
Abstract
Hydronephrosis is a common disease characterized by dilation of the renal pelvis and calices, resulting in loss of kidney function in the most severe cases. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces nonobstructive hydronephrosis in mouse neonates through upregulation of prostaglandin E2 (PGE2) synthesis pathway consisting of cyclooxygenase-2 (COX-2) and microsomal prostaglandin E synthase-1 (mPGES-1) by a yet unknown mechanism. We here studied possible involvement of cytosolic phospholipase A2α (cPLA2α) in this mechanism. To this end, we used a cPLA2α-null mouse model and found that cPLA2α has a significant role in the upregulation of the PGE2 synthesis pathway through a noncanonical pathway of aryl hydrocarbon receptor. This study is the first to demonstrate the predominant role of cPLA2α in hydronephrosis. Elucidation of the pathway leading to the onset of hydronephrosis using the TCDD-exposed mouse model will deepen our understanding of the molecular basis of nonobstructive hydronephrosis in humans.
Similar content being viewed by others


Introduction
Hydronephrosis, defined as dilation of the renal pelvis and calices, is a common disease, found in 1.5–3.3% of autopsies1. In the most severe cases, kidney function is lost as a result of complete destruction of the renal parenchyma. Obstruction of urine flow at any point along the urinary tract causes retention of urine and increased hydrostatic pressure in the renal pelvis and calices, which is one of the known causes of hydronephrosis. The prevalence of antenatal hydronephrosis was reported to be in the range of 1–5% of pregnancies diagnosed by the use of fetal ultrasonography2. While the underlying etiologies of antenatal hydronephrosis are considered to be diverse, most cases lack obstruction3,4. Nonobstructive hydronephrosis is clinically defined by the absence of urinary tract obstruction and vesicoureteral reflux. However, its molecular pathophysiology is unclear.
Certain types of chemical agents induce hydronephrosis. Administration of lithium chloride results in hydronephrosis in mouse neonates5. Drugs such as mitomycin7 and adriamycin8 are also known to produce hydronephrosis. Dioxins, a group of halogenated aromatic hydrocarbons, induce hydronephrosis in rodent fetuses and neonates6. They are produced during combustion processes and are ubiquitously present in the environment. They bioaccumulate in humans and wild animals9 and have a variety of toxic effects, including teratogenicity, reproductive toxicity, neurobehavioral disorders, immune dysfunction and carcinogenicity6. The most potent congener among dioxins is 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). TCDD administration induces hydronephrosis in rodents when they are exposed to it during gestation or via lactation10,11,12. A pathological characteristic of TCDD-induced hydronephrosis in mouse neonates is the absence of ureteral morphological obstruction13. Therefore, TCDD-induced hydronephrosis is a potential model of nonobstructive hydronephrosis in mammals. Another important characteristic is that the critical exposure period leading to the development of TCDD-induced hydronephrosis is confined to the perinatal period14,15, which makes TCDD-induced hydronephrosis a potential model of the perinatal hydronephrosis that is frequently found in human fetuses3,4.
The toxic effects of dioxins, including hydronephrosis, are mediated by aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor. AhR is essential for dioxin toxicity as is demonstrated by the absence of toxicity in AhR-null mice13,16,17,18. Several genes in addition to that for AhR have been shown to be involved in dioxin toxicity19. Among them, genes encoding enzymes for the production of prostaglandins are responsible for the onset of TCDD-induced neonatal hydronephrosis in mice13,20. AhR-dependent upregulation of COX-2 plays a critical role in the onset of hydronephrosis induced by lactational exposure to TCDD in mice. COX-2 upregulation by TCDD is AhR-dependent in vivo and the requirement for COX-2 was confirmed by the abrogation of TCDD-induced hydronephrosis by a COX-2-selective inhibitor13. COX-2 is an inducible form of cyclooxygenase, which converts arachidonic acid to prostaglandin H2 (PGH2). Microsomal prostaglandin E synthase-1 (mPGES-1) transforms PGH2 to prostaglandin E2 (PGE2) and is required for TCDD-induced hydronephrosis, as demonstrated by the lack of hydronephrosis in mPGES-1-null mice20. These studies indicate that PGE2 production, mediated by COX-2 and mPGES-1, plays a key role in the onset of TCDD-induced hydronephrosis.
