
Abstract
Targeted delivery of drugs and sensors into cells is an attractive technology with both medical and scientific applications. Existing delivery vehicles are generally limited by the complexity of their design, dependence on active transport and inability to function within cellular compartments. Here, we developed self-assembled nanofibrous hydrogel fibers using a biologically inert, low-molecular-weight amphiphile. Self-assembled nanofibrous hydrogels offer unique physical/mechanical properties and can easily be loaded with a diverse range of payloads. Unlike commercially available E. coli membrane particles covalently bound to the pH reporting dye pHrodo, pHrodo encapsulated in self-assembled hydrogel-fibers internalizes into macrophages at both physiologic (37°C) and sub-physiologic (4°C) temperatures through an energy-independent, passive process. Unlike dye alone or pHrodo complexed to E. coli, pHrodo-SAFs report pH in both the cytoplasm and phagosomes, as well the nucleus. This new class of materials should be useful for next-generation sensing of the intracellular milieu.
Similar content being viewed by others


Introduction
The transportation of therapeutic or diagnostic agents and reporter molecules across the cell membrane into multiple intracellular compartments remains a significant challenge due to the selective permeability and biological complexity of eukaryotic cell membranes. Active transport via endocytosis, phagocytosis and pinocytosis are thought to be the major mechanisms mediating uptake of particles. However, controversy over specific mechanisms that mediate internalization still exist1,2. Specialized mammalian cells associated with the immune response readily engulf solid particles with diameters > 750 nm through phagocytosis2,3. In humans, professional phagocytes such as macrophages, monocytes, neutrophils and dendritic cells display the most permissive internalization of large particles (>1 μm), while smaller particles are internalized via endocytosis by a wide range of cell types. Professional phagocytes engulf and digest bacteria and tissue debris, an essential element of their function in early and late immune responses, wound healing and cellular recycling4,5. The bactericidal effect of phagosomes relies on acidification and oxygen radicals to create a hostile environment for microbes. However, some intracellular pathogens are known to evade the phagosome by perturbing phagosomal membrane maturation and modulating acidification6,7,8. Although multiple types of pH sensing probes have been developed, efficient delivery of such probes into specific intracellular compartments still remains a significant challenge. Existing technologies for measuring phagosomal pH depend on active internalization of particles tagged with reporters that are amenable to chemical modification9,10,11,12,13,14 and are not capable of concomitant reporting of the nucleus and cytoplasm, or of multiplexed reporting of multiple intracellular parameters. These considerations emphasize the need for different kinds of reporter vehicles, capable of reporting activity across multiple cellular compartments and the cytoplasm.
Current delivery systems are reporter-specific and generally custom-made by covalently linking fluorescent reporters to particles (1–10 μm) such as latex beads or bacterial wall fragments11,12,13,14. For example, covalently attached pHrodo on bacterial-particles and dextran-particles have become a gold standard to track phagocytosis and endocytosis, respectively15,16. However, not all fluorescent dyes are amenable to covalent binding and chemical modification can compromise or eliminate the reporting properties of the dye, limiting the potential scope of this approach to a narrow range of molecules that retain their activity after binding. Alternative strategies are similarly limited. For example Modi et al recently reported17 the development of a DNA I-switch to monitor endosomal pH using fluorescence resonance energy transfer (FRET). Although those nanosensors function adequately, they are limited by cost and scalability given that new designs are required for each condition to be sensed. Non-covalent delivery systems have traditionally been hindered by multiple constraints. Physical encapsulation of dyes in a polymer matrix often generates heterogeneity in the system and the leaching of the sensor dyes causes instability, thus reducing the life time and reproducibility of the sensor18,19. Thus, there is a significant unmet need to develop a platform delivery vehicle that could be loaded with a wide range of reporters without covalent modification and that could be readily internalized into multiple intracellular compartments including the cytoplasm and nucleus. Ideally, once internalized the vehicle/reporter construct should remain stable and rapidly, reversibly and reproducibly respond to and report intracellular physiology in real-time.
To address these design criteria, we have explored the use of custom designed self-assembled gel fibers (SAFs) that can physically encapsulate reporter molecules. Intracellular compartments may contain high concentrations of proteolytic enzymes and reactive oxygen species; thus SAFs should be relatively inert to enable long-term stability following internalization. In addition, SAFs should not exhibit buffering capacity that could alter the pH of the microenvironment and interfere with a pH sensing dye. Additionally, the ideal delivery vehicle would permit the encapsulation of multiple reporters to enable multiplexed readouts, such that multiple intra-phagosomal functions could be tracked concurrently. To fulfill these criteria, we have designed and synthesized an amphiphilic derivative of the nonvolatile, odorless, color-stable, water-soluble and essentially non-toxic tri(hydroxymethyl)aminomethane (TRIS). The chemical structure of TRIS includes three primary alcohol functional groups that can aid in hydrogen bonding for self-assembly and a primary amine that can easily be functionalized to convert TRIS into an amphiphile.
The rational molecular design of self-assembled biomaterials enables the fabrication of supramolecular hydrogel structures that have the ability to entrap agents with high loading and encapsulation efficencies20,21. Diffusion coefficient studies have shown that the dynamic properties of water molecules in nanofibrous self-assembled hydrogels are almost unchanged compared to pure water22. Therefore, diffusion of protons and likely other analytes is highly feasible and unaltered in nanofibrous self-assembled hydrogels. This could be a major advantage of SAFs to sense the pH efficiently, as unperturbed proton diffusion and mobility is key to facilitate interaction between protons and encapsulated reporter dye to enable a precise readout. While typically it is a significant challenge to develop large particles that efficiently internalize into the cytoplasm and nucleus23,24,25 rather than being constrained within endosomes, we found that SAFs readily internalize into the cytoplasm, phagosomes and nucleus of cultured murine macrophages. The macrophage provides an ideal platform on which to study the uptake of particles into multiple compartments by both active internalization through both endocytosis and large particle phagocytosis and passive internalization under low energy conditions. pH regulation of the phagosome has been well characterized26,27 and both phagosome and cytosol pH can be experimentally modulated using common reagents.
