice were adoptively transferred with LN cells isolated from DNFB-sensitized WT or ASK1 KO donor mice, followed by treatment of the ears of recipient mice with DNFB 1 hr after the transfer. Forty-eight hr later, ear swelling was determined. Data are shown as the mean ± SEM (n = 7) and are representative of four experiments. (B) ASK1 is dispensable for DNBS-induced proliferation of sensitized LN cells. LN cells from DNFB-sensitized WT or ASK1 KO mice were cultured in the presence or absence of DNBS for 3 days. Relative cell numbers were measured as the relative fluorescence intensity using a Cell counting Kit-F. Data are shown as the mean ± SEM (n = 5). (C) ASK1 is required for DNBS-induced production of IL-17, but not of IFN-γ, in sensitized LN cells. LN cells from DNFB-sensitized WT or ASK1 KO mice were cultured in the presence or absence of DNBS for 3 days. The amounts of IFN-γ and IL-17 in the culture medium were measured by ELISA (left and right graphs, respectively). Data are shown as the mean ± SEM (n = 5) and are representative of four experiments. (D) Proportion of IL-17-producing LN cells are reduced in sensitized ASK1 KO mice. The proportion of IFN-γ-producing (IFN-γ+) or IL-17-producing (IL-17+) cells among all LN cells isolated from DNFB-sensitive mice is shown. Data are shown as the mean ± SEM (n = 5). (E) Expression of RORγt is reduced in CD4+ T cells isolated from sensitized ASK1 KO mice. Relative mRNA expression of the indicated transcription factors in CD4+ T cells isolated from DNFB-sensitive WT and ASK1 KO mice was examined by quantitative RT-PCR. Data are shown as the mean ± SEM (n = 5).
At this point, we could not discriminate the roles of ASK1 in the sensitization phase from those in the elicitation phase because in vitro treatment with DNBS somewhat mimics the response of rechallenge with DNFB in the elicitation phase. Therefore, we examined the proportion of IL-17-producing LN cells in the absence of in vitro treatment with DNBS to determine if ASK1 is involved in the differentiation of IL-17-producing cells during sensitization. When the intracellular IFN-γ and IL-17 were detected by immunofluorescence in combination with flow cytometric analysis, there was a lower percentage of IL-17-producing LN cells in ASK1 KO mice compared with WT mice, whereas the proportion of IFN-γ-positive cells was similar between WT and ASK1 KO mice (Fig. 2D). A source of IL-17 in CHS is a CD4+ helper T-cell subset called T-helper 17 (Th17) cells11. Because the steroid receptor-type nuclear receptor RORγt, which is a splicing variant of RORγ, is critical for Th17 differentiation, we examined the expression of RORγt mRNA in CD4+ T cells isolated from sensitized ASK1 KO mice. As shown in Fig. 2E, RORγt expression was significantly lower in CD4+ T cells from ASK1 KO mice than in those from WT mice, whereas expression of other transcription factors such as T-bet, GATA3 and FoxP3, which are critical for the differentiation of Th1 cells, Th2 cells and regulatory T cells, respectively, was not significantly different between WT and ASK1 KO CD4+ T cells. These results suggest that the attenuation of CHS in ASK1 KO mice is caused, at least in part, by the decreased differentiation of IL-17 cells during sensitization.
Selective suppression of ASK1 activity in the elicitation phase attenuates CHS response
Considering that the reduction of IL-17-producing cells in ASK1 KO mice was not robust (Fig. 2D), there was a possibility that the decreased differentiation of IL-17 cells during sensitization was not the primary cause of the attenuated CHS response in ASK1 KO mice. To address this issue, we took advantage of a chemical genetic approach, termed analog sensitive kinase allele (ASKA), in which the kinase activity of ASK1 is pharmacologically controlled by the administration of 1Na-PP1, a derivative of the general kinase inhibitor PP118.