In the present study, we hypothesized that there is at least one upstream mediator that is necessary for the TCDD-induced upregulation of COX-2 and/or mPGES-1. Such a mediator is thought to affect the pathogenesis of TCDD-induced hydronephrosis via an increase in the production of PGE2. Arachidonic acid, the substrate of COX-2, is one of the main precursors of eicosanoids, including leukotrienes, thromboxanes and prostaglandins. Phospholipase A2 (PLA2) enzymes catalyze the hydrolysis of membrane phospholipids to release arachidonic acid21. Of more than 30 enzymes with PLA2 activity, cytosolic phospholipase A2α (cPLA2α) is transcriptionally upregulated22,23 or enzymatically activated24 in response to TCDD exposure. Upregulation and/or activation of cPLA2α can modulate a number of biological processes through arachidonic acid-derived eicosanoids, including PGE2. Therefore, cPLA2α has the potential to mediate the toxic effects in response to TCDD exposure. The present study examined the possible role(s) of cPLA2α in the onset of TCDD-induced hydronephrosis and in the upregulation of COX-2 and mPGES-1.
Results
Role of cPLA2α in TCDD-induced hydronephrosis
To investigate the potential role of cPLA2α in the onset of TCDD-induced hydronephrosis in mouse neonates, cPLA2α KO mice were produced by mating heterozygous cPLA2α KO mice with each other. After delivery, dams were administered TCDD (20 μg/kg) or vehicle on PND 1 and consequently WT [cPLA2α (+/+)] and KO [cPLA2α (−/−)] pups were exposed to TCDD through lactation. The cPLA2α KO pups did not show any overt abnormality until PND 14 when they were sacrificed for analyses. The body weight of the TCDD-exposed KO pups and that of the WT pups were similar to each other on PND 14 (6.25 ± 0.41 g in female WT pups vs 6.30 ± 0.50 g in female KO pups and 6.39 ± 0.27 g in male WT pups vs 6.83 ± 0.25 g in male KO pups), which indicates that the KO pups were fed normally and suggests that these two groups of mice were exposed to a similar dose of TCDD via milk. This supposition was confirmed by the nearly identical expression levels of the AhR target genes, CYP1A125, AhRR26 and IGFBP-127, upon TCDD exposure between the groups of KO pups and WT pups (Fig. 1A–C and Supplementary Fig. 1A–C).

cPLA2α-dependent and -independent increases in gene expression in 7-day-old female pups lactationally exposed to TCDD.
CYP1A1 (A), AhRR (B), IGFBP-1 (C) and cPLA2α (D) mRNA abundances in the kidney after normalizing to the cyclophilin B mRNA level. Results are presented as mean ± SEM for 4–6 dams/group. Histograms with different letters indicate a statistically significant difference at P < 0.05 by one-way ANOVA with the post hoc test. No statistical comparison was performed for cPLA2α KO pups expressing truncated cPLA2α31.
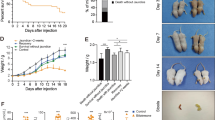
In histological examination, compared with the normal morphology of kidneys from vehicle-treated WT pups (Fig. 2A) and that of KO pups (Fig. 2C), the most severe degree of hydronephrosis was found in TCDD-exposed WT pups (Fig. 2B), while only modest (Fig. 2D) or no effects were observed in TCDD-exposed KO pups. Incidence and severity of hydronephrosis were examined in TCDD-exposed WT and KO pups and control pups on PND 14. All TCDD-exposed female (Table 1) and male (Supplementary Table 1) WT pups developed hydronephrosis by PND 14 and most mice of both sexes showed the most severe degree of hydronephrosis (Table 1 and Supplementary Table 1). By contrast, the incidence of hydronephrosis in TCDD-exposed female and male cPLA2α KO pups on PND 14 was 33% and 36%, respectively and none showed the most severe degree of hydronephrosis (Table 1 and Supplementary Table 1). No hydronephrosis was observed in female and male control pups (Table 1 and Supplementary Table 1). These results demonstrate that a lack of cPLA2α markedly reduces the incidence and severity of TCDD-induced neonatal hydronephrosis.

