
Abstract
Expression of a mesenchymal phenotype is often associated with invasive/metastatic behaviors of carcinoma cells. Acquisition of a mesenchymal phenotype by a carcinoma cell is known as epithelial-mesenchymal transition (EMT). The membrane-anchored matrix metalloproteinase-regulator RECK is abundant in normal mesenchymal cells. In aggressive carcinomas, however, RECK expression is often downregulated. This apparent paradox prompted us to clarify the relationship between EMT and RECK. We found that TGFβ-induced E-cadherin downregulation, a hallmark of EMT, is accompanied by RECK-upregulation in a non-tumorigenic epithelial cell line (MCF10A). In contrast, the loss of E-cadherin expression is uncoupled from RECK-upregulation in carcinoma-derived cell lines (MCF7, MDA-MB-231 and A549). When RECK was artificially expressed in A549 cells, it showed little effect on EMT but elevated the level of integrin α5 and attenuated cell proliferation and migration. These findings implicate RECK in the regulation of proliferation and migration of normal epithelial cells after EMT and suggest how the uncoupling between EMT and RECK-upregulation impacts on the fates and behaviors of carcinoma cells.
Similar content being viewed by others


Introduction
Cells are under the strong influence of a wide variety of microenvironmental cues including cytokines, extracellular matrix (ECM) and changes in oxygen concentration. One prominent change that epithelial cells exhibit in response to certain microenvironmental cues is a process called epithelial-mesenchymal transition (EMT)1. Through EMT, epithelial cells lose their robust cell-cell contacts and apico-basal polarity, exhibit spindle-shaped morphology and acquire motile or invasive properties. At the molecular level, EMT is characterized by down-regulation of epithelial markers such as E-cadherin (also known as CDH1) and induction of mesenchymal markers such as N-cadherin (CDH2), vimentin and fibronectin (FN): in this paper, we used the loss of E-cadherin as a hallmark of EMT. Although EMT is known to play crucial roles in animal development and tissue repair, aberrant EMT contributes to the invasive and metastatic dissemination of malignant cells during carcinoma progression2,3. Transforming growth factor-beta (TGFβ) is a potent inducer of EMT capable of upregulating certain ECM components [for instance, FN and type I collagen (COL)] as well as ECM-degrading enzymes [for instance, matrix metalloproteinases (MMPs)] in epithelial cells4,5.
RECK was first identified as a cDNA inducing flat reversion in v-K-ras-transformed NIH3T3 cells6. The RECK gene encodes a membrane-anchored glycoprotein capable of regulating several members of the MMP family (MMP2, MMP7, MMP9 and MT1-MMP)6,7,8,9 and some other extracellular metalloproteinases such as CD13/aminopeptidase N and ADAM108,10. RECK is expressed in multiple normal tissues, including developing vasculature, skeletal muscles, neuromuscular junctions, cartilage, fibroblasts and neural precursor cells7,10,11,12,13. In cultured mouse embryo fibroblasts, RECK was found to be required for proper cell-substrate adhesion and persistent directional migration14.
RECK expression is often downregulated in tumors of both mesenchymal and epithelial origin and the level of residual RECK expression in cancer tissues correlates with better prognoses15. Moreover, forced RECK-expression in tumor cells results in reduced angiogenesis, invasion and metastasis in xenograft models6,7, suggesting that reduction in the level of RECK may confer significant advantage to the cells during malignant growth and/or dissemination.
A large proportion of malignant neoplasms are of epithelial origin (namely, carcinomas). Although RECK is often downregulated in carcinomas, the functions of RECK in epithelial cells remain largely unknown. It is also unclear how to reconcile the fact that RECK is abundant in mesenchymal cells and with the fact that malignant progression often involves the expression of a mesenchymal phenotype (i.e., EMT). We therefore asked in this study whether induction of EMT affects RECK expression or vise versa. Our data indicate that RECK is induced after EMT induction in non-malignant epithelial cells but not in carcinoma cells and that RECK has little influence on EMT itself but does have a significant impact on cell behavior after EMT.
Results
Upregulation of RECK in a non-malignant epithelial cell line, MCF10A, after EMT
To understand the relationship between EMT and RECK, we first asked whether EMT had any effect on RECK expression in a non-malignant epithelial cell line, MCF10A (Fig. 1). This cell line shows typical epithelial (cobblestone-like) morphology when cultured in the medium termed MEBM, consisting of Mammary Epithelial Basal Medium with supplements (Fig. 1a, panel 1). In contrast, the cells showed some changes in morphology (spread-out and/or spindle-shaped) when incubated for 7 days in: (1) MEBM containing 5 ng/ml TGFβ, (2) Dulbecco's modified Eagle (DME) medium supplemented with 10% fetal bovine serum (GM), or (3) GM plus TGFβ (Fig. 1a, panels 2–4). Under these conditions, the mesenchymal marker N-cadherin was upregulated (Fig. 1b; compare lanes 4, 7 and 10 to lane 1). After longer incubation periods in MEBM containing TFGβ (20 to 40 days), however, N-cadherin returned to basal levels and E-cadherin downregulation was not apparent (Fig. 1b; lanes 5, 6). Hence, according to this criterion, the cells cultured in MEBM plus TGFβ did not undergo an EMT-like change. In GM, on the other hand, robust N-cadherin upregulation was found as early as 7 days incubation, but E-cadherin downregulation became apparent only after 40 days incubation in GM (Fig. 1b, lane 9). After 20 and 40 days incubation in GM plus TGFβ, the cells exhibited further induction of N-cadherin expression and a dramatic downregulation of E-cadherin and these changes were accompanied by RECK upregulation (Fig. 1b; compare lanes 11, 12 to lane 10) and prominent morphological changes (Fig. 1a, panel 6). Gradual unpregulation of RECK was found in cells incubated in GM alone (Fig. 1b, lane 9). Immunofluorescent staining (Fig. 1c) indicated clear switching from an E-cadherin-positive/N-cadherin-negative state to an E-cadherin-negative/N-cadherin-positive state after prolonged incubation of MCF10A cells in GM (Fig. 1c; compare right column to left column) and co-expression of both markers in cells after 7-day incubation in GM (Fig. 1c, central column). Henceforth, we call MCF10A cells maintained in GM for more than 30 days “m10A” in this report.

