
Abstract
Recent studies have shown that endogenous metabolites act via aryl hydrocarbon receptor (AhR) signalling pathway in tubulointerstitial fibrosis (TIF) pathogenesis. However, the mechanisms underlying endogenous metabolite-mediated AhR activation are poorly characterised. In this study, we conducted untargeted metabolomics analysis to identify the significantly altered intrarenal metabolites in a mouse model of unilateral ureteral obstruction (UUO). We found that the levels of the metabolite 1-methoxypyrene (MP) and the mRNA expression of AhR and its target genes CYP1A1, CYP1A2, CYP1B1 and COX-2 were progressively increased in the obstructed kidney at Weeks 1, 2 and 3. Furthermore, these changes were positively correlated with progressive TIF in UUO mice. In NRK-52E, RAW 264.7 and NRK-49F cells, MP dose-dependently upregulated the mRNA expression of AhR and its four target genes and the protein expression of nuclear AhR, accompanied by the upregulated protein expression of collagen I, α-SMA and fibronectin, as well as downregulated E-cadherin expression. Consistently, oral administration of MP in mice progressively enhanced AhR activity and upregulated profibrotic protein expression in the kidneys; these effects were partially inhibited by AhR knockdown in MP-treated mice and cell lines. In addition, we screened and identified erythro-guaiacylglycerol-β-ferulic acid ether (GFA), which was isolated from Semen plantaginis, as a new AhR antagonist. GFA significantly attenuated TIF in MP-treated NRK-52E cells and mice by partially antagonising AhR activity. Our results suggest that MP activates AhR signalling, thus mediating TIF through epithelial-mesenchymal transition and macrophage-myofibroblast transition. MP is a crucial metabolite that contributes to TIF via AhR signalling pathway.
Similar content being viewed by others


Introduction
The changes in genes, proteins and metabolites in tumour cells and tissues and the dynamic interactions between the tumour cells and the surrounding extracellular matrix (ECM), which is mainly governed and regulated by the tumour microenvironment, are the major driving factors for tumour development and progression [1]. The renal interstitium is a complex structure composed of multiple cells, ECM and various secreted or cell membrane-presented molecules [2, 3]. Intrarenal cellular and acellular components, such as different cells (e.g., epithelial cells, endothelial cells, macrophages, fibroblasts, lymphocytes, pericytes and neutrophils), signalling molecules (e.g., interleukin-15 and matrix metalloproteinase-9) and ECM (collagens, laminins and fibronectin), represent the global tubulointerstitial microenvironment that contributes to renal damage [3,4,5]. Among these components, excessive accumulation and deposition of ECM is a crucial factor in progressive tubulointerstitial fibrosis (TIF) [6,7,8], which is a complicated process that is the hallmark of renal damage in the most common kidney diseases [9]. TIF is characterised by tubular atrophy, excessive accumulation and deposition of ECM components and the accumulation of fibroblasts and myofibroblasts [10]. Early studies demonstrated that ECM mediated TIF by modulating cell–cell or cell–matrix crosstalk [11]. Bidirectional communication between intrarenal cells and ECM components, including collagens, laminins and fibronectin, as well as growth factors, cytokines, chemokines, and endogenous metabolites orchestrates cellular responses to both physiological and pathological conditions [12]. Although various cell types contribute to progressive TIF, the activation of fibroblasts is the critical pathogenic mechanism in TIF. Epithelial-mesenchymal transition (EMT), macrophage-myofibroblast transition (MMT) and endothelial-mesenchymal transition are three important processes in organ fibrosis [13]. Extensive studies have demonstrated that EMT is the most important ECM-associated process in TIF [14]. The latest evidence has shown that MMT also affects ECM in TIF [15, 16].