Representative pictures of cPLA2α-dependent hydronephrosis on PND 14 induced by lactational exposure to TCDD in male pups.
Dams were administered TCDD (20 μg/kg) on PND 1. Kidney of vehicle-exposed cPLA2α (+/+) (A) and cPLA2α (−/−) (B) mice and TCDD-exposed cPLA2α (+/+) (C) and cPLA2α (−/−) (D) mice on PND 14. The degree of hydronephrosis in (B) and (D) was diagnosed as 4 and 2, respectively, using the scale, reported previously34. Scale bars = 500 μm. Refer to Table 1 and Supplementary Table 1 for information on all pups examined on PND 14.
Role of cPLA2α in upregulation of the PGE2 production pathway
The possible role of cPLA2α in the TCDD-induced increase in PGE2 synthesis was examined on PND 7. This time point was chosen because TCDD-induced increase in urinary PGE2 and upregulation of enzymes responsible for the PGE2 production, COX-2 and mPGES-1, are prominent on PND 713,20. In female TCDD-exposed WT pups, COX-2 mRNA abundance, mPGES-1 mRNA abundance and urinary PGE2 concentration were significantly increased 5.5, 6.1 and 13.3 fold, respectively, compared with those in the control pups (Fig. 3A–C). In female TCDD-exposed KO pups, COX-2 mRNA abundance, mPGES-1 mRNA abundance and urinary PGE2 concentration were 1.9, 1.6 and 2.2 fold, respectively, compared with those in the vehicle-treated KO pups and none of these increase was significant (Fig. 3A–C). The TCDD-dependent fold induction of COX-2 mRNA, mPGES-1 mRNA and urinary PGE2 concentration in female cPLA2α KO pups was significantly reduced by 74%, 65% and 84%, respectively, compared with those in female WT mice. The degree of such reductions by the genetic ablation of cPLA2α was comparable to the degree of suppression in the incidence of TCDD-induced hydronephrosis (i.e., 67%). Essentially the same results were observed in TCDD-exposed male pups. TCDD exposure considerably increased abundance of COX-2 and mPGES-1 mRNAs and urinary PGE2 concentration in WT pups and such increases were suppressed to nearly the control level by genetic ablation of cPLA2α (Supplementary Fig. 2). These results demonstrate that cPLA2α plays a predominant role in TCDD-dependent increases in COX-2 and mPGES-1 mRNAs and urinary PGE2 excretion in WT C57BL/6J mouse pups.

cPLA2α-dependent increase in urinary PGE2 production in 7-day-old female pups lactationally exposed to TCDD.
COX-2 (A) and mPGES-1 (B) mRNA abundances in the kidney on PND 7 are shown after normalizing to the cyclophilin B mRNA level. (C) PGE2 concentration in urine. Results are presented as mean ± SEM for 4–6 dams/group. Histograms with different letters indicate a statistically significant difference at P < 0.05 by one-way ANOVA with a post hoc test.
Changes in gene expression in the kidney associated with TCDD-induced hydronephrosis
We analyzed expression of several genes the deficiency of which have been reported to induce hydronephrosis28,29,30 and that are downregulated in the kidney of mouse pups exposed to TCDD13,20. In female pups on PND 7, expression of mRNAs of electrolyte transporters, Na-K-Cl cotransporter 2 (NKCC2) and renal outer medullary K channel (ROMK), were significantly downregulated in the TCDD-exposed WT pups (46% and 32%, respectively) compared with those in the vehicle-treated WT pups (Figs. 4A and B). Expression of mRNA for a water channel, aquaporin 2 (AQP2), in the TCDD-exposed WT pups was also significantly reduced to 50% compared with the vehicle-control WT pups (Fig. 4C). In particular, no such decreases in NKCC2, ROMK and AQP2 mRNAs were observed in cPLA2α KO pups (Fig. 4). In male WT and cPLA2α KO pups (Supplementary Fig. 3), lactational exposure to TCDD led to results essentially identical to those observed in the above-described female pups (Fig. 4).

cPLA2α-dependent alterations in gene expression in 7-day-old female pups lactationally exposed to TCDD.