Induction of RECK expression after EMT in a nonmalignant epithelial cell line.
(a) Morphology of a normal epithelial cell line maintained in various media. MCF10A cells were cultured in MEBM (panels 1, 2) or GM (panels 3–6) without (left panels) or with 5 ng/ml TGFβ (right panels) for 7 days (panels 1–4) or 1 month (panel 5, 6). Scale bars: 100 μm. (b) Immunoblot detection of RECK and EMT markers in MCF10A cells maintained in different media. MCF10A cells were incubated for 7, 20, or 40 days in either MEBM or DME containing the indicated supplement(s). Media were renewed every 4–5 days. Cell lysates were subjected to immunoblot assay using antibodies against RECK, E-cadherin (E-cad), N-cadherin (N-cad), or α-tubulin (α-tub; loading control). The blots have been cropped focusing on the bands or interest; See Supplementary Fig. S8 for full-length blots. (c) Immunofluorescent double staining for E-cadherin (red, panels 4–6, 10–12) and N-cadherin (green, panels 7–12) in MCF10A cells either maintained in MEBM (panels 4, 7, 10) or exposed to GM for 7 days (panels 5, 8, 11) or maintained in GM for more than 40 days (panels 6, 9, 12). Panels 10–12 are merged images with nuclei counter-stained with DAPI (blue). Phase-contrast images of respective cultures are also shown (panels 1–3). Scale bars: 50 μm.
The slow upregulation of RECK in cells after exposure to GM implicates changes in cellular properties (such as those resulting from EMT), rather than extracellular stimuli itself (such as growth factor signaling), in this event. Another possibility that a subpopulation of cells has been selected during prolonged incubation seems unlikely, since when m10A cells were incubated in MEBM without TGFβ, phenotypic reversion occurred within a week as demonstrated by conversion to epithelial morphology (Fig. 2a), E-cadherin-upregulation (Fig. 2b; Fig. 2c, lanes 1, 3) and mild N-cadherin/vimentin-downregulation accompanied by RECK-downregulation (Fig. 2c, lanes 1, 3).

Relationships among E-cadherin, β-catenin and RECK in MCF10A cells.
(a) Morphology of MCF10A cells maintained in GM for one month (m10A) and then cultured in either GM (panel 1) or MEBM (panel 2) for one week. (b) Immunofluorescent double staining for E-cadherin (red) and N-cadherin (green) in m10A cells cultured for one week in the indicated media. Cell nuclei were visualized with DAPI (blue). Scale bars: (a) 100 μm, (b) 50 μm. (c) Immunoblot detection of RECK and EMT markers in m10A cells cultured as shown in (a). An additional EMT marker, vimentin (Vim), was included. Note the reappearance of epithelial phenotype in m10A cells after exposure to MEBM for one week (lane 1). (d) Relative intensity of the bands detected in (c). (e, f) Effects of E-cadherin siRNAs on the level of RECK mRNA (qRT-PCR, n = 3) (e) or RECK and E-cadherin proteins (immunoblot assay) (f) in MCF10A cells. MCF10A cells were transfected with a control (C1 or C2) or an E-cadherin (E1 or E2) siRNA and incubated for 2 days. (g, h) Effects of β-catenin inhibitors, FH535 and iCRT3, on RECK expression in m10A (g) or NIH3T3 (h) cells. Cells were treated for 48 h with indicated inhibitor at indicated concentration. (i) Effects of β-catenin inhibitors on Reck promoter activity in NIH3T3 cells. The cells co-transfected with the pRL-TK plasmid and the vacant pGL3-Basic vector or the vector containing a 4.1 kb Reck promoter fragment (pGL3-4110)19 were exposed, on the following day, to medium containing 50 μM inhibitor for 24 h and luciferase activities determined (n = 2). The data were normalized against the Renilla luciferase, vacant pGL3-Basic and vehicle (DMSO) treatment. (j) Effects of E-cadherin siRNAs and a β-catenin inhibitors, FH535, on RECK expression in MCF10A cells. MCF10A cells transfected with a control (C2) or an E-cadherin (E1) siRNA, incubated for 2 days and then exposed to medium without or with 50 μM FH535 for additional 24h. Numbers in blue in (f), (g), (h), (j) indicate relative band intensities as determined by densitometry. See Supplementary Fig. S9 for full-length blots. ** P < 0.01 as compared to control.
Possible involvement of E-cadherin-downregulation on induction of RECK expression in MCF10A cells
During the above study, we noted a clear inverse-correlation between the levels of E-cadherin and RECK (Fig. 1b, lanes 9, 11, 12; Fig. 2c, 2d). Previous studies indicate that direct E-cadherin-knockdown in epithelial cells may lead to the expression of some features of EMT16. We therefore employed this approach to examine the causal-effect relationship between RECK-upregulation and E-cadherin-downregulation. Treatment of MCF10A cells with two different E-cadherin siRNAs resulted in RECK-upregulation both at the mRNA (Fig. 2e, bars 4, 5) and protein levels (Fig. 2f, lanes 4, 5), while no such upregulation was found after treatment with 2 different control siRNAs (Fig. 2e, bars 2, 3; Fig. 2f, lanes 2, 3). Hence, E-cadherin-downregulation seems to be prerequisite for the RECK-upregulation after the EMT in this system.
β-catenin has been implicated as an essential mediator in the EMT induced by E-cadherin-knockdown16. We therefore tested whether β-catenin is involved in RECK upregulation in post-EMT epithelial cells (m10A) by using two small molecule inhibitors of Wnt/β-catenin signaling, FH53517 and iCRT318. These inhibitors have distinct mechanisms of actions: FH535 inhibits recruitment of β-catein and GRIP2 to PPARγ, while iCRT3 binds β-catenin and inhibits its interaction with TCF4. Both these inhibitors (at 75 μM) reduced the levels of RECK partially in m10A cells (Fig. 2g) and more clearly in NIH3T3 cells (Fig. 2h). They also suppressed Reck-promoter activity19 as assessed by luciferase-reporter assay in NIH3T3 cells (Fig. 2i). The inhibitor also suppressed the RECK-upregulation induced by E-cadherin-knockdown in MCF10A cells (Fig. 2j, lane 4). These findings suggest the possible involvement of β-catenin-mediated transactivation in RECK-upregulation after EMT (marked by E-cadherin-downregulation) in this system.