To confirm the location and function of SAFs, a pH-reporter fluorescent dye (pHrodo) was encapsulated into SAFs. pHrodo is a well-known fluorophore dye used for sensing changes in pH across the physiologic range (pH 4–8) in phagosomes and endosomes of mammalian cells through customized delivery vehicles28. To design effective SAFs, we have synthesized a low-molecular-weight amphiphilic molecule that forms a fibrous hydrogel through self-assembly and physically encapsulates dyes such as pHrodo without the need for covalent modification (Fig. 1a). To facilitate uptake through either endocytosis or a phagocytosis, we have developed a method to isolate SAF particles from the bulk hydrogel with an appropriate size to maximize internalization29,30.

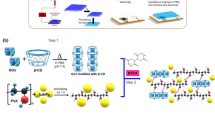
Synthesis and self-assembly of a TRIS 12 amphiphilic gelator (SAF).
(a), Single step synthesis of the Tris-12 amphiphile and schematic showing the formation of pHrodo-SAFs. (b), Optimized structure of Tris-12 using ab initio Hartree-Fock calculations and schematic representation of self-assembled amphiphiles. Dotted red lines indicate the hydrogen-bonding network. Scanning electron microscope images of (c), native self-assembled Tris-12 fibers (SAFs) and (d), pHrodo encapsulated fibers (pHrodo-SAFs).
Results
Design and characterization of self-assembled hydrogels
To generate a TRIS amphiphile that could assemble into a hydrogel, we conjugated dodecanoic acid to create Tris-12 (Fig. 1a), whereby the introduction of an aliphatic chain promotes self-assembly through van der Waals interactions (in addition to hydrogen bonding from TRIS hydroxyl groups). In addition, this amphiphile was engineered to resist digestion by esterases and reactive oxygen species and exhibit minimal to no buffering ability through converting the TRIS primary amine, which is required for buffering capacity, to an amide. We also anticipated that the hydrogen-bonding interactions of the carbonyl groups of amides would aid in the assembly process. Importantly, unlike synthesis of many amphiphilic hydrogelators, the synthesis of the Tris-12 amphiphile was obtained through a single high-yielding step (see Methods). Furthermore, the crude products showed comparable self-assembling ability to the purified form of Tris-12, which may permit the development of these amphiphiles on an industrial scale for a wide range of applications which is often a roadblock in self-assembled hydrogels31,32,33.
Tris-12 exhibited excellent self-assembling ability in multiple solvents including aqueous (acidic and basic) and organic solvents (for detailed self-assembly characterization see Supplementary Table S1). Typically, 1–4% (wt/v) solid Tris-12 in phosphate buffered saline (PBS) was heated to 80°C until dissolved and upon cooling to ambient temperature Tris-12 self-assembled into bulk hydrogels. Repeating a similar procedure in the presence of pHrodo dissolved in PBS (40 μg) produced a pHrodo-encapsulated self-assembled hydrogel. Subsequently, SAFs were isolated from the bulk hydrogel (see Methods). Importantly, the presence of pHrodo altered neither the self-assembling ability of the amphiphile nor the morphology of the fibers (Fig. 1c,d).
Through a series of ab initio calculations, X-ray diffraction (XRD) and Fourier Transform Infrared Spectroscopy (FT-IR), the geometry of Tris-12 was optimized in the gaseous state and lengths of single amphiphile molecules and possible lamellar assemblies were calculated. In XRD experiments, xerogel of Tris-12 showed long distance spacing of 3.1 nm, which is larger than the molecular length (1.86 nm from the optimized geometry calculations) and less than double the extended molecular length of Tris-12 (Fig. 1b, & see Supplementary Information). This is likely explained by interdigitated molecular packing as shown in Fig. 1b. In this model, hydrophilic hydroxyl groups are exposed to the outer solvent while hydrophobic chains are highly interdigitated, which is consistent with earlier reported polyhydroxyl based self-assembled amphiphiles34. In addition, in FT-IR spectra of the Tris-12 amphiphile the carbonyl stretching value of the amide group in the self-assembled gel (1629 cm−1) was lower than in a methanolic solution of Tris-12 (1672 cm−1). This significant shift of 42 cm−1 could be attributed to the strong hydrogen-bonding interactions of the carbonyl groups of amides in the self-assembled state, which clearly suggests that hydrogen-bonding enhances the self-assembly of these amphiphiles to form fibrous structures as depicted in Fig. 1b.
Buffering capacity of self-assembled fibers
To be an effective vehicle for reporter dyes, the carrier should not exhibit buffering capacity under a wide range of pH. A series of experiments were performed to confirm the absence of buffering capacity by the SAFs (without pHrodo). The pH of a concentrated solution of SAFs was continuously monitored during the serial addition of small volumes of dilute acid (HCl) and base (NaOH). The SAFs solution exhibited a linear pH change over the pH range of 4 to 10.5, which suggests that fibers do not have buffering capacity (Supplementary Fig. S1). Thus, we envisioned that the encapsulated dye should accurately report the pH of the compartment without interference from the SAFs vehicle.
Encapsulation of pHrodo dye in SAFs
Encapsulation of pHrodo in SAFs was characterized via UV-vis absorption spectrophotometry (absorption and emission maxima for pHrodo are 530 and 590 nm, respectively (Fig. 2a and b)). As illustrated in Fig. 2a, Tris-12 SAFs do not have a discrete absorbance peak between 300 and 700 nm, allowing encapsulated pHrodo to be detected by absorption spectroscopy without interference from its vehicle. Physical encapsulation of pHrodo dye did not significantly alter its inherent spectral properties (absorption maximum shifted a marginal 5 nm, while emission spectra remained similar, Fig. 2a and b).