NKCC2 (A), ROMK (B) and AQP2 (C) mRNA abundances in the kidney (after normalizing to the cyclophilin B mRNA level) are shown. Results are presented as mean ± SEM for 4–6 dams/group. Histograms with different letters indicate a statistically significant difference at P < 0.05 by one-way ANOVA with the post hoc test.
CYP1A1, AhRR and IGFBP-1 mRNAs were quantified to monitor AhR transactivation activity and found to be significantly increased by TCDD exposure not only in female WT pups but also in female cPLA2α KO pups (Fig. 1A–C). The expression level of these AhR dependent genes were used as surrogate marker of TCDD exposure as already described above. Essentially, the same result was observed in male pups (Supplementary Fig. 1), although the difference in IGFBP-1 mRNA abundance was not statistically significant probably due to a low induction level by TCDD. The abundance of cPLA2α mRNA was increased by TCDD exposure in female and male WT pups (Fig. 1D and Supplementary Fig. 1D, respectively), but was barely detected in female and male KO pups, possibly reflecting the expression of a truncated cPLA2α lacking enzymatic activity31.
Discussion
This study was performed to elucidate the possible roles of cPLA2α, a member of PLA2 enzyme family, in the pathogenesis of hydronephrosis in mouse neonates caused by lactational exposure to TCDD. Heterozygous female and male cPLA2α KO mice were mated and the resulting dams were given TCDD on PND 1 at a dose sufficient to induce hydronephrosis in all WT pups by PND 14. In contrast to WT pups, only 33% of female and 36% of male cPLA2α KO pups showed signs of hydronephrosis (Table 1 and Supplementary Table 1). This marked reduction in the incidence of hydronephrosis by genetic ablation of cPLA2α, as well as the concurrent reduction in PGE2 excretion in the urine, i.e., 84% in KO females and 72% in KO males, compared with the WT pups, demonstrates a significant role of cPLA2α in the onset of TCDD-induced neonatal hydronephrosis and suggests that cPLA2 is the major player among many other PLA2 enzymes in this role.
Approximately one-third of female and male cPLA2α KO pups developed hydronephrosis upon TCDD exposure, although the degree of hydronephrosis was much less severe than those in female and male WT pups (Table 1 and Supplementary Table 1). The mechanism by which TCDD-exposure developed hydronephrosis in cPLA2α KO neonates is thought to be dependent on PGE2 because of the following two reasons. (1) Genetic ablation of mPGES-1, a PGE2 synthase, completely blocks the onset of hydronephrosis in TCDD-exposed mouse neonates20, which demonstrates PGE2 is essential for the pathogenesis. (2) Synthesis of PGE2 is not completely blocked in the cPLA2α KO pups (Fig. 3C and Supplementary Fig. 2C) and the minor increase in PGE2 could elicit onset of the less severe hydronephrosis.
Another important finding of this study is that cPLA2α is necessary for the TCDD-induced upregulation of the expression of COX-2 and mPGES-1. The indispensable roles of these two enzymes in the TCDD-induced neonatal hydronephrosis were previously demonstrated by pharmacological inhibition of COX-213 and by genetic ablation of mPGES-120; each of these treatments completely abolished TCDD-induced hydronephrosis. These studies also revealed that TCDD exposure upregulates the mRNA expression of both COX-2 and mPGES-1 in vivo, but did not clarify the component that mediates upregulation of the two genes. The present study showed that cPLA2α has a role to mediate TCDD-induced upregulation of COX-2 and mPGES-1 expression, because the TCDD-induced upregulation of COX-2 and mPGES-1 mRNAs was markedly reduced in the absence of cPLA2α (Fig. 3 and Supplementary Fig. 2). It has been established that the expression of CYP1A125, AhRR26 and IGFBP-127 is induced by a canonical AhR signaling pathway (also referred to as a genomic action pathway of AhR) because they require the transactivation activity of AhR for their expression in response to TCDD. TCDD-induced increase in mRNA abundance of CYP1A1, AhRR and IGFBP-1 was not influenced by the presence or absence of cPLA2α (Fig. 1 and Supplementary Fig. 1), while that of COX-2 and mPGES-1 was dependent on cPLA2α, indicating that the upregulation mechanism of these three genes is distinct from that of COX-2 and mPGES-1.