We first generated cDNA encoding of an ASKA of ASK1 (ASK1ASKA) (Fig. S1) and found that the basal activity of the ASK1ASKA protein, as detected by a phospho-ASK1 antibody that monitors the state of activating phosphorylation of ASK119, was comparable to that of WT ASK1 (Fig. 3A, lanes 1 & 2). The activity of ASK1ASKA, but not of WT ASK1, was strongly inhibited by 1Na-PP1, whereas the inhibitory effect of 1NM-PP1, another derivative of PP1, on ASK1ASKA was marginal (Fig. 3A, lanes 3–10). We then generated knock-in mice harboring ASK1ASKA (Fig. 3B) and found that the H2O2-induced activation of endogenous ASK1 in the absence of 1Na-PP1 was similar in bone marrow-derived macrophages (BMDMs) from homozygous ASK1ASKA mice and WT mice (Fig. 3C). We also found that the H2O2-induced activation of ASK1 was inhibited by 1Na-PP1 in ASK1ASKA mice in a dose-dependent manner but was not inhibited in WT mice, demonstrating the establishment of a chemical genetic tool suitable for elucidating the biological significance of ASK1 activity in vivo. Interestingly, the selective inhibition of ASK1 activity during elicitation by the administration of 1Na-PP1 to ASK1ASKA mice strongly suppressed CHS to a level similar to that of ASK1 KO mice [Fig. 3D, 1Na-PP1 (d5), cf. Fig. 1B], whereas the administration of 1Na-PP1 during sensitization did not suppress CHS [Fig. 3D, 1Na-PP1 (d0/1)]. Thus, the inhibition of ASK1 activity during elicitation appeared to be sufficient to suppress CHS. To further examine the requirement of ASK1 activity for CHS in the elicitation phase, we used K811, an inhibitory compound against ASK120. Administration of K811 to WT mice only during elicitation suppressed CHS to a similar extent to that of mice administered K811 during both sensitization and elicitation (Fig. 3E). Taken together, these results suggest that ASK1 activity in the elicitation phase is more relevant to the overall CHS response than that in the sensitization phase.

Selective suppression of ASK1 activity in the elicitation phase attenuates CHS response.
(A) ASK1ASKA, but not wild type ASK1, is inhibited by 1Na-PP1 and to a lesser extent by 1NM-PP1. Flag-tagged WT ASK1 (W), ASK1ASKA (A; V745L/M761A/S828A), or kinase-negative (KN; K716R) ASK1 was transiently expressed in HEK293 cells. Twenty-three hr after transfection, cells were further cultured in the presence of 10 μM (+) or 20 μM (++) 1Na-PP1 or 1NM-PP1 for 1 hr and then lysed. Cell lysates were subjected to immunoblot analysis using the indicated antibodies. (B) Targeting strategy for knock-in mice harboring ASK1ASKA. Grey vertical bars indicate exons 16 and 18 including ASKA mutations. (C) H2O2-induced activation of endogenous ASK1 is inhibited by 1Na-PP1 in BMDMs from homozygous ASK1ASKA mice. BMDMs isolated from WT and ASK1ASKA mice pretreated with the indicated final concentration of 1Na-PP1 were stimulated with 0.3 mM H2O2 for 5 min and then lysed. Cell lysates were subjected to immunoblot analysis using the indicated antibodies. (D) Administration of 1Na-PP1 prior to the elicitation phase attenuates CHS response in sensitized ASK1ASKA mice. ASK1ASKA mice were intravenously injected with 1Na-PP1 on day 0 and day 1 [1Na-PP1 (d0/1)] or on day 5 [1Na-PP1 (d5)]. Injection of 1Na-PP1 or vehicle was performed 5 min prior to treatment of the ear with DNFB. Twenty-four hours after the DNFB application, ear swelling was determined. Data are shown as the mean ± SEM (n = 8) and are representative of six experiments. (E) Administration of K811, an inhibitory compound against ASK1, prior to the elicitation phase attenuates CHS response in sensitized mice. DNFB-sensitized WT mice were orally administered vehicle (–), K811 twice on day 5 (d5), or K811 twice a day from day 0 to day 5 (d0-5). Twenty-four hours after the final DNFB application, ear swelling was determined. Data are shown as the mean ± SEM (n = 10) and are representative of six experiments.