Stability of pHrodo in SAFs and its response to pH modulation.
(a), Absorption and (b), emission spectra of pHrodo in SAFs and pHrodo free dye in dimethyl sulfoxide (DMSO) solvent. (c), Stability of pHrodo-SAFs at a wide range of pH (2.1–8.3) in the presence and absence of esterase enzyme (100 U/mL, after a 72 hr incubation). Inset of (c), shows that at pH 4 the dye did not leak into the solution and thus it is anticipated that non-specific pinocytosis of dye released from extracellular pHrodo-SAFs is minimal. (d), pHrodo dye encapsulated within self-assembled fibrous-particles, in DMSO solvent and covalently conjugated to E. coli bacterial particles exhibit similar responses to a wide range of pH.
Stability of encapsulated pHrodo in SAFs and its response to pH
The stability of pHrodo encapsulated SAFs (pHrodo-SAFs) was investigated over a pH range from 2 to 8.5. To ensure pHrodo-SAFs are stable in a broad range of pH and pHrodo remains confined within the fibers and does not leak the dye over time, pHrodo-SAFs were incubated in solutions of varying pH. To mimic the presence of proteolytic enzymes, fibers were also incubated with or without esterase enzyme (300 U/mL) at multiple pH conditions. Data in Fig. 2c indicates that pHrodo-SAFs are stable and the dye remains within the self-assembled fibers over a wide range of pH. In the presence degradative enzymes over 3 days, >85% of the dye remained stable within the self-assembled fibers and no further loss was observed beyond this time-point. As demonstrated in Fig. 2c, negligible release of the dye was observed at pH 4. In addition, pHrodo-SAFs were lyophilized and stored at room temperature. After regular intervals (1, 4, 8 and 10 weeks), re-dispersion of pHrodo-SAFs in PBS did not release the dye (confirmed with UV-vis spectra, data not shown).
The responsiveness of pHrodo was examined in its solubilized form, in its entrapped form in SAFs and in its covalently bound form with commercially available pHrodo-conjugated E. coli cell wall particles (pHrodo-E.coli) at specific pH's ranging across the physiologic spectrum from 4–7.5. As shown in Fig. 2d, the fluorescence response of the entrapped dye closely mirrored that of the free-solubilized form of pHrodo and the pHrodo covalently conjugated to E. coli particles (Fig. 2d). These results suggest that encapsulation of pHrodo in SAFs did not compromise its ability to sense changes in pH.
Uptake of pHrodo-SAFs by macrophages and sensing of intracellular pH changes through pHrodo-SAFs
Micron sized particles are known to internalize into macrophages through endocytosis and phagocytosis29,30; thus it was anticipated that micron-sized pHrodo-SAFs (Fig. 1d) would be internalized by macrophages through phagocytosis. As shown in Supplementary Fig. S2, uptake of pHrodo-SAFs into acidified intracellular compartments at 37°C was demonstrated by discrete, punctate regions of high fluorescence within the cell suggesting that pHrodo-SAFs are localized in phagosomes. In addition to discrete punctate signals and unlike E.coli-bound pHrodo, fluorescence was also observed throughout the cytoplasm and was modulated by changing cytoplasmic pH, suggesting that pHrodo-SAFs also deliver pHrodo into the cytoplasm. Unlike phagosomal uptake, this effect was noted at both 37°C and 4°C, suggesting that cytoplasmic delivery occurs by an energy-independent process.
Localization of pHrodo-SAFs in intracellular compartments
Under physiologic conditions, pHrodo-SAFs reported pH in multiple cell compartments, with dominant concentration in acidified phagosomes and the cytoplasm (Fig. 3a,b). After clamping the intracellular pH to 5, to maximize pHrodo fluorescence, cells were fixed and labeled with DiO (cell membrane staining dye, ex/em 484/501 nm), V450 anti-mouse CD107a (lysosome staining, ex/em 406/450 nm) and TO-PRO®-3 (nucleus staining, ex/em 642/661 nm). Co-localized confocal scanning fluorescence images (Fig. 3a,b and Supplementary Fig. S3,4,5,6) suggest that cells loaded with pHrodo-SAFs (internalized at 37°C) localized pHrodo predominantly in the cytoplasm while a significant amount of dye was also observed in the phagosomes, with a minute quantity of pHrodo consistently localized in the nucleus. On the contrary, pHrodo-E.coli particles were confined to phagosomes. A modest amount of free dye was internalized (Fig. 3a,b), presumably via endocytosis of media. Transmission electron microscope experiments should be performed to visualize the location of the fibers.

Confocal laser scanning fluorescence images of pHrodo-SAFs, pHrodo-E.coli and free-dye pHrodo internalized into the macrophages at 37 and 4°C.
(a), Two-dimensional and (b), orthogonal images of pHrodo-SAFs, pHrodo-E.coli and free-pHrodo internalized into macrophages at 37°C. Significant amounts of pHrodo-SAFs were localized in the cytoplasm, whereas pHrodo-E. coli particles remain localized in lysosomal compartments. (c), Two-dimensional and (d), orthogonal images of pHrodo-SAFs, pHrodo-E.coli and free-pHrodo internalized into macrophages at 4°C. Images suggest that pHrodo-SAFs were efficiently internalized by macrophages at low temperature compared to pHrodo-E.coli and free-pHrodo that did not enter into the cells at these conditions. Representative images are shown in this figure and in Supplementary figures S3–S6.