Two distinct cPLA2α pathways in AhR signaling can mediate responses to TCDD. The first pathway, referred to as the genomic action pathway24, is well established and is the basis of a dogma to explain TCDD toxicity. This response requires translocation of the TCDD-AhR complex into the nucleus to form a heterodimer with the AhR nuclear translocator (ARNT)25, which upregulates a number of genes, including CYP1A125, AhRR26 and IGFBP-127. The cPLA2α gene is also expected to be a target of the genomic action pathway of AhR for the following reasons: the gene has an AhR binding site in its intron23 and the TCDD-induced increase in cPLA2α mRNA is dependent on AhR in hepatoma-derived cell lines22,23. The second pathway of AhR, on the other hand, does not require dimerization of AhR with ARNT or binding of the AhR-ARNT-ligand complex to the promoter region of its target genes; it is named the nongenomic AhR pathway24. This response requires phosphorylation of cPLA2α through activation of protein kinases upon Ca2+ influx, as has been demonstrated in MMDD1 cells32 and several other cell lines24. As a logical consequence, a question arises about the dominance of either the genomic or the nongenomic AhR pathway in the pathogenesis of TCDD-induced hydronephrosis. However, the question has not been explored yet because an appropriate experimental model to differentiate the two mechanisms on TCDD toxicity via cPLA2α is unavailable. The present study demonstrated for the first time that TCDD-induced neonatal hydronephrosis is a suitable model to explore the question because this toxicity phenotype was revealed to be dependent on cPLA2α, which mediates both genomic and nongenomic actions of AhR.
Three enzymes, cPLA2α, COX-2 and mPGES-1, are the main components of the PGE2 synthesis pathway. In this pathway, cPLA2α catalyzes the hydrolysis of membrane phospholipids to release arachidonic acid, which is then converted to PGH2 by COX-2. PGH2 is converted to PGE2 by mPGES-1. TCDD activates this pathway. These three enzymes were upregulated in the kidneys of mouse pups exposed to TCDD and the PGE2 concentration in the urine was increased (Figs. 1 and 3 and Supplementary Figs. 1 and 2). The indispensable roles of COX-213 and mPGES-120 and the crucial role of cPLA2α (demonstrated in the present study) clearly indicate that PGE2 elicits TCDD-induced neonatal hydronephrosis. The next step in elucidating the pathogenesis pathway of TCDD-induced neonatal hydronephrosis will be to identify a receptor for PGE2 responsible for the onset of hydronephrosis and to characterize the biological components downstream of the PGE2 receptor. Electrolyte transporters NKCC2 and ROMK and a water channel AQP2 are presumably the candidate components, since the lack of them can result in polyuria leading to the onset of hydronephrosis28,29,30.
A variety of biological phenomena, such as reproduction, cancer, immune responses and atherosclerosis, are influenced by cPLA2α through the production of eicosanoids, such as prostaglandins, thromboxanes and leukotrienes, which are metabolites of arachidonic acid21. Using hydronephrosis as a model, we demonstrate for the first time that cPLA2α is involved in the pathogenesis of TCDD toxicity. Given that the expression of cPLA2α is deregulated by TCDD (Fig. 1D and Supplementary Fig. 1), other cPLA2α-related phenomena could be disrupted through changes in eicosanoid levels. TCDD exposure was recently reported to alter the levels of many eicosanoids in several organs33, although the impact of the alterations on a variety of the endpoints of TCDD toxicity remains to be identified.
In conclusion, we have shown that cPLA2α predominantly affect the onset of TCDD-induced neonatal hydronephrosis and is responsible at least partly for the TCDD-induced increase in mRNAs of COX-2 and mPGES-1. The identification and characterization of downstream components of the cPLA2α/COX-2/mPGES-1 pathway will clarify the molecular mechanisms underlying the pathogenesis not only of TCDD-induced neonatal hydronephrosis, but also human nonobstructive hydronephrosis induced by yet uncharacterized mechanisms.