Uncoupling between E-cadherin-downregulation and RECK-upregulation in carcinoma-derived cell lines
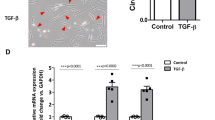
Next, we examined the effects of EMT on RECK expression in three cell lines of carcinoma origin: two cell lines derived from mammary adenocarcinomas (MCF7 and MDA-MB-231) and one from a lung adenocarcinoma (A549). MCF7 and A549 cells maintained in GM retained an epithelial phenotype: namely, cobble stone-like morphology (Fig. 3a, panels 1, 5) and E-cadherin expression (Fig. 3b, lanes 2, 6). In MCF7 cells, TGFβ showed some cytotoxicity (Fig. 3a, panel 2) but it downregulated E-cadherin (Fig. 3b, E-Cad, lane 3; Fig. 3c, panel 2) and upregulated fibronectin (Fig. 3b, FN, lane 3) to some extent. In A549 cells, TGFβ induced robust morphological changes (Fig. 3a, panel 6) and E-cadherin-downregulation (Fig. 3b, E-cad, lane 7; Fig. 3c, panel 10) accompanied by upregulation of two mesenchymal markers, fibronectin and N-cadherin (Fig. 3b, FN and N-cad, lane 7; Fig. 3c, panel 12). In these cell lines, however, E-cadherin-downregulation was not accompanied by RECK-upregulation (Fig. 3b, RECK, lanes 3, 7). Direct knockdown of E-cadherin was also ineffective in upregulating RECK in these cell lines (Fig. 3d, lanes 3, 6).

Uncoupling between E-cadherin downregulation and RECK-upregulation in carcinoma-derived cell lines.
(a, b) Effect of TGFβ on the morphology (a) of and the expression of RECK and EMT-markers (b) in, the carcinoma-derived cell lines, MCF-7, MDA-MB-231 (231) and A549, incubated for one week in growth medium without or with TGFβ. m10A was included as a control (lane 1). (c) Immunofluorescent double staining for E-cadherin (E-cad; red) and N-cadherin (N-cad; green) in MCF7, MDA-MB-231, or A549 cells cultured for one week in growth medium without or with TGFβ. Scale bars: (a) 100 μm, (b) 50 μm. (d) Effect of E-cadherin knockdown on RECK expression in MCF7 and A549 cells. Cells were transfected with either a control (C1) or an E-cadherin (E1) siRNA and incubated for 2 days. (e) The level of RECK mRNA (qRT-PCR, n = 3). MCF10A cells were incubated in MEBM (−), MEBM plus TGFβ (T), GM(F), or GM plus TGFβ(TF), for 7, 20, or 40 days (bars 1–12). Three carcinoma-derived cell lines were cultured in GM without (−) or with TGFβ (T) for 1 week (bar 13–18). MRC5 (normal human fibroblasts) RNA was used as a positive control (bar 19). * P < 0.05, ** P < 0.01 as compared to the medium control (−). (f) Effects of ΜΕΚ inhibitor and PI3K inhibitor on the levels of RECK mRNA (qRT-PCR, n = 3) in MCF7 and MDA-MB-231 cells. The cells plated 18 h earlier were exposed to medium containing PD98059 (PD) or LY294002 (LY) at the indicated concentration for 30 h. * P < 0.05, ** P < 0.01 as compared to the vehicle control (V). (g) Immunoblot assay for RECK protein in MDA-MB-231 cells treated with PD or LY (at the indicated concentration) or both (at 10 μM) for 30 h. (h) Effects of flavone on the levels of RECK mRNA (qRT-PCR, n = 3) in A549 cells. The cells were cultured in medium containing 50 μM flavone (FL) and/or TGFβ for 48 h. * P < 0.05 as compared to the culture without flavone. (i) Immunoblot assay for RECK and α-tubulin proteins in similarly treated cells. See Supplementary Fig. S10 for full-length blots.
MDA-MB-231 cells maintained in GM showed a mesenchymal morphology and low E-cadherin expression (Fig. 3a, panel 3; Fig. 3b, E-cad, lane 4; Fig. 3c, panel 5). RECK-expression was undetectable and was not induced by TGFβ in these cells (Fig. 3b, RECK, lane 5). Thus, an inverse correlation between the levels of E-cadherin and RECK was not found in any of the carcinoma-derived cell lines tested.
At the mRNA level, RECK expression in non-malignant MCF10A cells was appreciably upregulated within a week when cultured in GM plus TGFβ (Fig. 3e, bar 4). Consistent with our observations at the protein level (Fig. 1b), RECK mRNA was progressively upregulated after prolonged culture in this medium (Fig. 3e, bars 8, 12). In MCF7 cells, on the other hand, RECK mRNA was barely detectable when cultured in GM plus TGFβ for 1 week (Fig. 3e, bar 14). In MDA-MB-231 and A549 cells, RECK mRNA was detectable (Fig. 3e, bars 15, 17), despite the difficulty in detecting RECK protein (Fig. 3b, RECK, lanes 4, 6). Nevertheless, TGFβ failed to upregulate, or even downregulated, RECK mRNA in these cell lines (Fig. 3e, bars 16, 18). Thus, the levels of RECK mRNA do not inversely correlate with the levels of E-cadherin in these carcinoma-derived cell lines.
A wide variety of oncogenes, including activated RAS, have been found to downregulate Reck in fibroblasts6,19. The KRAS gene is mutated in the MDA-MB-231 and A549 cell lines20,21. We therefore asked whether deregulated RAS-signaling plays any role in the E-cadherin-RECK uncoupling, described above, using inhibitors of two downstream signaling molecules, MEK (PD98059) and PI3-kinase (LY294002). Both inhibitors elevated the levels of RECK mRNA in MDA-MB-231 cells but not so much in MCF7 cells (Fig. 3f). In MDA-MB-231, upregulation of RECK could be detected at the protein level as well (Fig. 3g), supporting the involvement of RAS-signaling in RECK-repression in this cell line. In A549, although the effect of PD98059 on RECK expression was mild and not cooperative with TGFβ (data not shown), a broad-spectrum kinase inhibitor, flavone, was found to cooperate with TGFβ in upregulating RECK at the mRNA and protein levels (Fig. 3h, i). These results demonstrate multiple modes of RECK-downregulation among different tumors.