Endogenous small molecule metabolites are cellular building blocks that regulate signalling molecules, especially receptors, and exert their biological effects through interactions with macromolecules such as proteins [17]. Based on metabolomics, numerous studies have identified the metabolites that are implicated in various diseases, such as cancer, immune disorders, cardiovascular disease (CVD) and chronic kidney disease (CKD) [17]. Recent studies have confirmed that a number of metabolites are ligands that mediate various diseases, such as cancer, immune disorders, CVD and CKD [18,19,20]. Among them, the cytoplasmic receptor aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates the activation of downstream target genes, such as cytochrome P450, family 1, member 1 A (CYP1A1); cytochrome P450, family 1, member 2 A (CYP1A2); cytochrome P450, family 1, subfamily B (CYP1B1) and cyclooxygenase-2 (COX-2) [21]. AhR was first identified as a mediator of the anti-toxic responses to ubiquitous environmental pollutants such as halogenated aromatic hydrocarbons, polycyclic aromatic hydrocarbons (PAHs) and coplanar polychlorinated biphenyls, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [21]. Recent evidence points to the role of ligand-activated AhR in the pathogenesis of various diseases, such as cancer, immune disorders, CVD and CKD [20,21,22]. Recently, increasing interest has been focused on a large number of AhR ligands whose chemical structures and physiochemical characteristics are different from those of classic TCDD ligands [18, 22,23,24]. A recent study demonstrated that endogenous 1-aminopyrene in the remnant kidney was a novel mediator of progressive CKD via AHR activation in a 5/6 nephrectomized rat model [18]. Upregulated mRNA expression of AhR and its downstream target genes, including CYP1A1, CYP1A2 and CYP1B1, was observed in kidney tissues of 1-aminopyrene-induced mice, and pathological analysis showed that 1-aminopyrene mediated renal fibrosis [18]. Despite strong evidence of AhR activation by endogenous PAHs such as 1-aminopyrene, the underlying mechanisms by which PAHs mediate AhR activation are poorly characterised, and it is not clear how to leverage these findings to develop renal disease-specific therapeutics. A number of compounds isolated from natural products, including flavonoids such as barleriside A, rhoifolin, 5,6,7,8,3′,4′-hexamethoxyflavone and 5,7,3,4′,5′-pentahydroxyflavanone, and lignins such as matairesinol could ameliorate renal fibrosis by inhibiting the AhR signalling pathway [18, 25]. Using a mouse model of unilateral ureteral obstruction (UUO), we first identified the endogenous metabolite 1-methoxypyrene (MP) by using ultra-performance liquid chromatography coupled with a high-definition mass spectrometry (UPLC-HDMS)-based untargeted metabolomics. We further demonstrated that MP mediated renal fibrosis through EMT and MMT by activating AhR signalling pathways and revealed the mechanism of MP-induced TIF. In addition, we screened and identified the AhR antagonist erythro-guaiacylglycerol-β-ferulic acid ether (GFA), which is isolated from Semen plantaginis and can slow TIF by inhibiting MP-induced AhR activity.
Materials and methods
Animals and experimental protocol
Male C57BL/6 mice (20–25 g) were purchased from the Central Animal Breeding House of Xi’an Jiaotong University (Xi’an, Shaanxi, China). Animal protocols were approved by the Northwest University Institutional Animal Care and Use Committee (No. SYXK2010–004). All procedures followed the ARRIVE Guidelines for Reporting Animal Research. The mice were randomised into two groups (n = 32/group): the sham and UUO groups. The UUO procedure was performed as previously described [26]. At Weeks 0, 1, 2 and 3, eight mice from the sham and UUO groups were selected randomly and then anaesthetised with 10% urethane. The obstructed kidneys were immediately removed and processed for subsequent analysis.
To study the effect of MP on AhR activity and ECM components, C57BL/6 mice were randomised into the control group and groups that were treated with 100, 200, 300 and 400 mg/kg MP at Week 8 (n = 6/group). C57BL/6 mice were randomised into two groups (n = 24/groups): the control and MP-treated groups. MP-treated group mice were given 300 mg/kg MP by oral gavage every day, while the control mice received the same volume of normal saline. Six mice were selected from each groups, randomly anaesthetised with 10% urethane, and sacrificed at Weeks 0, 4, 8 and 12. Serum and kidneys were collected for subsequent analysis.