Methods
Animals and treatment
A TCDD solution (50 μg/mL in n-nonane) was purchased from Cambridge Isotope Laboratory (Andover, MA, USA) and diluted in corn oil (Wako Pure Chemicals, Osaka, Japan). Corn oil containing 2% n-nonane was used as vehicle.
C57BL/6J strain mice were purchased from CLEA Japan (Tokyo, Japan). cPLA2α-null mice were generated as described previously31 and backcrossed more than 12 times in the laboratory of T. Shimizu and more than 4 times in the laboratory of C. Tohyama. Mice were housed at 23 ± 1°C and 50 ± 10% humidity on a 12/12 h light–dark cycle. Laboratory rodent chow (Labo MR Stock; Nosan, Yokohama, Japan) and distilled water were provided ad libitum. The study protocols were approved by the Animal Care and Use Committee of the Graduate School of Medicine, The University of Tokyo.
Heterozygous cPLA2α knockout (KO) mice on the C57BL/6J background were mated to produce WT [cPLA2α (+/+)] and homozygous KO [cPLA2α (−/−)] mice. Parturition was checked twice daily and the day of birth was designated postnatal day 0 (PND 0). Dams were orally administered either a single dose of TCDD (20 μg/kg, 20 mL/kg body weight) or an equivalent volume of vehicle on PND 1. Consequently, pups were exposed to TCDD via lactation and were sacrificed on PND 7 or PND 14 to collect the kidneys and urine from the bladder. The numbers of dams and pups used for gene expression analysis on PND 7 were 6 and 4–6, respectively, for the vehicle groups and 5 and 4–5, respectively, for the TCDD groups. The pups used for histological analysis on PND 14 are described in Tables 1 and 2. Data derived from individual pups of the same sex, age, genotype and TCDD dose were averaged within a litter and then averaged across litters.
The genotypes of the mice (WT or cPLA2α KO) were determined using PCR to amplify genomic DNA collected from tail snips using the primer sequences CTCTGGTGTGATGAAGGCACTCTATGAGTC and CCCTACCTACAATGTTCACCCAAAACTAGC for WT [cPLA2α (+/+)] and TCGTGCTTTACGGTATCGCCGCTCCCGATT and CCCTACCTACAATGTTCACCCAAAACTAGC for KO [cPLA2α (−/−)] mice.
Histology of the kidney
Kidney specimens were fixed in 10% neutral-buffered formalin, cryoprotected in 20% sucrose solution, embedded in O.C.T. compound (Sakura Finetek Japan, Tokyo, Japan) and snap-frozen in liquid nitrogen. Tissues were sectioned (5-μm thick sections) and stained with hematoxylin and eosin.
Hydronephrosis severity scores were assigned using a previously described scoring system, ranging from ‘0 = no hydronephrosis’ to ‘+4 = the most severe hydronephrotic kidney’34. For determining the severity score, blind scoring was performed for all samples (vehicle/TCDD and WT/KO). Incidence of hydronephrosis in the pups was calculated as the percentage of pups with a severity score ≥2 on either the left or right kidney, in accordance with previous reports34,35.
Quantitative RT-PCR
Total RNA was isolated from the kidneys using the RNeasy Mini Kit (Qiagen, Tokyo, Japan). cDNA synthesis of a given mRNA was performed using an oligo-dT20 primer and SuperScript III (Invitrogen, Carlsbad, CA, USA).
Gene expression levels were quantitatively determined using a LightCycler System (Roche Molecular Biochemicals, Indianapolis, IN, USA) with the Thunderbird SYBR qPCR Mix (Toyobo, Osaka, Japan). Primers for the genes (Table 2) were designed from the respective cDNA or mRNA sequences using Primer3 software36. No-template controls were analyzed in every PCR to monitor cross-contamination. To verify amplification specificity, melting curve analyses of the products were performed for every PCR. The mRNA expression levels were calculated using the ΔΔCt method and normalized to cyclophilin B expression.
PGE2 measurement
Urine was collected from the bladders of 7-day-old mice using a syringe. Urinary PGE2 concentrations were measured using a Prostaglandin E2 EIA kit (Cayman Chemical, Ann Arbor, MI, USA).