ASK1 activity in sensitized CD4+ T cells is required for IL-17 production
We finally examined the cell type in which ASK1 regulates IL-17 expression. When the LN cells from DNFB-sensitized ASK1ASKA mice were treated with 1Na-PP1, DNBS-induced activation of ASK1 and the downstream p38 MAPK was detected (Fig. 4A, left panels). The activation of ASK1 and p38 sustained at least until 2 hr after DNBS stimulation and was strongly suppressed by treatment with 1Na-PP1 (Fig. 4A, right panels). Under these experimental conditions, DNBS-induced IL-17 production, but neither DNBS-induced IFN-γ production nor cell proliferation, was suppressed by 1Na-PP1 (Fig. 4B–D), suggesting that ASK1 is required in the elicitation phase for IL-17 production. We have previously shown that ASK1 plays a critical role in the cytokine response to lipopolysaccharide (LPS) in DCs16 and thus raised the possibility that ASK1 in DCs and/or T cells contributes to IL-17 production. To address this possibility, we isolated CD4+ T cells and CD11c+ DCs separately from DNFB-sensitized WT or ASK1ASKA mice and co-cultured them in vitro in the presence of DNBS. When WT CD4+ T cells and DCs were reconstituted, DNBS-dependent IL-17 production was clearly induced (Fig. 4E). Intriguingly, 1Na-PP1 suppressed DNBS-induced IL-17 production when ASK1ASKA CD4+ T cells and WT CD11c+ DCs were reconstituted. However, 1Na-PP1 did not suppress IL-17 production upon reconstitution of WT CD4+ T cells and ASK1ASKA DCs, suggesting that the ASK1 activity in CD4+ T cells, rather than that in DCs, is required for the CHS response (Fig. 4F). Although it has been shown that CD8+ T cells are also critically involved in the CHS response21,22, selective inhibition of the ASK1 activity in CD8+ T cells did not affect the DNBS-induced IL-17 production (Fig. 4G). Taken together, CD4+ T cells appear to be the primary cells in which ASK1 promotes IL-17 production during CHS.

ASK1 activity in sensitized CD4+ T cells is required for IL-17 production.
(A) DNBS-induced activation of ASK1 is inhibited by 1Na-PP1 in LN cells from sensitized ASK1ASKA mice. LN cells isolated from mice sensitized with DNFB were pretreated with (+) or without (−) 1 µM 1Na-PP1 and then stimulated with DNBS for the indicated time periods. Cells were lysed and cell lysates were subjected to immunoblot analysis using the indicated antibodies. (B, C) DNBS-induced production of IL-17, but not IFN-γ, is inhibited in 1Na-PP1-treated LN cells from sensitized ASK1ASKA mice. LN cells isolated from DNFB-sensitized mice were pretreated with (+) or without (−) 1 µM 1Na-PP1 and then stimulated with DNBS for 3 days. The amounts of IFN-γ (B) and IL-17 (C) in the culture medium were measured by ELISA. Data are shown as the mean ± SEM (n = 6) and are representative of four experiments. (D) Proliferation of LN cells from sensitized ASK1ASKA mice is not affected by 1Na-PP1. Relative numbers of LN cells isolated and treated as shown in (B, C) were measured as relative fluorescence intensity using a Cell Counting Kit-F. Data are represented as the mean ± SEM (n = 3). (E, F, G) ASK1 activity in DNFB-sensitized CD4+ T cells is required for DNBS-dependent IL-17 production. CD4+ and CD8+ T cells were isolated separately from regional LNs of DNFB-sensitized WT or ASK1ASKA mice. CD11c+ DCs were isolated from spleens of DNFB-sensitized mice. CD4+ T cells (E, F) or CD8+ T cells (G) and CD11c+ DCs were co-cultured in the presence (+) or absence (−) of 1 µM 1Na-PP1. Thirty minutes after the treatment with 1Na-PP1, cells were stimulated with DNFB. After 24 hr of co-culture, the amounts of IL-17 in the culture medium were measured by ELISA. Data are shown as the mean ± SEM (n = 7) and are representative of five experiments.
Discussion
In this study, we showed that ASK1 in the elicitation phase is more relevant to the overall CHS response than that in the sensitization phase, although ASK1 also plays a role in the differentiation of Th17 cells during sensitization. In the elicitation phase, ASK1 in sensitized CD4+ T cells appears to play a major role in CHS by facilitating IL-17 production, consistent with the reported findings that deficiencies in IL-17 or the IL-17 receptor or the neutralization of IL-17 in WT mice all induce an attenuated CHS response8,9,21. Whereas the critical involvement of CD8+ T cells in the elicitation of CHS has been proposed21,22, ASK1 does not appear to be involved in the role of CD8+ T cells in DNFB-dependent IL-17 production in our reconstitution experiments using CD8+ T cells and DCs. However, the observed strong attenuation of CHS under ASK1-deficient or ASK1-inactivated conditions still suggest that ASK1 functions not only in CD4+ T cells but also in CD8+ T cells and therefore, this phenomenon requires further investigation.