Effects of forced RECK expression on the phenotype and behavior of carcinoma cells
If E-cadherin-downregulation is not accompanied by RECK-upregulation in carcinoma cell, then what happens if RECK alone is artificially expressed in such cells? To address this issue, we chose to use adenovirus-mediated gene transfer (Ad-V), which allows us to assess the effects of acute RECK expression in cultured cells22. We also focused on A549 cells, since they show a robust EMT-like phenotypic conversion after TGFβ-treatment (see Fig. 3). Efficient expression of RECK protein could be achieved after infection of A549 cells with the RECK-expressing virus (Ad-RECK or labeled “R” in short) (Fig. 4a, RECK, lanes 3, 6) as compared to the cells infected with the control virus (Ad-Z or “Z” in short) (Fig. 4a, RECK, lanes 2, 5). The RECK expression, however, had little effect on the levels of EMT markers either in the absence or presence of TGFβ (Fig. 4a, lanes 3, 6). Similar results were obtained in the mammary epithelial cell line MCF10A and the two mammary carcinoma-derived cell lines MCF7 and MDA-MB-231 (Supplementary Fig. S1). Thus, RECK seems to have little effect on the induction of EMT itself.

Effects of RECK on proliferation and migration of A549 cells.
(a–c) Effects of RECK, TGFβ, or both on the expression of EMT markers (a), morphology (b) and proliferation (c). A549 cells either uninfected (N), infected with Ad-Z (Z), or infected with Ad-RECK (R) were incubated for 24 h in growth medium (GM), followed by incubation for 48 h (a, b) or 72 h (c) in GM (−) or GM containing TGFβ. Relative cell number was estimated by SF assay (c). Bar: mean ± s.e.m. (n = 3). (d) Effects of RECK and TGFβ on random migration. A549 cells infected with Ad-V and incubated for 48–96 h were replated onto glass-bottom dishes coated with FN or COL. After 4-h incubation, the cells were subjected to time-lapse recording for 6 h (see Supplementary Movies 1 and 2 for typical examples) to estimate the speed and directional persistence of migration. Bar: mean ± s.e.m (n ≥ 25 cells). (e) Effects of RECK, TGFβ and FN substrate on the levels of FN, FN-receptor (ITGα5, ITGβ1), phospho-FAK (pFAK) and total FAK in cell lysates. A549 cells infected with Ad-V and incubated in medium without (−) or with (+) TGFβ for 1 day were replated on either regular dishes (lanes 1–4) or FN-coated dishes (lanes 5–8) and incubated for an additional 24 h in medium without or with TGFβ. The experiments were repeated three times with similar results. See Supplementary Fig. S3 for densitometric quantification. (f, g) Collagen gel culture. A549 cells infected with Ad-V were embedded in a collagen gel without or with TGFβ and incubated for 4 days (f). Proportion of the cells with motile morphology (arrows; see Supplementary Movie 3) in TGFβ-containing cultures (panels 3, 4) were determined ((g); n ≥ 570). (h, i) Matrigel invasion assay. Cells infected with Ad-V and incubated without or with TGFβ for 2 days were subjected to Matrigel invasion assay (n = 4). The cells migrated across the Matrigel layer (h) were counted (i). Scale bars in (b), (f) and (h): 100 μm. In bar graphs: *P < 0.05, **P < 0.01. See Supplementary Fig. S11 for full-length blots.
Of note, infection of A549 cells with Ad-RECK resulted in altered cell morphology (Fig. 4b and Supplementary Fig. S2a, panels 2, 4) and reduced proliferation (Fig. 4c, bar 2). While TGFβ alone reduced proliferation (Fig. 4c, bar 3) and increased spreading of A549 cells (Supplementary Fig. S2b, panel 3; Fig. S2c, bar 3), Ad-RECK apparently enhanced these effects (Fig. 4c, bar 4; Supplementary Fig. S2b, c).
Next, we tested whether RECK had any effect on the behavior of A549 cells before and after TGFβ-treatment. Our previous study14 indicated that in fibroblasts (i.e., mesenchymal cells), Reck reduces their migration speed and augments their directional persistence, two important parameters of cell migration defined by Dunn23. In the present study, A549 cells were first infected with Ad-Z (Z) or Ad-RECK (R), then incubated in the absence or presence of TGFβ for 1 to 3 days and subjected to migration assays on dishes coated with FN or COL (Fig. 4d). TGFβ stimulated the migration of these cells on FN (Fig. 4d, compare bar 3 to bar 1), while RECK slowed the migration (Fig. 4d, compare bars 2, 4 to bars 1, 3). RECK also slowed A549 migration on COL (Fig. 4d, compare bar 6 to bar 5). Importantly, a small but significant increase in the directional persistence of A549 cells' migration was observed after Ad-RECK infection only when the cells were plated on FN and treated with TGFβ (Fig. 4d, bars 9 and 10; Supplementary Movies 1 and 2).
Previous findings in mesenchymal cells (e.g., mouse embryo fibroblasts and HT1080 fibrosarcoma cells) suggest that FN is a critical molecule protected by RECK9,14. We therefore performed immunoblot assays focusing on some of the proteins involved in cell-FN interactions (Fig. 4e; see Supplementary Fig. S3 for densitometric data). On regular dishes, Ad-RECK and TGFβ showed similar effects to increase the levels of FN-receptor (especially its ligand-specific subunit, integrin α524) and the proportion of phospho-FAK (representing FAK-activation25) (Fig. 4e; Supplementary Fig. S3, panels c and g, bars 2 and 3). The integrin α5 upregulation by Ad-RECK probably reflects a post-transcriptional mechanism, since no increase in the level of mRNA was detected (Fig. S4, bar 2 in panel g). On the other hand, TGFβ increased the levels of integrin α5 mRNA (Fig. S4, bar 3 in panel g), suggesting that RECK and TGFβ achieve the same goal via different mechanisms. On FN-coated dishes, on the other hand, the effects of Ad-RECK and TGFβ to increase the levels of FN-receptor and phospho-FAK were attenuated (Fig. 4e; Supplementary Fig. S3, panels c and g, bars 6 and 7), probably reflecting some negative feedback mechanisms to adjust the extent of cell-FN interactions to a certain range. Of note, under these conditions, Ad-RECK and TGFβ cooperate to increase the levels of integrin α5 and phospho-FAK, giving rise to abundant FN in the cell lysate (Fig. 4e; Supplementary Fig. S3, panel b, bar 8). As described above, A549 cells exhibit increased directional persistence in migration when the cells were treated with both TGFβ and Ad-RECK on FN-coated dishes (Fig. 4d, bar 10; see above). These results, together with the observations with HT1080 cells treated with RGD peptide (see Discussion), suggest the importance of RECK's activity to sustain integrin α5 expression for proper migration of epithelial cells after EMT.