GFA treatment was further used to investigate the effects on AhR activity and ECM components in MP-induced mice. Mice were randomised into six groups (n = 6–8/group): the control, MP and MP + GFA groups. GFA (5, 10, 20 and 40 mg/kg) was given to MP-induced mice by intragastric administration from the 9th week to the 12th week. All animals were provided access to water and food ad libitum. Control, MP and MP + GFA mice were sacrificed at Week 12. The optimal dose of GFA was chosen based on α smooth muscle actin (α-SMA) mRNA expression levels. Blood, serum and kidneys were collected for subsequent analysis.
Knockdown of AhR in mice
Lentivirus carrying shRNA against AhR and lentivirus containing nonspecific shRNA (scramble) were used. After anaesthesia and surgery, mice were laparotomized and injected with recombinant adenovirus vector. Then, 100 μL of saline or lentivirus cocktail (1 × 105 IU/µL) was injected into the kidney.
Cell culture
Three different cell types were used in the present study. The normal rat kidney proximal tubular epithelial cell line (NRK-52E), murine macrophage cell line (RAW 264.7) and normal rat kidney interstitial fibroblast line (NRK-49F) were cultured in DMEM-F12 supplemented with 10% foetal bovine serum (Gibco, Carlsbad, CA, USA) at 37 °C with 5% CO2. RAW 264.7 cells were cultured in RPMI (Gibco, Carlsbad, CA, USA) with 10% foetal bovine serum and 1% penicillin–streptomycin.
Cell viability analysis by CCK-8 assays
NRK-52E cells (1 × 104) were cultured in 96-well plates and treated with different concentrations of MP (0, 25, 50, 100, 200, 400 nM) for 24 h or 10 nM MP for different times (0, 6, 12, 24, 36, 48 h). In addition, NRK-52E cells were treated with different concentrations of GFA (0, 5, 10, 20, 40, 80, 160 nM) for 24 h or 10 μM GFA for different time points (0, 3, 6, 12, 24, 36, 48, 72 h) under the same conditions. A CCK-8 kit was used to measure cell viability based on the manufacturer’s protocol. The absorbance at 450 nm was determined by a microplate reader. The cell viability assay was repeated six times for each concentration.
Cell treatments
NRK-52E, RAW 264.7 and NRK-49F cells were stimulated with 5, 10 and 20 nM MP for 24 h or 10 nM MP for 6, 12 and 24 h. In addition, NRK-52E cells were stimulated with 10 nM MP for 24 h in the absence or presence of GFA (10 μM) and CH223191 (10 μM). The treated cells were collected for subsequent analysis.
Knockdown of AhR in NRK-52E, RAW 264.7 and NRK-49F cells
NRK-52E, RAW 264.7 and NRK-49F cells were transfected with 3 μL of 10 μM shRNA AhR or scramble per well using Lipofectamine RNAiMAX (Invitrogen, New York, USA). Cell samples were processed by quantitative reverse transcriptase-polymerase chain reaction (qRT–PCR) and Western blot analysis.
Sample preparation and UPLC-HDMS analysis for metabolomics
Intrarenal metabolites were examined using UPLC-HDMS based on untargeted metabolomics. The metabolomic procedures, including tissue preparation, sample separation, metabolite detection, data processing and statistical analysis for metabolite identification, were performed based on our previous protocols with minor modifications [27].
Model construction for metabolite selection
Data for positive and negative ion modes were analysed separately. LASSO-based variable selection was performed using the R package glmnetcr based on our recent publication [28]. The lambda values were chosen based on the Bayesian Information Criterion. All procedures were implemented in the package glmnetcr. Heatmap and unsupervised cluster analyses were conducted using the R package “heatmap3”. Support vector machine analysis was performed using the R package “e1071”.
Histological analysis
Masson’s trichrome staining was performed as previously described [28]. Immunohistochemical analysis of the obstructed kidney was performed as previously described [28]. After immunohistochemical analysis, Image-Pro Plus software version 6.0 was used to analyse the optical density of the images. Immunofluorescence staining for NRK-52E cells was performed as described previously [29].