In contrast, under hypothermic (4°C) conditions that inhibit active transport free dye and pHrodo-E.coli particles did not enter into cells, whereas large quantities of pHrodo-SAFs continued to internalize into the cytoplasm (Fig. 3c,d and Supplementary Fig. S5, S6). pHrodo-SAFs internalized by macrophages at 4°C co-localized to both the cytoplasm and phagosomes (Fig. 3c, d and Supplementary Figure S5, S6).
Function of internalized pHrodo-SAFs
To quantify the response of internalized dye to intracellular pH modulation in a population of cells and to test the utility of pHrodo-SAFs in high-throughput, plate-reader based experiments, pHrodo-SAFs and pHrodo-E.coli particles were internalized into macrophages at 37°C (in independent experiments) and nigericin treatment was used to clamp the pH of the cytoplasm, nuclei and intracellular compartments to the pH of the extracellular solution35. Intracellular pH was subjected to pH cycling between 5 and 7 by exchange of nigericin solutions at each pH. A 1.5-fold increase in fluorescence was observed for internalized pHrodo-SAFs when the intracellular pH was clamped from native pH 7.4 to acidic pH 5 (Fig. 4a) suggesting that pHrodo-SAFs were present within the cytoplasm. On the contrary, upon clamping the pH to 5, a similar result was not observed in pHrodo-E.coli internalized-macrophages (Fig. 4a). These observations suggest that pHrodo-E.coli particles were confined into already acidic phagosomes but not present in the cytoplasm or nucleus, which is in agreement with the previous reports11,36. Returning the pH from 5 to 7 caused a drop in fluorescence that indicates that pHrodo physically encapsulated in SAFs, like pHrodo bound to E. coli particles, retain their ability to respond to changes in microenvironmental pH.

Probing the response of pHrodo-SAFs and pHrodo-E.coli to pH modulation in macrophages and the mechanism mediating the internalization of SAFs.
Quantified fluorescence of macrophages loaded with pHrodo-SAFs and pHrodo-E.coli at (a), 37°C and (b), 4°C. Cell density was 100,000 cells/well and results are average value from 9 wells. Intracellular pH was clamped from native pH to 5 then to 7.4. Changing the cytoplasmic and nuclear pH from native (7.4) to 5 led to an increase in signal from pHrodo-SAFs loaded macrophages suggesting that pHrodo-SAFs reside in the cytoplasm. The absence of a similar response for pHrodo-E.coli loaded macrophages suggests that pHrodo-E.coli particles localized in phagosomes. The response observed at 4°C (b) verifies that pHrodo-SAFs internalized into cells at lower temperature while pHrodo-E.coli particles did not internalize at these conditions. (c), Pretreatment of RAW 264.7 macrophages with either latrunculin A or cytochalasin D for 30 min prior to incubation with SAFs did not affect the internalization of pHrodo-SAFs at 37°C and 25°C. (d), On the contrary, pretreatment of RAW 264.7 macrophages with either latrunculin A or cytochalasin D inhibited the internalization of pHrodo-E.coli at 37°C and 25°C. These results suggest that internalization of pHrodo-SAFs into macrophages is an energy-independent, non-phagocytosis passive process. * = p < 0.001 and ** = p < 0.002.
pHrodo-E.coli particles are known to internalize through an energy-dependent phagocytosis11,36. On the contrary, uptake of pHrodo-SAFs into macrophages at 4°C was comparable with the uptake at 37°C (Fig. 4b and a, respectively), suggesting an energy-independent process. To further characterize the mechanism for uptake of pHrodo-SAFs and pHrodo-E.coli particles we inhibited endocytosis and actin-function with latrunculin A and cytochalasin D37 respectively, at both 37°C and 25°C These inhibitors blocked the uptake of pHrodo-E.coli particles while internalization of pHrodo-SAFs was not affected (Fig. 4c, d). These results suggest that pHrodo-SAFs can enter cells in an energy-independent, non-endocytotic, passive manner. Importantly, internalization of pHrodo-SAFs did not affect the viability of macrophages compared to untreated control cells up to 48 hr (see Supplementary Fig. S7). To demonstrate that change in the observed fluorescence is due to pHrodo dye responding to the change in pH, DiO (a dye inert to pH change, ex/em 484/501 nm) was co-encapsulated with pHrodo in SAFs and internalized into macrophages at 37°C. Intracellular pH was subjected to pH cycling between 5 and 7 by exchange of nigericin solutions at each pH. Change in fluorescence emission at 590 nm (pHrodo) and 501 nm (DiO) were quantified (see Supplementary Fig. S8). As anticipated, pHrodo emission at 590 nm changed in response to a pH change, while DiO emission at 501 nm did not show any change. This suggests that the observed fluorescence change throughout this study is indeed due to the response of pHrodo dye to changes in micro-environmental pH and shows the potential for multiplex readouts of SAFs encapsulating multiple dyes simultaneously.
Live cell monitoring of intracellular pH
Following uptake of pHrodo-SAFs by macrophages at 37°C, intracellular pH was studied by quantification of fluorescence of individual cells using real-time fluorescence microscopy (ex/em 530/590 nm). In Fig. 5a, individual lines represent the signal from a single cell in a repre-sentative experiment. These results indicate that pHrodo-SAFs report the change in intracellular pH in an efficient, reproducible and rapid manner in the range that pHrodo is known to sense (pH 4 to 7.5). The response to incremental changes in pH over time was measured (Fig. 5b, characteristic sigmoidal graph) showing that the ability to dynamically sense incremental changes in pH over multiple cycles (i.e. reversibility) was preserved.

Live cell imaging of response of pHrodo-SAFs internalized within macrophages to cyclical modulation of pH and response to a proton pump inhibitor.