Data analysis
To normalize possible litter effects, the data for individual pups of the same sex, age, genotype and TCDD dose were averaged within a litter and then averaged across litters. All data are expressed as the mean ± SEM for the indicated numbers of litters. Differences in means were analyzed by one-way analysis of variance (ANOVA) followed by Tukey-Kramer post hoc analysis. The statistical significance level was set at <0.05.
References
Frøkiær, J. & Zeidel, M. L. Urinary Tract Obstruction. in The Kidney (eds Taal, M. W. et al.) Ch. 37, 1383–1410 (Elsevier Saunders, 2011).
Lee, R. S., Cendron, M., Kinnamon, D. D. & Nguyen, H. T. Antenatal hydronephrosis as a predictor of postnatal outcome: a meta-analysis. Pediatrics 118, 586–593 (2006).
Yamacake, K. G. & Nguyen, H. T. Current management of antenatal hydronephrosis. Pediatr Nephrol 28, 237–243 (2013).
Woodward, M. & Frank, D. Postnatal management of antenatal hydronephrosis. BJU Int 89, 149–156 (2002).
Yoshioka, W. et al. Severe toxicity and cyclooxygenase (COX)-2 mRNA increase by lithium in the neonatal mouse kidney. J Toxicol Sci 34, 519–525 (2009).
Pohjanvirta, R. & Tuomisto, J. Short-term toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals: effects, mechanisms and animal models. Pharmacol Rev 46, 483–549 (1994).
Molyneux, G. et al. The haemotoxicity of mitomycin in a repeat dose study in the female CD-1 mouse. Int J Exp Pathol 86, 415–430 (2005).
Goncalves, A., Franca, W. G., Moraes, S. G., Pereira, L. A. & Sbragia, L. Adriamycin-induced fetal hydronephrosis. Int Braz J Urol 30, 508–513 (2004).
Schecter, A. & Gasiewicz, T. A. Dioxins and Health. 2 edn, (Wiley, 2003).
Nishimura, N., Yonemoto, J., Nishimura, H. & Tohyama, C. Localization of cytochrome P450 1A1 in a specific region of hydronephrotic kidney of rat neonates lactationally exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology 227, 117–126 (2006).
Moore, J. A., Gupta, B. N., Zinkl, J. G. & Vos, J. G. Postnatal effects of maternal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Environ Health Perspect 5, 81–85 (1973).
Courtney, K. D. & Moore, J. A. Teratology studies with 2,4,5-trichlorophenoxyacetic acid and 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 20, 396–403 (1971).
Nishimura, N. et al. Critical role of cyclooxygenase-2 activation in pathogenesis of hydronephrosis caused by lactational exposure of mice to dioxin. Toxicol Appl Pharmacol 231, 374–383 (2008).
Couture-Haws, L., Harris, M. W., McDonald, M. M., Lockhart, A. C. & Birnbaum, L. S. Hydronephrosis in mice exposed to TCDD-contaminated breast milk: identification of the peak period of sensitivity and assessment of potential recovery. Toxicol Appl Pharmacol 107, 413–428 (1991).
Couture, L. A., Harris, M. W. & Birnbaum, L. S. Characterization of the peak period of sensitivity for the induction of hydronephrosis in C57BL/6N mice following exposure to 2,3,7, 8-tetrachlorodibenzo-p-dioxin. Fundam Appl Toxicol 15, 142–150 (1990).
Schmidt, J. V., Su, G. H., Reddy, J. K., Simon, M. C. & Bradfield, C. A. Characterization of a murine Ahr null allele: involvement of the Ah receptor in hepatic growth and development. Proc Natl Acad Sci U S A 93, 6731–6736 (1996).
Gonzalez, F. J. & Fernandez-Salguero, P. The aryl hydrocarbon receptor: studies using the AHR-null mice. Drug Metab Dispos 26, 1194–1198 (1998).
Mimura, J. et al. Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 2, 645–654 (1997).
Yoshioka, W., Peterson, R. E. & Tohyama, C. Molecular targets that link dioxin exposure to toxicity phenotypes. J Steroid Biochem Mol Biol 127, 96–101 (2011).