In CHS models, not only DNFB and FITC that we used in this study, but also oxazolone (Ox) and trinitrochlorobenzene (TNCB), are used as typical haptens10. These haptens induce similar responses but probably through different mechanisms. For instance, it has been reported that TNCB- and DNFB-induced, but not Ox-induced, CHS is attenuated in IL-4-deficient mice23,24,25,26, whereas CHS induced either by TNCB, DNFB, or Ox is attenuated in IL-17-deficient mice8,27. ASK1 may thus be differently involved in CHS induced by Ox or TNCB, prompting us to further analyze the response of ASK1 KO mice to these haptens for comprehensive understandings of the roles of ASK1 in CHS.
In addition to the established roles of the ASK1-p38 MAPK pathway in innate immune cells, this study proposes a role for ASK1 in acquired immunity through IL-17 production in lymphocytes. From the viewpoint of IL-17-related diseases, it is intriguing that p38 has been shown to promote IL-17 synthesis in CD4+ T cells and to be required for the development and progression of experimental allergic encephalomyelitis (EAE), the principal autoimmune model of multiple sclerosis28. In the same model, ASK1 KO mice have also been reported to exhibit attenuated inflammatory symptoms29, suggesting that the ASK1-p38 MAPK pathway is a key player in various IL-17-related inflammatory diseases. In this study, the ASKA technology constituted a chemical genetic tool that contributed not only to elucidate the phase-specific relevance of ASK1 but also to demonstrate the inhibitory effect of the ASK1 inhibitor K811 on CHS. The combined usage of genetic and chemical tools will be a valuable strategy for the development of ASK1 inhibitors as therapeutic agents against various IL-17-related inflammatory diseases.
Methods
Cell culture, antibodies and reagents
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 4.5 mg/ml glucose and 100 units/ml penicillin G. A crude population of BMDMs was generated in vitro from mouse bone marrow as described30. PE-conjugated antibodies specific for MHC class II (M5/114.15.2), B220 (RA3-6B2) and IL-17 (TC11-18H10) were purchased from BD PharMingen. Phycoerythrin (PE)-Cy5-, PE- and FITC-conjugated antibodies against CD4 (GK1.5) and IFN-γ (XMG1.2) were purchased from eBioscience. Phospho-ASK1 antibody that recognizes the activating phosphorylation of ASK1 was described previously19. DNFB and DNBS were purchased from Sigma-Aldrich. FITC was purchased from Thermo Scientific. 4-Amino-1-tert-butyl-3-(1′-naphthyl)pyrazolo[3,4-d]pyrimidine (1Na-PP1) and 4-amino-1-tert-butyl-3-(1′-naphthylmethyl)pyrazolo[3,4-d]pyrimidine (1NM-PP1) were purchased from Calbiochem. The ASK1 inhibitor K811 was provided by Kyowa Hakko Kirin Co., Ltd20.
Generation of ASK1ASKA
We employed the analog-sensitive kinase allele (ASKA) approach18 to establish a system by which ASK1 activity can be manipulated in vitro and in vivo without affecting the ASK1 protein expression. This aspect is important for the precise investigation of ASK1 because deficiencies of ASK1 proteins have been shown to lead to the degradation of another MAP3K molecule, ASK2, which is stabilized only by forming a heteromeric complex with ASK131. In this approach, a subtle but unique structural distinction between the catalytic domain of one kinase and all other kinases is created by introducing some mutations in the ATP-binding pocket of the kinase, allowing inhibition by ATP analog compounds without affecting the intrinsic kinase activity using native ATP. Based on a variety of successfully established ASKAs reported thus far32, two key residues that should be modified in WT kinases to allow the recognition of ATP analogs modified at the N6 position have been proposed; in the case of murine ASK1, M761 and S828 corresponded to these key residues and were named gatekeeper (GK) and DFG(–1), respectively. In addition, V745, named GK(–16), might be in close contact with the N6 amino group of bound ATP analogous with CaMKII reported previously33. We therefore generated cDNA that encodes ASK1 carrying triple mutations in V745L, M761A and S828A and designated this as ASK1ASKA. The kinase activity and 1Na-PP1 sensitivity of ASK1ASKA was evaluated in comparison with ASK1 carrying double mutations (M761A and S828A) as follows: WT ASK1, ASK1ASKA (V745L/M761A/S828A), or ASK1-M761A/S828A with a C-terminal V5 epitope tag were expressed in HEK293 cells and proteins immunoprecipitated with a V5 antibody were subjected to the in vitro kinase assay using recombinant kinase-inactive MKK4 (Merck Millipore) as a substrate as described34.