RNA extraction and qRT–PCR analysis
Total RNA was extracted from the renal cortex or cells by using TRIzol reagent according to the manufacturer’s instructions (Roche, Germany). The qRT–PCR analysis was performed as previously described [26]. The specific primers are listed in Supplementary Table S1. The primers were designed based on their GenBank sequences and were synthesised by Sangon Biotech (Shanghai, China). The quantitative results were normalised by using the housekeeping gene β-actin as a reference, and the fold-increase over control values was analysed by the 2−ΔΔCt relative quantification method.
Western blot analysis
Western blot analysis was performed as previously described [26]. Primary antibodies against AhR, CD3, collagen I, α-SMA, fibronectin and E-cadherin were purchased from Abcam or Santa Cruz. The blots were visualised by using ECL reagent, and protein levels were normalised to GAPDH or α-tubulin expression. Specific bands of the target proteins were analysed using ImageJ 1.48 v software.
Stable reporter luciferase assay
AhR activity was examined in NRK-52E, RAW 264.7 and NRK-49F cells by using a promoter-reporter plasmid as described previously [29]. NRK-52E, RAW 264.7 and NRK-49F cells stably expressing Cignal Lenti Reporter XRE tethered to the luciferase reporter were seeded into 24-well plates and treated with MP with or without GFA and the AhR antagonist CH223191 for 24 h. The cells were lysed, and luciferase activity was determined using luciferase detection system substrate; the results are plotted relative to the protein concentration of the lysates according to the manufacturer’s instructions.
Molecular ligand docking and binding energy calculations
The chemical structures of potent metabolites were created with the ACD/ChemSketch tool, and energy minimisation of both target AhR and metabolites was optimised in the Molecular Operating Environment (MOE 2014.0 09). The PAS domain of AhR was the ligand-binding domain (LBD). The structures of several PAS domains are currently available in the Protein Data Bank, and the PAS domain structure of hypoxia-inducible factor 2R (HIF-2R) (PDB id: 1p97) was selected to establish a homology model of the mouse AhR-LBD according to its structural similarity with AhR in this region [30]. Docking analyses were performed using the program AutoDock 4.0 to determine the conformation and orientation of the ligand on the surface of HIF-2R. Each of the AhR LBDs was generated by using the homology model protocol as described previously [31]. Following the establishment of the model, the mouse AhR-LBD homology model was used to characterise the ligand-binding pocket. Then, this AhR-LBD homology model was used to structurally analyse the effects of potent metabolites on AhR. The lower equilibrium dissociation constant (Kd) was calculated.
Statistics analysis
The data are expressed as the mean ± SEM. Statistical analyses were performed with SPSS 22.0 software, R 2.15.0 and GraphPad Prism v6.0. A two-tailed unpaired Student’s t test was used for comparisons between the two groups. Statistically significant differences among more than two groups were analysed using one-way analysis of variance (ANOVA) followed by Dunnett’s post-hoc tests. P < 0.05 was considered significant. PCA was performed using SIMCA-P software to cluster the sample plots across different groups.
Results
UUO leads to impaired renal function and TIF
The obstructed kidney exhibited interstitial fibrosis, inflammatory cell infiltration, and tubular dilatation. As shown in Fig. 1a, b, UUO resulted in increased interstitial oedema, inflammatory cell infiltration, tubular dilation, tubular atrophy and epithelial cell necrosis at different stages. The tubulointerstitial collagen deposition score was determined by Masson’s trichrome staining, while the vimentin expression score was examined by immunohistochemical staining and showed similar results. Collagen deposition and vimentin expression (a myofibroblastic marker) in the obstructed kidney steadily increased during Weeks 1, 2 and 3. Compared to that in the sham group, the interstitial infiltration of CD3-positive lymphocytes steadily increased at Weeks 1, 2 and 3.

a Images of Masson’s trichrome staining and immunohistochemical staining of CD3 and vimentin expression at Weeks 1, 2 and 3. b The histograms refer to the relative expression of Masson’s trichrome staining, as well as vimentin and CD3 expression, in each group as indicated. c Visualisation of changes in the metabolic trajectories in UUO mice. PCA score plots of metabolic profiles for the different UUO groups at Weeks 1, 2 and 3 in positive ion mode. d Visualisation of changes in the metabolic trajectories in UUO mice. PCA score plots of metabolic profiles for the different UUO groups at Weeks 1, 2 and 3 in negative ion mode. e Heatmap of 53 metabolites in the sham and UUO groups at Weeks 1, 2 and 3. **P < 0.01 compared with the sham group.