(a), Spectra signal from three individual macrophages shows response to pH cycling of pHrodo-SAFs internalized in murine macrophages upon alternate addition of acid and base (time of addition shown by black arrows). (b), A plot of emission of pHrodo at 590 nm as a function of time while exposed to incremental changes in pH. This reversible cycle of pH produced a mirror response. Each line in the spectra represents the signal from an individual macrophage. (c) and (d), Effect of bafilomycin (inhibitor for the vacuolar ATPase) addition on pH change in macrophage phagosomes. Clamping of pH by addition of bafilomycin and nigericin was followed via fluorescence emission of pHrodo dye (encapsulated within SAFs). Each line in (d) in the spectra represents the signal from an independent macrophage (n = 6).
To demonstrate that internalized pHrodo-SAFs are capable of reporting physiologic changes in intraphagosomal pH, fluorescence was measured during exposure to modulators of phagosomal pH (Fig. 5c). Bafilomycin A is a known selective inhibitor of the proton pump vacuolar ATPase (V-ATPase) that is responsible for the acidification of phagosomes38. Thus, addition of bafilomycin-A increases phagosomal pH. Upon addition of bafilomycin-A (200 nM) to pHrodo-SAFs loaded cells, pHrodo fluorescence decreased by 35% within 30 mins (±4%, n = 4, P < 0.05, (Fig. 5c) indicating that pHrodo-SAFs internalized within the phagosomes specifically report changes in phagosomal pH. Real-time fluorescence microscopy of individual cells (pHrodo-SAFs internalized cells, Fig. 5d) reveals that upon addition of bafilomycin-A, there was a moderate decrease in fluorescence. Whereas cells subsequently treated with nigericin and clamped the pH to 5 followed by 7.5 triggered a sudden increase and decrease in fluorescence intensity, respectively (Fig. 5d). These results reveal that the majority of SAFs were localized within the cytoplasm.
To delineate the retention of SAFs in intracellular compartments, a kinetic study was performed by quantification of fluorescence intensity of pHrodo at different time points (0.1, 12, 24 and 48 hr) during post-fiber internalization. pHrodo-SAFs were internalized into RAW 264.7 macrophages and at each time point intracellular pH was clamped to pH 5.2 and fluorescence intensity was measured. Results in Supplementary Fig. S9 suggest that although the total fluorescence decreased over time, a significant amount of pHrodo-SAFs remain inside cells at least for 48 hr. The decrease is likely due to exocytosis given that gels remain stable when incubated in the presence of degradative enzymes or over a wide range of pH (Fig. 5). The percentage of total fluorescence corresponding to pHrodo dye (ex:530/em:590 nm) was 100, 40, 31 and 22.5% at 0.1, 12, 24 and 48 hr, post fiber internalization, respectively (n = 9). While a 50–60% decrease in the fluorescence of dye loaded SAFs was observed during first 12 hr, likely due to SAF exocytosis, ~25% of SAFs remain inside the cells for at least 48 hr.
SAFs as a sensing platform for multiple reporter dyes
Given that pHrodo is confined within the lamellar assembled structures of SAFs through physical encapsulation and SAFs have an ability to efficiently internalize into macrophages at a range of temperatures, we examined the potential of this platform to encapsulate a wide range of dyes. Specifically, we showed that the Tris-12 amphiphile efficiently encapsulated diverse dyes such as metal (Zinc) sensors39 FluoZin-3, 6-Methoxy-(8-p-toluenesulfonamido)quinoline and the intracellular pH sensor lysosensor40 DND-160 (Fig. S10 and Table S2) as well as DiO as described above. Thus, self-assembled hydrogel fibers loaded with dyes could be used to sense many other biological phenomena in the intracellular environment.
Discussion
There is a significant need to develop materials that target intracellular compartments for delivery of a wide range of payloads, from fluorescent reporters of physiologic states as described here to small-molecular drugs, antibodies, or gene-silencing RNA and DNA41. Covalent binding to micron-scale particles such as E.coli cell walls can reliably deliver a limited range of payloads to phagosomes. However, few approaches are available to target similarly large payloads to the cytoplasm and nucleus, or to deliver molecules that are not amenable to covalent binding23,42. We anticipated that self-assembled fibers formed from amphiphilic small molecules with an overall neutral surface charge could efficiently cross the cell membrane into the cytoplasm, bypassing the process of endocytosis and delivering a pH-reporting dye in an energy-independent manner.
pHrodo-SAFs were efficiently internalized by macrophages at both 37°C and at 4°C and simultaneously localized predominantly into the cytoplasm, phagosomes and nucleus, while conventional pHrodo-E. coli particles did not internalize into either the cytoplasm or nucleus. Physically encapsulated pHrodo in SAFs dynamically responded to modulation of pH in these intracellular compartments at both 37 and 4°C. This new class of fibers should be useful to encapsulate and deliver a wide range of sensors or therapeutic agents for potential biomedical applications. Our results further suggest that two or more reporters can be simultaneously loaded into SAFs, providing a multiplexed readout of physiologic states. Although SAFs load multiple compartments within the cell, careful selection of the reported states allows for selection of signal from individual compartments. For example, using pHrodo, which has minimal fluorescence at cytoplasmic pH (~7) but strong signal at phagosomal pH, cells in their native state provide a readout of phagosomal acidity, while artificial clamping of cytoplasmic pH at phagosomal levels (pH 5) reveals the true degree of loading with the dye across all compartments. Thus the same dye, with the same delivery vehicle, is capable of reporting multiple cell states and potentially providing internal controls for differences of loading between cells.
In summary, SAFs offer the opportunity to deliver desired agents into the intracellular environment at lower temperatures or other metabolically inactive states. SAFs further permit encapsulation of dye without the necessity of covalent binding to a delivery vehicle, while not impacting the function of the payload. We have demonstrated their ability to report pH using a dye, pHrodo, otherwise deliverable only by covalent binding to bacterial particles and thus limited to the phagosome. pH changes across multiple cellular compartments are measurable in both in live cell experiments and in high-throughput plate reader based assays.