Mice
A phage genomic DNA library from C57BL/6 mice was screened with a cDNA probe containing exons 15 to 20 of the MAP3K5 gene. From a resulting genomic clone containing a 16.5-kb sequence, a 2.7-kb fragment upstream of exon 16 containing the GK and GK(–16) mutations and 5.7-kb fragments downstream of exon 18 containing the DFG(–1) mutation were taken as the 5′ and 3′ homology arms for the targeting vector, in which a neomycin resistance gene cassette flanked by loxP sites was inserted between exons 17 and 18. The linearized targeting vector was electroporated into C57BL/6N-derived CMT2 ES cells. Five out of 3,800 G418-resistant clones were identified by Southern blot analysis with flanking 5′ and 3′ probes and a neomycin cassette probe followed by confirmation of the three mutations by sequencing the cDNA from each clone. Transient transfection with a Cre recombinase expression vector was used to remove the selection cassette. One of the resulting clones was injected into C57BL/6N blastocysts and passed through the germline of chimeras to generate the ASK1ASKA mouse line by Merck Serono S.A. Homozygous mutant mice were obtained by F1 heterozygous intercrosses. ASK1 KO mice, in which the ASK1-encoding MAP3K15 gene was homozygously deleted, have been described30,35. The mice were housed in a specific pathogen-free facility. Female mice at 8–10 weeks of age were used for all experiments, which were performed in accordance with protocols approved by the Animal Research Committee of the Graduate School of Pharmaceutical Sciences at the University of Tokyo (Tokyo, Japan).
CHS model
The abdomens of mice were epicutaneously sensitized twice with 25 µl of 0.5% DNFB at a 24-hr interval (days 0 and 1) and the ears were challenged with 20 µl of 0.3% DNFB on day 5. Twenty-four hours later, the ear swelling was measured using a thickness gauge (Mitutoyo) and determined as the difference between the ear thickness of the DNFB-treated side and the vehicle-treated side of the same mouse. For FITC-induced CHS, 400 µl of 0.2% FITC in 1:1 acetone/dibutylphthalate (Ac/DBP) was applied to the shaved abdominal skin and the mice were challenged with 20 µl 0.2% FITC/Ac/DBP on day 6. For the adoptive transfers, 5 × 106 LN cells isolated from the LNs of WT or ASK1 KO mice sensitized with DNFB for 5 days were injected intravenously into naive WT or ASK1 KO mice. The ear was immediately challenged with DNFB and its thickness was measured 48 hr later. For ASK1ASKA mice, 1Na-PP1 (476.1 ng per g) or the vehicle was injected intravenously. K811 was administered orally at a dose of 100 mg/kg per day as described20.
Histology and immunohistochemistry
For morphological analysis of the DNFB-induced CHS response, the mice were killed at day 6 and sections (6 μm) of fresh–frozen ear were stained with hematoxylin and eosin. The FITC-based migration assay was performed as described36. Briefly, 24 hr after the application of 25 μl of 1% FITC in acetone/dibutyl phthalate to the pre-shaved arm or flank, the brachial and inguinal LNs were analyzed morphologically and the isolated LN cells were stained with an antibody against MHC class II before flow cytometric analysis using a FACSCaliber (BD Biosciences). For observation of LCs, the ears were excised and placed in dishes containing 250 units/ml of dispase (Godoshusei) in PBS overnight at 4°C. The epidermal sheets were peeled from the underlying dermis and stained with an MHC class II antibody. The numbers of MHC class II-positive cells were counted in random fields for each sheet and used to calculate the LC density.