Urinary tract obstruction alters the intrarenal metabolic profile
To determine the effect of urinary tract obstruction on intrarenal metabolites, UPLC-HDMS-based metabolomics was performed in positive and negative ion modes. A two-predictive component PCA was performed on the kidney tissue metabolites from UUO and sham mice at Weeks 1, 2 and 3. Unsupervised PCA score plots were divided into two clusters, indicating that the tissue metabolic pattern was significantly altered in obstructed kidneys (Supplementary Fig. S1a, b). The PCA score plots further indicated the good fitness and high predictability of this model at Weeks 1, 2 and 3 with high statistical R2 and Q2 values in both positive and negative ion modes (Supplementary SFig. 1a, b). The trajectories indicated that the UUO group (1st week) moved away from the sham group position along the t [1] axis from sham mice onwards, and there was a maximum shift by the 3rd week in both positive and negative ion modes (Fig. 1c, d). These changes demonstrate that UUO leads to significantly altered intrarenal metabolites.
Altered metabolites are associated with progressive TIF
To further identify the UUO-induced changes in intrarenal metabolites, variables were selected between the UUO (at weeks 1, 2 or 3) and sham groups according to the fold changes and statistical analysis. The metabolites were identified using our previously reported methods [32,33,34]. A total of 231 metabolites were identified based on MS, MSE fragment information and i-FIT values compared to the reported literature, database resources or authentic standards. Compared to those in the sham group, 53 of the 231 metabolites showed significant differences at all three time points in the UUO group (Table 1 and Supplementary Table S2). Except for docosahexaenoic acid, aspartic acid and taurine, 50 of the 53 metabolites were significantly increased at all three time points in UUO mice. As shown in Fig. 1e, heatmap analysis showed clear separation between the sham and UUO groups at the three time points. Furthermore, UUO caused the gradual increase in the levels of these metabolites from the sham to the 1st week in the UUO and the 3rd week in the UUO group, paralleling the progression of TIF. This finding indicates that progressive TIF is accompanied by the accumulation of intrarenal metabolites, and these metabolites are associated with progressive TIF. In addition, 178 of the 231 metabolites showed significant differences at least at one time point in the UUO group compared to the sham group (Supplementary Table S3).
Elevated MP levels in UUO mice and patients with tubulointerstitial nephropathy
Based on the association of bioactivity with chemical structure, we further classified the metabolites and found that the metabolites mainly had aromatic hydrocarbons, indoles, heterocycles and fatty acids. Therefore, these metabolites were classified as 60 aromatic hydrocarbons (2 PAHs and 58 one-benzene-ring-containing metabolites), 12 indoles and 15 heterocycles (Table 1 and Supplementary Tables S2, S3). The remaining 158 metabolites could not be classified with specific structures (Supplementary Tables S2, S3). We hypothesised that aryl hydrocarbon metabolites have a critical role in progressive TIF. Aryl hydrocarbon compounds, including aromatic hydrocarbons, indoles and polyheterocyclic compounds, are ligands of AhR, which is a ligand-activated basic helix-loop-helix transcription factor [35]. Interestingly, the relative intensities of 10 metabolites showed an increasing trend from the sham to UUO group at Week 3 (Supplementary Table S2). Furthermore, four metabolites (MP, indoxyl sulfate, 6-hydroxymelatonin and 5-methoxytryptamine) showed high fold change values and significant differences in obstructed kidney tissues and the serum of mice at the different time points (Fig. 2a–c). Taken together, these results suggest that the increased MP levels are associated with TIF.

a Relative intensities of four significantly altered metabolites in the obstructed kidney tissues of UUO mice at Weeks 1, 2 and 3. b Chemical structures of the four metabolites. c Relative intensities of the four significantly altered metabolites in the serum of UUO mice at Weeks 1, 2 and 3. **P < 0.01 compared with the sham group; ^P < 0.05, ^^P < 0.01 compared with the UUO group at Week 1; #P < 0.05, ##P < 0.01 compared with the UUO group at Week 2.