Methods
Reagents and solutions
Except where noted, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). pHrodo dye was purchased from Invitrogen (Carlsbad, CA). Calcium Ringers was prepared with the following composition: NaCl 145 mM, KH2PO4 2.5 mM, MgSO4 1 mM, CaCl2 1 mM, D-Glucose 10 mM, HEPES 10 mM. High-K+ Ringers: NaCl 105 mM, KCl 40 mM, KH2PO4 2.5 mM, MgSO4 1 mM, CaCl2 1 mM, D-Glucose 10 mM, HEPES 10 mM.
Synthesis of Tris-12 amphiphile
To a solution of 2-Amino-2-hydroxymethyl-propane-1,3-diol (1.21 g, 10 mM) in anhydrous dimethyl sulfoxide (DMSO, 10 mL), 1 equivalent of methyl dodecanoate (2.14 g, 10 mM) and 2 equivalents of anhydrous K2CO3 (2.76 g, 20 mM) was added and stirred at room temperature. Following a 48 h reaction that was quenched with ice cold water (40 mL), stirring continued for 30 mins. White precipitate that was formed during quenching was filtered and washed with cold water (5 × 10 mL). Drying under vacuum produced Tris-12 amphiphile in its pure form (2.84 g, 94% yield).
Encapsulation of pHrodo in self-assembled fibrous particles
Tris-12 (8 mg) was placed in a scintillation glass vial with 20 μl of DMSO solution of pHrodo (40 μg) was added, subsequently after addition 180 μl of water (either PBS, or double-distilled water which was passed through Chelex 100 resin), the vial was tightly closed with a screw-cap and heated to 70°C for 2 mins to dissolve the Tris-12. As the vial cooled to room temperature the fibers self-assembled within 30 mins. After one hour, 0.5 ml of water was added to the vial and the gel was uniformly dispersed through mixing with a pipette-tip, then transferred to an eppondorf tube, which was subjected to centrifugation (15,000 rpm for 7 min). Following removal of the supernatant, fibers were rinsed with 1 mL of water and subjected to centrifugation. This process was repeated our times to completely remove non-encapsulated pHrodo. The final concentration of the dye in the fibers was established by dissolving the fibers in DMSO and measuring the absorbance against a standard curve of known dye concentrations. The concentration of pHrodo in the fibers was calculated to be 20 μg of pHrodo in 2 mg of fibers.
Scanning electron microscopy (SEM)
To examine the morphology of the fibers, 20 μl of fibers-dispersed water was placed on carbon-tape attached to an aluminium grid and dried overnight under ambient conditions, followed by coating with a thin-layer of gold (30 nm) using a sputtering machine. Those aluminum grids were directly imaged with an environmental SEM (FEI/Philips XL30 FEG-ESEM, FEI, Hillsboro, Oregon) using 10 kV.
X-ray diffraction (XRD)
XRD measurements were acquired using a Bruker AXS D-8 Discover with GADDS diffractometer using graded d-space elliptical side-by-side multilayer optics, monochromated Cu-Kα radiation (40 kV, 40 mA) and imaging plate.
UV-vis spectroscopy
UV-visible spectra of the amphiphiles was acquired with a CARY100BIO spectrophotometer using a quartz cuvette of 1-cm path length.
Spectroscopy and in vitro fluorescence measurements
Absorbance of 50 μl of 10% Tris-12 fibers solution, with or without 20 μg/ml pHrodo, was measured in 1 nm increments using a 96-well microplate absorbence spectrometer (Synergy 4, BioTek Inc, Winooski, VT). To measure fluorescence, the same plate reader and nanofiber solutions were used. 5 μl of nanofiber solution was added to wells containing 100 μl buffered Calcium Ringer's solution titrated to pHs from 4 to 10 using HCl and NaOH. Fluorescence was measured using excitation and emission filters of 530(+/−25) nm and 590 (+/−35) nm, respectively. All experiments were performed in triplicate.
Cell culture
RAW 264.7 murine macrophages (ATCC) were maintained on plastic Petri dishes at 37°C and 5% CO2 in culture media consisting of RPMI 1640 supplemented with 5% heat-inactivated fetal bovine serum, 1 mM glutamine, penicillin/strepromycin/fungizone, 10 mM HEPES buffer, 100 μM nonessential amino acids and 2.5 × 10−5 M 2-mercaptoethanol (Invitrogen Corp., Carlsbad, CA). Cells were passaged every three days.
pH modulation
RAW 264.7 macrophages on glass coverslips in 3 mL of cell culture media were incubated with pHrodo-SAFs (0.4 μg of pHrodo dye, MW ~ 1000) for three hours. Selected individual cells that showed particle uptake were imaged using a Nikon TE-2000U inverted real-time fluorescence microscope (Nikon, Inc, Melville, NY). Digital images were captured using a digital CCD camera (Hamamatsu ORCA-ER, Bridgewater, NJ). Pixel intensity was measured using image-processing software (Universal Imaging Corp., Downington, PA). Images were acquired every 30 s, with an exposure time of 0.3 s. After equilibrating in a Calcium Ringer's solution for five minutes, the solution was exchanged for alternating pH solutions of high-K+ Ringer's containing 10 μM nigericin at pH 5 and pH 7.4. For high-throughput measurements of physiologic pH changes, cells were plated on black fluorescence microplates (Corning Costar) at a density of 25 × 104 cells/well. After allowing cells to settle for one hour at 37° and 5% CO2, culture media was exchanged for Calcium Ringer's solution containing 20 μL of concentrated fiber solution/mL. Cells were placed in a humidified room air incubator at 37° for 3 hr, subsequently washed once with Calcium Ringer's before addition of Calcium Ringer's with or without the vacuolar H+-ATP-ase inhibitor bafilomycin-A (200 nM). Results were read on a microplate reader (Synergy 2, BioTek, Winooski, VT) using excitation filters of 530 nm and emission filters of 590 nm.