Proliferation and cytokine assays
Single-cell suspensions (1 × 106 cells/ml RPMI 1640 medium containing 10% FBS) prepared from the inguinal and axillary LNs of mice sensitized with DNFB for 5 days were cultured in the presence or absence of 100 µg/ml DNBS for 3 days. The relative cell numbers were determined as the relative fluorescence intensity using a Cell Counting Kit-F (Dojindo). The amounts of IFN-γ and IL-17 in the culture supernatants were measured using Quantikine ELISAs according to the manufacturer's instructions (R&D Systems). For the reconstitution assay, CD4+ or CD8+ T cells (1 × 106/well) and CD11c+ DCs (1 × 105/well) purified from DNFB-sensitized mice using the MACS system (Miltenyi Biotec; purity: >95% and 70%, respectively) were co-cultured in the presence of 100 µg/ml DNBS for 24 hr and the amounts of IFN-γ or IL-17 in the culture supernatants were measured. For the measurement of the proportion of IFN-γ- or IL-17-producing cells among all LN cells, the cells were stained with FITC-conjugated IFN-γ and PE-conjugated IL-17 antibodies using a Mouse Intracellular Cytokine Staining Starter Kit according to the manufacturer's instructions (BD Biosciences) and were analyzed by flow cytometry.
Quantification of mRNA expression
Total RNA was isolated from CD4+ T cells purified from draining LN cells using ISOGEN (Nippon Gene) and reverse-transcribed with the QuantiTect Reverse Transcription Kit (Qiagen). Quantitative PCR was performed with SYBR Green PCR Master Mix using the ABI PRISM 7000 Sequence Detection System (Applied Biosystems). To normalize the relative expression of T-bet, GATA3, RORγt and Foxp3 to the GAPDH control, standard curves were prepared for GAPDH and for each gene in each experiment.
Immunoblot analysis
The cells were lysed with a buffer containing 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 10 mM EDTA, 1 mM phenylmethylsulfonyl fluoride and 5 μg/ml aprotinin. The lysates were resolved by SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes. After blocking with 5% skim milk in TBS-T (50 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.05% Tween 20), the membranes were probed with antibodies. The antibody/antigen complexes were detected using the ECL system (GE Healthcare).
Statistical analyses
Student's t-test (unpaired, two-tailed) was used to compare two groups of independent samples.
References
Grabbe, S. & Schwarz, T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Immunol Today 19, 37–44 (1998).
Peiser, M. et al. Allergic contact dermatitis: epidemiology, molecular mechanisms, in vitro methods and regulatory aspects. Current knowledge assembled at an international workshop at BfR, Germany. Cell Mol Life Sci 69, 763–781 (2012).
Bursch, L. S. et al. Identification of a novel population of Langerin+ dendritic cells. J Exp Med 204, 3147–3156 (2007).
Ginhoux, F. et al. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med 204, 3133–3146 (2007).
Honda, T. et al. Compensatory role of Langerhans cells and langerin-positive dermal dendritic cells in the sensitization phase of murine contact hypersensitivity. J Allergy Clin Immunol 125, 1154–1156 e1152 (2010).
Kissenpfennig, A. et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22, 643–654 (2005).
Noordegraaf, M., Flacher, V., Stoitzner, P. & Clausen, B. E. Functional redundancy of Langerhans cells and Langerin+ dermal dendritic cells in contact hypersensitivity. J Invest Dermatol 130, 2752–2759 (2010).
Nakae, S. et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17, 375–387 (2002).
He, D. et al. IL-17 and IFN-gamma mediate the elicitation of contact hypersensitivity responses by different mechanisms and both are required for optimal responses. J Immunol 183, 1463–1470 (2009).
Iwakura, Y., Nakae, S., Saijo, S. & Ishigame, H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev 226, 57–79 (2008).
Korn, T., Bettelli, E., Oukka, M. & Kuchroo, V. K. IL-17 and Th17 Cells. Annu Rev Immunol 27, 485–517 (2009).
Arrighi, J. F., Rebsamen, M., Rousset, F., Kindler, V. & Hauser, C. A critical role for p38 mitogen-activated protein kinase in the maturation of human blood-derived dendritic cells induced by lipopolysaccharide, TNF-alpha and contact sensitizers. J Immunol 166, 3837–3845 (2001).
Takanami-Ohnishi, Y. et al. Essential role of p38 mitogen-activated protein kinase in contact hypersensitivity. J Biol Chem 277, 37896–37903 (2002).
Aiba, S. et al. p38 Mitogen-activated protein kinase and extracellular signal-regulated kinases play distinct roles in the activation of dendritic cells by two representative haptens, NiCl2 and 2,4-dinitrochlorobenzene. J Invest Dermatol 120, 390–399 (2003).