The effect of RECK on carcinoma migration could be demonstrated more dramatically when the cells were embedded in a collagen gel. A549 cells stayed round and immobile irrespective of RECK expression under these conditions (Fig. 4f, panels 1, 2). In the presence of TGFβ, however, a significant fraction (~25%) of the cells became elongated (Fig. 4f, panel 3), which represented actively migrating cells as evidenced by time-lapse recording (Supplementary Movie 3). Importantly, the proportion of such motile cells was significantly reduced after Ad-RECK-infection (Fig. 4f, panel 4; Fig. 4g, bar 2), documenting the strong suppressive effect of RECK on the motility of post-EMT carcinoma cells in a 3D-collagen matrix. We also employed matrigel invasion assay and found that TGFβ had an activity to stimulate A549 cells' invasion through matrigel layer while Ad-RECK diminished this activity (Fig. 4h, i).
Discussion
It is difficult to tell whether a given cell is epithelial or mesenchymal just by looking at their morphology; however, it is possible to compare the levels of a molecular marker between two cells. In this study we used E-cadherin (in addition to the “epithelial” morphology) as a marker of an epithelial nature and the loss of E-cadherin expression (in addition to the acquisition of “mesenchymal” morphology) as a marker for EMT. Based on these criteria, TGFβ is an effective inducer of EMT in the non-tumorigenic epithelial cell line MCF10A and some carcinoma-derived cell lines, such as MCF7 and A549. Another carcinoma-derived cell line, MDA-MB-231, is already mesenchymal according to those criteria. If we use other “mesenchymal” markers, such as N-cadherin, vimentin and fibronectin, the definition of EMT becomes less clear. For instance, N-cadherin is upregulated when MCF10A cells were simply cultured in DMEM containing fetal bovine serum (Fig. 1b, third panel, lanes 7–12) and its levels do not show consistent correlation with E-cadherin levels (Fig. 1b, second panel). Likewise, these mesenchymal markers behave independently from each other among different cell lines (Fig. 3b, panels FN, N-cad and Vim). We therefore concluded that E-cadherin is the more useful marker, at least for the present purpose. This conclusion is consistent with the previously proposed notion that the loss of E-cadherin expression is a hallmark of EMT both in embryonic development and in cancer progression1,2,3,26.
In the case of MCF10A cells, a slow decline of E-cadherin expression was accompanied by RECK-upregulation (Fig. 5a). Although the reason for this long latency period is presently unclear, the level of RECK mRNA seems to increase gradually (Fig. 3e). Our hypothesis that E-cadherin suppresses RECK transactivation mediated by β-catenin (Fig. 5c) was supported by the experiments using multiple E-cadherin siRNAs and small molecule β-catenin inhibitors (Fig. 2e–j). This hypothesis is also supported by two sets of transcriptome data16,27 in the database (GEO Profiles, NCBI) obtained with another non-malignant mammary epithelial cell line, HMLE. Indirect support comes from the study by Taube et al. (16): Cells expressing TGFβ1 or one of several EMT-inducing transcription factors (TWIST, GSC, or SNAI1), which tend to result in reduction of E-cadherin expression, exhibited significant upregulation of RECK mRNA (probe: 205407_at) (Supplementary Fig. S5a). More direct evidence comes from the study by Onder et al (28): Cells expressing E-cadherin shRNA or a dominant-negative E-cadherin mutant also exhibited significant upregulation of RECK mRNA (probe: 205407_at) (Supplementary Fig. S5b). In the latter case, the level of RECK mRNA was reduced when β-catenin shRNA was co-expressed with E-cadherin shRNA (Supplementary Fig. S5b, 3rd bar), providing further evidence that β-catenin may play a role in the RECK-upregulation caused by E-cadherin reduction. The exact molecular pathways, cis-elements and transactivators involved need to be elucidated in future studies. Lee et al. previously reported upregulation of RECK by TGFβ in pancreatic stellate cells28, a cell type with mesenchymal character29. In that system, however, the mechanism of RECK-induction was proposed to be post-transcriptional28 and hence likely to be distinct from the mechanism in the present system.

Summary and a model.
(a) In the non-malignant epithelial cell line MCF10A, TGFβ leads to E-cadherin-downregulation and concomitant RECK-upregulation after a long latency period. (b) In carcinoma-derived cell lines, E-cadheirn expression is either minimal or rapidly downregulated upon TGFβ-treatment, but RECK expression stays low. (c) A model consistent with our findings. In normal epithelial cells, TGFβ downregulates E-cadherin, leading to RECK-upregulation and subsequent suppression of peri-cellular proteolysis as well as TGFβ-responsive pathways and hence, to suppression of malignant behavior. In carcinoma cells, oncogenic signals suppress both E-cadherin and RECK expression, leading to deregulated gene expression and proteolysis [targeting substrates present in the vicinity of cell surface, such as ECM components, ECM receptors (ECM-R), growth factor receptors (GF-R)] and to consequent malignant behavior.