Determination of cell viability
Cell viability was assessed by monitoring uptake and intracellular conversion of nonfluorescent calcein-AM to fluorescent calcein, using a modification of previously reported method43. pHrodo-SAFs were internalized into RAW264.7 cells were, subsequently plated on three different plates for 4, 24 and 48 hr time points. At each time point, wells were gently rinsed with Ringer's buffer and solutions were replaced with identical solutions containing calcein-AM (8 μM) for 40 min, after which fluorescence from each well [excitation (Ex.) 485 nm, emission (Em.) 528 nm] was recorded.
Confocal laser scanning microscope and cell labeling
Cells were scraped from the cell culture plate, seeded at a density of 200,000 cells/well on FN pre-treated cover-slips (12 mm, Bellco Glass) in 24-well plates and maintained in an incubator for one hour. Cells were then washed with Ringer's buffer and either pHrodo-SAFs (40 μg/ml), pHrodo-E.coli particles (10 μg/ml) or pHrodo free-dye (10 μg/ml) was added in Calcium Ringer's buffer to the wells. After a 3 hour incubation, cells were washed with Calcium Ringer' buffer 2 times before being fixed for 15 minutes in 3.7% formaldehyde solution at 37°C. Cells were then rinsed 3 times with PBS and permeated with 0.1% Triton X-100 in PBS for 5 min at room temperature. After rinsing 2 times with Ringer' buffer, cell membranes were stained with DiO (10 μM in Clacium Ringer's buffer) for 20 mins at 37°C. Then cells were washed with Ringer's buffer twice and cell endosomes were stained with V450 Anti-mouse CD107a (0.4 ug, BD Horizon) in 0.5 mL Calcium Ringer's buffer for 30 min at room temperature. Finally, after rinsing three times with Calcium Ringer's buffer, nuclei were stained with 1 μM TO-PRO®-3 dye in Calcium Ringer's buffer for 15 minutes. After rinsing 3 times with Ringer's buffer and once with DI water, cover-slips were mounted on glass slides in ProLongGold(Invitrogen) anti-fading medium.
Retention time of pHrodo-SAFs inside cells
RAW 264.7 macrophages were seeded into 96 well plates (Corning) at the density of 100,000 cells/well. One hour after seeding, cell media was re-placed with 100 μl Ca2+ Ringer's buffer (pH = 7.2) containing 0.1 mg/ml pHrodo-SAFs. After a 3 hour incubation at 37°C, buffer was removed and cells were washed twice with PBS and plated in culture media at 37°C. Following 0.1, 12, 24 and 48 hours, cells were washed with PBS thrice prior to treatment with K+ Ringer's buffer (pH = 5.2). The fluorescent signal was measured with ex/em: 530 nm/590 nm by SpectraMaxR microplate reader (Molecular Devices, Sunnyvale, CA).
References
Zhao, F., Zhao, Y., Liu, Y., Chang, X. & Chen, C. Cellular uptake, intracellular trafficking and cytotoxicity of nanomaterials. Small 7, 1322–1337 (2011).
Ishimoto, H. et al. Single-cell observation of phagocytosis by human blood dendritic cells. Jpn J Infect Dis 61, 294–297 (2008).
Liu, Y. et al. The effect of Gd@C82(OH)22 nanoparticles on the release of Th1/Th2 cytokines and induction of TNF-alpha mediated cellular immunity. Biomaterials 30, 3934–3945 (2009).
Silva, M. T. When two is better than one: macrophages and neutrophils work in concert in innate immunity as complementary and cooperative partners of a myeloid phagocyte system. J Leukoc Biol 87, 93–106 (2010).
Haas, A. The phagosome: compartment with a license to kill. Traffic 8, 311–330 (2007).
Flannagan, R. S., Cosio, G. & Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7, 355–366 (2009).
Huynh, K. K. & Grinstein, S. Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev 71, 452–462 (2007).
Jabado, N. et al. Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J Exp Med 192, 1237–1248 (2000).
Rybicka, J. M., Balce, D. R., Chaudhuri, S., Allan, E. R. & Yates, R. M. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J 31, 932–944 (2012).
Rybicka, J. M., Balce, D. R., Khan, M. F., Krohn, R. M. & Yates, R. M. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A 107, 10496–10501 (2010).
Deriy, L. V. et al. Disease-causing mutations in the cystic fibrosis transmembrane conductance regulator determine the functional responses of alveolar macrophages. J Biol Chem 284, 35926–35938 (2009).
Hackam, D. J. et al. Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K+-ATPases and vacuolar-type H+-atpases. J Biol Chem 272, 29810–29820 (1997).
VanderVen, B. C., Yates, R. M. & Russell, D. G. Intraphagosomal measurement of the magnitude and duration of the oxidative burst. Traffic 10, 372–378 (2009).
Yates, R. M., Hermetter, A. & Russell, D. G. Recording phagosome maturation through the real-time, spectrofluorometric measurement of hydrolytic activities. Methods Mol Biol 531, 157–171 (2009).
Miksa, M., Komura, H., Wu, R., Shah, K. G. & Wang, P. A novel method to determine the engulfment of apoptotic cells by macrophages using pHrodo succinimidyl ester. J Immunol Methods 342, 71–77 (2009).
Strunnikova, N. V. et al. Loss-of-function mutations in Rab escort protein 1 (REP-1) affect intracellular transport in fibroblasts and monocytes of choroideremia patients. PLoS One 4, e8402 (2009).
Modi, S. et al. A DNA nanomachine that maps spatial and temporal pH changes inside living cells. Nat Nanotechnol 4, 325–330 (2009).
Basabe-Desmonts, L., Reinhoudt, D. N. & Crego-Calama, M. Design of fluorescent materials for chemical sensing. Chemical Society reviews 36, 993–1017 (2007).