Takeda, K., Noguchi, T., Naguro, I. & Ichijo, H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu Rev Pharmacol Toxicol 48, 199–225 (2008).
Matsuzawa, A. et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol 6, 587–592 (2005).
Ichijo, H. et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90–94 (1997).
Shokat, K. & Velleca, M. Novel chemical genetic approaches to the discovery of signal transduction inhibitors. Drug Discov Today 7, 872–879 (2002).
Tobiume, K., Saitoh, M. & Ichijo, H. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J Cell Physiol 191, 95–104 (2002).
Hayakawa, Y. et al. Apoptosis signal-regulating kinase-1 inhibitor as a potent therapeutic drug for the treatment of gastric cancer. Cancer Sci 103, 2181–2185 (2012).
He, D. et al. CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol 177, 6852–6858 (2006).
Christensen, A. D. & Haase, C. Immunological mechanisms of contact hypersensitivity in mice. APMIS 120, 1–27 (2012).
Berg, D. J. et al. Interleukin 10 but not interleukin 4 is a natural suppressant of cutaneous inflammatory responses. J Exp Med 182, 99–108 (1995).
Dieli, F., Sireci, G., Scire, E., Salerno, A. & Bellavia, A. Impaired contact hypersensitivity to trinitrochlorobenzene in interleukin-4-deficient mice. Immunology 98, 71–79 (1999).
Traidl, C., Jugert, F., Krieg, T., Merk, H. & Hunzelmann, N. Inhibition of allergic contact dermatitis to DNCB but not to oxazolone in interleukin-4-deficient mice. J Invest Dermatol 112, 476–482 (1999).
Weigmann, B. et al. Diminished contact hypersensitivity response in IL-4 deficient mice at a late phase of the elicitation reaction. Scand J Immunol 45, 308–314 (1997).
Oboki, K., Ohno, T., Saito, H. & Nakae, S. Th17 and allergy. Allergol Int 57, 121–134 (2008).
Noubade, R. et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood 118, 3290–3300 (2011).
Guo, X. et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol Med 2, 504–515 (2010).
Iriyama, T. et al. ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. EMBO J 28, 843–853 (2009).
Takeda, K. et al. Apoptosis signal-regulating kinase (ASK) 2 functions as a mitogen-activated protein kinase kinase kinase in a heteromeric complex with ASK1. J Biol Chem 282, 7522–7531 (2007).
Zhang, C. et al. A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat Methods 2, 435–441 (2005).
Wang, H. et al. Inducible protein knockout reveals temporal requirement of CaMKII reactivation for memory consolidation in the brain. Proc Natl Acad Sci U S A 100, 4287–4292 (2003).
Saitoh, M. et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17, 2596–2606 (1998).
Tobiume, K. et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2, 222–228 (2001).
Tang, H. L. & Cyster, J. G. Chemokine Up-regulation and activated T cell attraction by maturing dendritic cells. Science 284, 819–822 (1999).
Acknowledgements
The ASK1ASKA mouse line was generated and provided by Merck Serono S.A. The authors thank Kyowa Hakko Kirin Co., Ltd for providing K811 and all of the members of Cell Signaling Laboratory for critical discussions. This work was supported by KAKENHI from JSPS and MEXT, Global Center of Education and Research for Chemical Biology of the Diseases, the Uehara Memorial Foundation, the Cosmetology Research Foundation, the Tokyo Biochemical Research Foundation and the Mitsubishi Foundation.
Author information
Authors and Affiliations
Contributions
J.M. designed and performed the experiments and wrote the manuscript. T.S. performed experiments. M.C., H.J., T.R. and D.S. generated the ASK1ASKA mouse line. R.T. designed the studies. K.T. designed the studies and wrote the manuscript. H.I. designed the studies, supervised the work and collaborations and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Mizukami, J., Sato, T., Camps, M. et al. ASK1 promotes the contact hypersensitivity response through IL-17 production. Sci Rep 4, 4714 (2014). https://doi.org/10.1038/srep04714
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04714
This article is cited by
-
ASKA technology-based pull-down method reveals a suppressive effect of ASK1 on the inflammatory NOD-RIPK2 pathway in brown adipocytes
Scientific Reports (2021)
-
ASK1 promotes uterine inflammation leading to pathological preterm birth
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