Yoshioka, W. et al. Critical role of microsomal prostaglandin E synthase-1 in the hydronephrosis caused by lactational exposure to dioxin in mice. Toxicol Sci 127, 547–554 (2012).
Murakami, M. et al. Recent progress in phospholipase A research: from cells to animals to humans. Prog Lipid Res 50, 152–192 (2011).
Kinehara, M., Fukuda, I., Yoshida, K. & Ashida, H. Aryl hydrocarbon receptor-mediated induction of the cytosolic phospholipase A(2)alpha gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin in mouse hepatoma Hepa-1c1c7 cells. J Biosci Bioeng 108, 277–281 (2009).
Kinehara, M., Fukuda, I., Yoshida, K. & Ashida, H. High-throughput evaluation of aryl hydrocarbon receptor-binding sites selected via chromatin immunoprecipitation-based screening in Hepa-1c1c7 cells stimulated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Genes Genet Syst 83, 455–468 (2008).
Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem Pharmacol 77, 608–626 (2009).
Mimura, J. & Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim Biophys Acta 1619, 263–268 (2003).
Mimura, J., Ema, M., Sogawa, K. & Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev 13, 20–25 (1999).
Murray, I. A. & Perdew, G. H. Omeprazole stimulates the induction of human insulin-like growth factor binding protein-1 through aryl hydrocarbon receptor activation. J Pharmacol Exp Ther 324, 1102–1110 (2008).
Rojek, A., Fuchtbauer, E. M., Kwon, T. H., Frokiaer, J. & Nielsen, S. Severe urinary concentrating defect in renal collecting duct-selective AQP2 conditional-knockout mice. Proc Natl Acad Sci U S A 103, 6037–6042 (2006).
Lorenz, J. N. et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter's syndrome. J Biol Chem 277, 37871–37880 (2002).
Takahashi, N. et al. Uncompensated polyuria in a mouse model of Bartter's syndrome. Proc Natl Acad Sci U S A 97, 5434–5439 (2000).
Uozumi, N. et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390, 618–622 (1997).
Dong, B., Nishimura, N., Vogel, C. F., Tohyama, C. & Matsumura, F. TCDD-induced cyclooxygenase-2 expression is mediated by the nongenomic pathway in mouse MMDD1 macula densa cells and kidneys. Biochem Pharmacol 79, 487–497 (2010).
Bui, P., Solaimani, P., Wu, X. & Hankinson, O. 2,3,7,8-Tetrachlorodibenzo-p-dioxin treatment alters eicosanoid levels in several organs of the mouse in an aryl hydrocarbon receptor-dependent fashion. Toxicol Appl Pharmacol 259, 143–151 (2012).
Bryant, P. L., Schmid, J. E., Fenton, S. E., Buckalew, A. R. & Abbott, B. D. Teratogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the expression of EGF and/or TGF-alpha. Toxicol Sci 62, 103–114 (2001).
Theobald, H. M. & Peterson, R. E. In utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-rho-dioxin: effects on development of the male and female reproductive system of the mouse. Toxicol Appl Pharmacol 145, 124–135 (1997).
Rozen, S. & Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132, 365–386 (2000).
Acknowledgements
This work was supported by a grant for Environmental Risk Research from the Ministry of the Environment to C.T.
Author information
Authors and Affiliations
Contributions
C.T. raised the initial idea of this study. W.Y. and C.T. designed experiments. W.Y., T.K., N.F. and K.A.Y. performed experiments. W.Y., C.T. and F.M. wrote and edited the paper. T.S. provided cPLA2α-null mice. All the authors discussed the results and provided comments on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yoshioka, W., Kawaguchi, T., Fujisawa, N. et al. Predominant Role of Cytosolic Phospholipase A2α in Dioxin-induced Neonatal Hydronephrosis in Mice. Sci Rep 4, 4042 (2014). https://doi.org/10.1038/srep04042
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04042
This article is cited by
-
Significance of AHR nuclear translocation sequence in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced cPLA2α activation and hydronephrosis
Archives of Toxicology (2019)
-
Roles of cytosolic phospholipase A2α in reproductive and systemic toxicities in 2,3,7,8-tetrachlorodibenzo-p-dioxin-exposed mice
Archives of Toxicology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