Zhan, W., Seong, G. H. & Crooks, R. M. Hydrogel-based microreactors as a functional component of microfluidic systems. Analytical chemistry 74, 4647–4652 (2002).
Palmer, L. C., Velichko, Y. S., de la Cruz, M. O. & Stupp, S. I. Supramolecular self-assembly codes for functional structures. Philos Transact A Math Phys Eng Sci 365, 1417–1433 (2007).
Palmer, L. C. & Stupp, S. I. Molecular self-assembly into one-dimensional nanostructures. Acc Chem Res 41, 1674–1684 (2008).
Bastrop, M. et al. Water dynamics in bolaamphiphile hydrogels investigated by 1H NMR relaxometry and diffusometry. J. Phys. Chem. B 115, 14–22 (2011).
Porter, A. E. et al. Direct imaging of single-walled carbon nanotubes in cells. Nat Nanotechnol 2, 713–717 (2007).
Oyelere, A. K., Chen, P. C., Huang, X., El-Sayed, I. H. & El-Sayed, M. A. Peptide-conjugated gold nanorods for nuclear targeting. Bioconjug Chem 18, 1490–1497 (2007).
Pantarotto, D., Briand, J. P., Prato, M. & Bianco, A. Translocation of bioactive peptides across cell membranes by carbon nanotubes. Chem Commun (Camb), 16–17 (2004).
Grinstein, S., Nanda, A., Lukacs, G. & Rotstein, O. V-ATPases in phagocytic cells. J Exp Biol 172, 179–192 (1992).
Willemsen, R. H., de Kort, S. W., van der Kaay, D. C. & Hokken-Koelega, A. C. Independent effects of prematurity on metabolic and cardiovascular risk factors in short small-for-gestational-age children. J Clin Endocrinol Metab 93, 452–458 (2008).
Bernardo, J., Long, H. J. & Simons, E. R. Initial cytoplasmic and phagosomal consequences of human neutrophil exposure to Staphylococcus epidermidis. Cytometry A 77, 243–252 (2010).
Champion, J. A. & Mitragotri, S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A 103, 4930–4934 (2006).
Sahay, G., Alakhova, D. Y. & Kabanov, A. V. Endocytosis of nanomedicines. J Control Release 145, 182–195 (2010).
Zhang, S. Hydrogels: Wet or let die. Nat Mater 3, 7–8 (2004).
Vemula, P. K., Cruikshank, G. A., Karp, J. M. & John, G. Self-assembled prodrugs: an enzymatically triggered drug-delivery platform. Biomaterials 30, 383–393 (2009).
Vemula, P. K., Li, J. & John, G. Enzyme catalysis: tool to make and break amygdalin hydrogelators from renewable resources: a delivery model for hydrophobic drugs. J Am Chem Soc 128, 8932–8938 (2006).
Vemula, P. K., Aslam, U., Mallia, V. A. & John, G. In situ synthesis of gold nanoparticles using molecular gels and liquid crystals from vitamin-C amphiphiles. Chem. Mater. 19, 138–140 (2007).
Grinstein, S. & Furuya, W. Assessment of Na+-H+ exchange activity in phagosomal membranes of human neutrophils. Am J Physiol 254, C272–285 (1988).
Berger, S. B. et al. SLAM is a microbial sensor that regulates bacterial phagosome functions in macrophages. Nat Immunol 11, 920–927 (2010).
Wakatsuki, T., Wysolmerski, R. B. & Elson, E. L. Mechanics of cell spreading: role of myosin II. J Cell Sci 116, 1617–1625 (2003).
Swallow, C. J., Grinstein, S. & Rotstein, O. D. A vacuolar type H(+)-ATPase regulates cytoplasmic pH in murine macrophages. J Biol Chem 265, 7645–7654 (1990).
Domaille, D. W., Que, E. L. & Chang, C. J. Synthetic fluorescent sensors for studying the cell biology of metals. Nat Chem Biol 4, 168–175 (2008).
DePedro, H. M. & Urayama, P. Using LysoSensor Yellow/Blue DND-160 to sense acidic pH under high hydrostatic pressures. Anal Biochem 384, 359–361 (2009).
Lee, S. H., Chung, B. H., Park, T. G., Nam, Y. S. & Mok, H. Small-Interfering RNA (siRNA)-Based Functional Micro- and Nanostructures for Efficient and Selective Gene Silencing. Acc Chem Res (2012).
Wang, Y. et al. Direct imaging of titania nanotubes located in mouse neural stem cell nuclei. Nano Res 2, 543–552 (2009).
Adler, M., Shafer, H., Hamilton, T. & Petrali, J. P. Cytotoxic actions of the heavy metal chelator TPEN on NG108-15 neuroblastoma-glioma cells. Neurotoxicology 20, 571–582 (1999).
Acknowledgements
The authors thank Kimberley Faldetta for technical assistance. This work was supported by a Harvard Institute of Translational Immunology/Helmsley Trust Pilot Grants in Crohn's Disease to J.M.K. NIH DE023432 to J.M.K. and the American College of Surgeons Resident Research Fellowship and T32 DK007754 (J.E.K.); R01 DK069929 (DIS).
Author information
Authors and Affiliations
Contributions
J.M.K., D.I.S., P.K.V. and J.E.K. conceptualized the idea, conceived experiments, analyzed the results and wrote the manuscript. A.B., M.W., C.X., L.C. and S.R.J. conducted experiments. G.J. helped to analyze data, wrote manuscript and all authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported license. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Vemula, P., Kohler, J., Blass, A. et al. Self-assembled hydrogel fibers for sensing the multi-compartment intracellular milieu. Sci Rep 4, 4466 (2014). https://doi.org/10.1038/srep04466
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04466
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