We found two common features among the carcinoma-derived cell lines that are not found in the non-tumorigenic MCF10A cell line. First, E-cadherin-expression is either absent (in MDA-MB-231) or present but rapidly downregulated after TGFβ-treatment (in MCF7 and A549) (Fig. 3b, c). Second, an inverse correlation between the levels of E-cadherin and RECK is not found (Fig. 3b, d; Fig. 5b). Thus, in the carcinoma-derived cell lines used in the present study, E-cadherin downregulation is not sufficient for RECK upregulation. Molecular mechanisms of RECK-silencing in cancer cells are not clear at the moment, but it is evident that multiple mechanisms are involved. Previous studies indicated that RECK expression is a common target of repression by various oncogenic signals (such as HER2, Ras, Src, PI3K and hypoxia) via multiple mechanisms (such as transcriptional repression, DNA methylation and microRNAs)6,19,30,31,32,33,34. The results of our present study suggest that activated oncogenic signaling is likely to be involved in RECK-repression in MDA-MB-231 cells (Fig. 3f, g). However, inhibition of oncogenic signaling in MCF7 cells did not relieve RECK-suppression in these cells; a preliminary study indicated that the RECK CpG island is extensively methylated in MCF7 cells (unpublished data). The low level of RECK expression in A549 cells, despite the substantial level of RECK mRNA detected (Fig. 3b, e), suggests post-transcriptional regulation, for instance, by oncogenic microRNAs33. The activity of flavone to cooperate with TGFβ in upregulating RECK may provide clues to additional mechanisms of RECK-repression operating in carcinoma cells. Flavonoids, a group of dietary polyphenols, are known to act on a variety of targets, including multiple protein kinases and metabolic enzymes and to have beneficial bioactivities including anticancer, antimicrobial, antiviral, anti-inflammatory, immunomodulatory and antithrombotic activities35,36. The exact mechanism of RECK-upregulation by flavone in the presence of TGFβ in A549 cells warrants further studies.
Efficient and acute expression of RECK using adenoviral vectors provides an opportunity to detect adverse effects of RECK on cell proliferation (Fig. 4b, c) which could be difficult to demonstrate by stable transfection. Our previous study with a colon adenocarcinoma cell line, SW620, indicated that RECK can suppress cell proliferation by modulating gene expression through ECM-integrin signaling22. In particular, SKP2-downregulation and consequent p27-stabilization were implicated in growth suppression. The mechanism of RECK-mediated inhibition of A549 proliferation, however, does not seem to involve SKP2-p27 axis (unpublished) and needs to be addressed in future studies.
Adenovirus-mediated gene transfer also enables us to study the effects of RECK on cell migration in various cell systems. For instance, we could detect the dramatic effect of RECK on migration of A549 cells treated with TGFβ in 3D-collagen gels (Fig. 4f, g) as well as on Matrigel invasion of these cells (Fig. 4h, i). Experiments in 2D-culture revealed that RECK has similar effects on migration speed and directional persistence between carcinoma (A549) cells after TGFβ-treatment (Fig. 4d) and sarcoma (HT1080) cells (Supplementary Fig. S6 and ref. 14) suggesting that some of the findings in sarcoma cells may be extapolated to carcinoma cells that have undergone EMT.
Our previous findings6,7,8,9,10,14 and the results of the present study suggest two possible mechanisms through which RECK brings about its biological activities: (1) regulation of peri-cellular proteolysis and (2) modulation of gene and/or protein expression (Fig. 5c). These mechanisms are not mutually exclusive, since altered peri-cellular proteins affect gene/protein expression via cell surface receptors and downstream signaling pathways and proteolysis is modified by altered expression of proteases and/or their regulators. Multiple lines of evidence suggest that RECK modulates peri-cellular proteolysis and that fibronectin and its receptor (integrins) are among the critical targets protected by RECK (Fig. 4e; Supplementary Fig. S3; Fig. 5c)9,14. Our findings with an MMP inhibitor (GM6001), MT1-MMP overexpression and an integrin inhibitor (RGD peptide) in HT1080 cells (Supplementary Fig. S6) also support this model. These findings with malignant cells, together with previous findings in Reck-null mouse embryo fibroblasts14, support the idea that normal epithelial cells acquire mesenchymal properties with balanced expression of proteases (such as MMPs) and their regulators (such as RECK) so that well-regulated, ECM-dependent migration becomes possible; in carcinoma, however, the balance is tilted toward excessive proteolysis due in part to suppression of RECK expression by constitutive activation of oncogenic signaling pathways (Fig. 5c).
In an attempt to assess the contribution of second mechanism (modulation of gene expression), we performed a transcriptome study using A549 cells treated in four different ways as shown in Figure 4b and found a significant impact of RECK to reduce the number as well as the repertoire of genes and pathways affected by TGFβ (Supplementary Fig. S7). Hence, RECK seems to attenuate TGFβ-signaling in these cells (Fig. 5c) and this raises an interesting possibility that RECK may play a role in the context-dependent responses to TGFβ observed in various systems37. RECK-overexpression, on the other hand, showed little effect on the levels of EMT-associated genes, many of which respond to TGFβ (Supplementary Fig. S4), further suggesting that RECK does not regulate EMT itself.
In summary, we found that TGFβ-induced EMT (as defined by E-cadherin downregulation) is accompanied by RECK-upregulation in the non-tumorigenic epithelial cell line MCF10A; in the three carcinoma-derived cell lines tested, the coupling between E-cadherin-downregulation and RECK-upregulation was lost. Forced expression of RECK in the carcinoma-derived cell line A549 does not affect EMT but may affect cell proliferation and migration after EMT. The mechanism of RECK's actions in these cells likely involves regulation of peri-cellular proteolysis (critical targets include fibronectin and its receptor) and modulation of gene expression.
Methods
Cell culture and reagents
MCF10A cells were cultured in MEBM (Lonza) containing 52 μg/ml Bovine Pituitary Extract (BPE), 500 ng/ml hydrocortisone, 10 ng/ml hEGF, 5 μg/ml insulin (MEGM SingleQuot, Lonza) and 100ng/ml cholera toxin (Wako). MCF-7, MDA-MB-231, A549 and HT1080 cells were maintained in Dulbecco's modified Eagle (DME) medium supplemented with 10% fetal bovine serum (GM). We obtained TGFβ1 from R&D systems iCRT3 from Sigma-Aldrich, FH535, PD98059 and LY294002 from Calbiochem and flavone (2-phenylchromone) from Nacalai Tesque. Cell proliferation was estimated using the colorimetric cell counting reagent WST-8 (SF reagent; Nacalai Tesque) in triplicate cultures.
Immunoblot assay
Immunoblot assays were performed as described previously (Oh et al., 2001.). The primary antibodies used were as follows: RECK (5B11D12)6, E-cadherin (610181, BD Biosciences), N-cadherin (610920, BD Biosciences), FN (610077, BD Biosciences), α-tubulin (CP06, Calbiochem), vimentin (V5255, Sigma), phospho-FAK (Y397) (611722, BD Biosciences), FAK (sc-557, Santa Cruz), integrin α5 (610633, BD Biosciences) and integrin β1 (610467, BD Biosciences).
Immunofluorescent staining
Cells were plated onto coverslips, cultured for the indicated period of time and then fixed in 4% paraformaldehyde in PBS pH7.4. Cells were permeabilized with 0.1% Tween-20 in PBS containing 1% BSA before staining. The cells were then stained following standard protocol using monoclonal anti-RECK, anti-E-cadherin (24E10, Cell Signaling) and anti-N-cadherin antibody as the primary antibody and CF488A goat anti-mouse IgG (H + L) or CF555 goat anti-rabbit IgG (H + L) (Biotium) as the secondary antibody. Nuclei were counterstained with DAPI. The stained cells were recorded using a fluorescence microscope (BZ-9000, KEYENCE).
Image and statistic analyses
Densitometry and various image analyses were performed using Image J software unless stated otherwise. Significance of differences were assessed using Student's t-test using Excel software (one-tailed, two-sample unequal variance) and p-values less than 0.05 are considered significant (*) and values less than 0.01 highly significant (**).
Quantitative real-time PCR
Total RNA was extracted using RNeasy Mini Kit (QIAGEN). The levels of specific mRNA were determined using SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit (Invitrogen) with the Mx 3005P Real-Time OCR System and Mx Pro software (Stratagene). Primers for human RECK were 5′-GCTGGCAATTTGGTGTGCTCTA-3′ and 5′-GGGTAAGTGCGCCCATTCTG -3′. Primers for the control human hypoxanthine phosphoribosyl-transferase 1 (HPRT1) were 5′-CCAGACAAGTTTGTTGTAGG-3′ and 5′-TCCAAACTCAACTTGAACTC-3′. The reaction consisted of an initial reverse transcription [50°C, 5min; 95°C, 5 min] followed by 45 cycles of PCR [94°C, 15 s; 54°C (HPRT) or 64°C (hRECK), 40 s; 72°C, 20 s].
Adenovirus infection
The recombinant adenoviruses, Ad-Z (abbreviated C in Figures) and Ad-RECK (abbreviated R in Figures), have been described elsewhere22. MCF10A (1.5 × 105/well), MCF-7 (2.0 × 105/well), MDA-MB-231 (2.0 × 105/well), or A549 cells (1.0 × 105/well) were plated onto 6-well plates and infected on the following day with Ad-Z and Ad-R at 600 pfu/cell (for MCF-7 and MDA-MB-231 cells) or 300 pfu/cell (for MCF10A and A549 cells). The cells were subjected to various assays between 48 and 96 h after infection.
Transfection
Dual Luciferase assay using the pRL-TK control vector and the pGL3-Basic vector (Promega) containing the 4.1 kb mouse Reck promoter fragment was performed as described previously19 except that Lipofectamine 2000 (Invitrogen) was used for transfection. siRNAs targeting E-cadherin [Ambion: 146381 (E1) and s2769 (E2)] or control siRNAs [Dharmacon: D-001210-01 (C1) and D-001210-03 (C2)] were transfected using RNAiMAX reagent (Invitrogen).
Motility assay
For microscopic observation of random migration, cells (7 × 102) were plated onto multi-well glass-bottom dishes (Matsunami glass) coated with 10 μg/ml fibronectin (Sigma) or acidified type-I collagen (Cellmatrix I-C, Nitta Gelatin) and containing 200 μl Leibovitz's L-15 medium (Gibco) containing 10% FBS. After a pre-incubation period, the cells in 5 fields were time-lapse recorded for 4-8 h as described previously (Morioka et al., 2009). The migration of 5 well-isolated, non-dividing cells per field was tracked on a monitor and their speed and directional persistence were calculated from a time series of coordinates using Dunn's formula23. For 3D-culture in collagen gel, cells suspended in 0.25 ml DME containing 2.4 mg/ml type I collagen (Cellmatrix I-A, Nitta Gelatin) freshly reconstituted on ice were layered onto a basal layer (0.25 ml, the same composition as top layer except the cells) preset in 12-well plates and the plates were incubated in a CO2 incubator at 37°C. The gel was overlaid with 1 ml growth medium after 30 min.
Matrigel invasion assay
FluoroBlok Transwell insert of 8 μm pore size (Corning) were coated by adding 100 μl diluted Matrigel (BD Biosciences; 250 μg/ml) and air-dried overnight. The coated inserts were placed on a 24-well plate containing DMEM supplemented with 5% FBS as a chemo-attractant. The cells (2.5 × 104) pre-labeled for 30 min with CellTracker Green CMFDA (Life Technologies) were suspended in DMEM containing 0.1% FBS without or with 5 ng/ml TGFβ and plated onto the insert. After 48-h incubation, the cells invaded to the lower side of the membrane were recorded using an inverted fluorescent microscope (KEYENCE BZ-9000).
References
Yang, J. & Weinberg, R. A. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14, 818–829 (2008).
Polyak, K. & Weinberg, R. A. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9, 265–273 (2009).
Thiery, J. P., Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 (2009).
Massague, J. TGFbeta in Cancer. Cell 134, 215–230 (2008).
Heldin, C. H., Landstrom, M. & Moustakas, A. Mechanism of TGF-beta signaling to growth arrest, apoptosis and epithelial-mesenchymal transition. Curr Opin Cell Biol 21, 166–176 (2009).
Takahashi, C. et al. Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK. Proc Natl Acad Sci U S A 95, 13221–13226 (1998).
Oh, J. et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell 107, 789–800 (2001).
Miki, T. et al. The reversion-inducing cysteine-rich protein with Kazal motifs (RECK) interacts with membrane type 1 matrix metalloproteinase and CD13/aminopeptidase N and modulates their endocytic pathways. J Biol Chem 282, 12341–12352 (2007).
Omura, A. et al. RECK forms cowbell-shaped dimers and inhibits matrix metalloproteinase-catalyzed cleavage of fibronectin. J Biol Chem 284, 3461–3469 (2009).
Muraguchi, T. et al. RECK modulates Notch signaling during cortical neurogenesis by regulating ADAM10 activity. Nat Neurosci 10, 838–845 (2007).
Echizenya, M. et al. The membrane-anchored MMP-regulator RECK is a target of myogenic regulatory factors. Oncogene 24, 5850–5857 (2005).
Kondo, S. et al. Dual effects of the membrane-anchored MMP regulator RECK on chondrogenic differentiation of ATDC5 cells. J Cell Sci 120, 849–857 (2007).
Kawashima, S. et al. Localization of the membrane-anchored MMP-regulator RECK at the neuromuscular junctions. J Neurochem 104, 376–385 (2008).
Morioka, Y. et al. The membrane-anchored metalloproteinase regulator RECK stabilizes focal adhesions and anterior-posterior polarity in fibroblasts. Oncogene 28, 1454–1464 (2009).
Noda, M. & Takahashi, C. Recklessness as a hallmark of aggressive cancer. Cancer Sci 98, 1659–1665 (2007).
Onder, T. T. et al. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68, 3645–3654 (2008).
Handeli, S. & Simon, J. A. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Mol Cancer Ther 7, 521–529 (2008).
Gonsalves, F. C. et al. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A 108, 5954–5963 (2011).
Sasahara, R. M., Takahashi, C. & Noda, M. Involvement of the Sp1 site in ras-mediated downregulation of the RECK metastasis suppressor gene. Biochem Biophys Res Commun 264, 668–675 (1999).
Valenzuela, D. M. & Groffen, J. Four human carcinoma cell lines with novel mutations in position 12 of c-K-ras oncogene. Nucleic Acids Res 14, 843–852 (1986).
Kozma, S. C. et al. The human c-Kirsten ras gene is activated by a novel mutation in codon 13 in the breast carcinoma cell line MDA-MB231. Nucleic Acids Res 15, 5963–5971 (1987).
Yoshida, Y., Ninomiya, K., Hamada, H. & Noda, M. Involvement of the SKP2-p27(KIP1) pathway in suppression of cancer cell proliferation by RECK. Oncogene 31, 4128–4138 (2012).
Dunn, G. A. Characterising a kinesis response: time averaged measures of cell speed and directional persistence. Agents Actions Suppl. 12, 14–33 (1983).
Barczyk, M., Carracedo, S. & Gullberg, D. Integrins. Cell Tissue Res 339, 269–280 (2010).
Mitra, S. K., Hanson, D. A. & Schlaepfer, D. D. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 6, 56–68 (2005).
Huber, M. A., Kraut, N. & Beug, H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 17, 548–558 (2005).
Taube, J. H. et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107, 15449–15454 (2010).
Lee, H. et al. TGF-beta signaling preserves RECK expression in activated pancreatic stellate cells. J Cell Biochem 104, 1065–1074 (2008).
Erkan, M. et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 61, 172–178 (2012).
Hsu, M. C., Chang, H. C. & Hung, W. C. HER-2/neu represses the metastasis suppressor RECK via ERK and Sp transcription factors to promote cell invasion. J Biol Chem 281, 4718–4725 (2006).
Hatta, M., Matsuzaki, T., Morioka, Y., Yoshida, Y. & Noda, M. Density- and serum-dependent regulation of the Reck tumor suppressor in mouse embryo fibroblasts. Cell Signal 21, 1885–1893 (2009).
Jeon, H. W. & Lee, Y. M. Inhibition of histone deacetylase attenuates hypoxia-induced migration and invasion of cancer cells via the restoration of RECK expression. Mol Cancer Ther 9, 1361–1370 (2010).
Loayza-Puch, F. et al. Hypoxia and RAS-signaling pathways converge on and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs. Oncogene 29, 2638–2648 (2010).
Hill, V. K. et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res 71, 2988–2999 (2011).
Nijveldt, R. J. et al. Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr 74, 418–425 (2001).
Kim, H. P., Son, K. H., Chang, H. W. & Kang, S. S. Anti-inflammatory plant flavonoids and cellular action mechanisms. J Pharmacol Sci 96, 229–245 (2004).
Massague, J. TGFbeta signalling in context. Nat Rev Mol Cell Biol 13, 616–630 (2012).
Acknowledgements
We are grateful to Drs. Yoko Morioka, Tomoko Matsuzaki and Hitoshi Kitayama for technical advice, Drs. Kiyotsugu Yoshikawa, Junichi Tsuji, Kazuki Matsui and Junji Ichihara for discussion, Dr. Hiroyuki Nakagawa for a part of the GEO data analysis and Dr. David B. Alexander for critical reading of the manuscript. We also thank Hai-Ou Gu, Aiko Nishimoto and Emi Nishimoto for technical assistance and Aki Miyazaki for secretarial assistance. This work was supported by KAKENHI [Grants-in-Aid for JSPS Fellows, Creative Scientific Research, Scientific Research on Priority Areas and Scientific Research on Innovative Areas].
Author information
Authors and Affiliations
Contributions
K.Y. conceived and designed the experiments; K.Y. and R.I. carried out the experiments; and K.Y., Y.Y., R.I. and M.N. analysed the data. K.Y. and M.N. wrote the manuscript and K.Y., Y.Y., R.I., H.H. and M.N. discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Movie 1
Supplementary Information
Supplementary Movie 2
Supplementary Information
Supplementary Movie 3
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yuki, K., Yoshida, Y., Inagaki, R. et al. E-cadherin–downregulation and RECK-upregulation are coupled in the non-malignant epithelial cell line MCF10A but not in multiple carcinoma-derived cell lines. Sci Rep 4, 4568 (2014). https://doi.org/10.1038/srep04568
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04568
This article is cited by
-
Suppression of tumor metastasis by a RECK-activating small molecule
Scientific Reports (2022)
-
Self-assembly of tessellated tissue sheets by expansion and collision
Nature Communications (2022)
-
Tumorigenic mesenchymal clusters are less sensitive to moderate osmotic stresses due to low amounts of junctional E-cadherin
Scientific Reports (2021)
-
Pathway crosstalk enables cells to interpret TGF-β duration
npj Systems Biology and Applications (2018)
-
Corepressor metastasis-associated protein 3 modulates epithelial-to-mesenchymal transition and metastasis
Chinese Journal of Cancer (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