AhR activation and battery gene expression show a time-dependent association with progressive TIF
To investigate whether ureteral obstruction directly activates the AhR signalling pathway, the changes in AhR and its target gene mRNA expression were quantitated in sham and UUO mice at Weeks 1, 2 and 3. UUO mice showed time-dependent upregulation of AhR mRNA expression (Fig. 3a), which was accompanied by the upregulated expression of AhR target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in a time-dependent manner (Fig. 3a). A significant increase in AhR protein expression in the nucleus was accompanied by a significant decrease in the cytoplasm in a time-dependent manner (Fig. 3b, c). In addition, immunohistochemical staining showed nuclear translocation of AhR in the obstructed kidneys of UUO mice (Fig. 3d, e). These findings indicate that urinary tract obstruction leads to AhR signalling activation.

a Time course of the mRNA expression of AhR and its target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in the obstructed kidney tissues of UUO mice at Weeks 1, 2 and 3. b Protein expression of AhR in the nucleus and cytoplasm of the obstructed kidneys of UUO mice at Weeks 1, 2 and 3. c Quantitative analysis of AhR in the nucleus and cytoplasm of the obstructed kidneys of UUO mice at Weeks 1, 2 and 3. d Immunohistochemical analysis with anti-AhR in the obstructed kidneys of UUO mice. e Quantitative results of AhR expression in the obstructed kidneys of UUO mice. f Ramachandran plot of the modelled mouse AhR-LBD. g The predicted binding mode of the four metabolites and TCDD docked into AhR-LBD (grey ribbon form). Stick structures represent the amino acid residues of AhR-LBD. *P < 0.05, **P < 0.01 vs the sham group.
Docking analysis demonstrates AhR activation by aryl hydrocarbon metabolites in TIF
We next examined whether AhR could be activated by four endogenous metabolites. Molecular docking was performed to investigate the interactions between the four identified metabolites and AhR. The Ramachandran plot (88.1% in the favoured region) verified the viability of the established model (Fig. 3f). Molecular docking was performed to investigate the interactions between the four identified metabolites and the AhR model. According to cluster analysis of 100 docking runs, the minimum energy (−7.60, −8.60, −6.49, −6.16, and −5.52 kcal/mol) with the highest cluster (100, 100, 68, 39, and 53 times) of conformations was chosen as the best binding orientation of these metabolites in the PAS-B domain of murine AhR (Fig. 3g). The results revealed that the Ki values of TCDD, MP, indoxyl sulfate, 6-hydroxymelatonin and 5-methoxytryptamine were 2.67, 0.49, 17.37, 55.51 and 89.94 μM, respectively. MP had a lower Ki value than the other metabolites, suggesting that PAHs have stronger affinities with AhR than other metabolites. The indole derivatives produced by tryptophan metabolism, including 6-hydroxymelatonin, indoxyl sulfate and 5-methoxytryptamine, showed significant interactions with AhR.
Data on the docking interactions of the four metabolites with AhR are shown in Fig. 3g. MP docked in the active site of AhR was surrounded by hydrophobic residues (Pro291, Leu309, Phe318, Phe345, Leu347 and Val357) and charged residues (Lys312). Both indoxyl sulfate and 6-hydroxymelatonin interacted with hydrophobic residues (Phe289 and Pro291) and charged residues (Lys312), while 5-methoxytryptamine bound with the residues His285, Phe289, Ile292, Lys312 and His320. Collectively, these data revealed that all four metabolites could bind to the ligand-binding site of AhR, suggesting that AhR signalling could be activated by these metabolites.
Toxicity of MP on NRK-52E cells
Based on the P values and molecular docking results, the effect of MP was investigated in subsequent experiments. We first investigated the effect of MP on NRK-52E cell viability. Cell viability was reduced with increasing MP concentrations, and cell viability was significantly inhibited at 36 h (Fig. 4a).

a The viability of NRK-52E cells treated with increasing concentrations of MP (0–400 nM) for 24 h and 10 nM MP for different times (0–48 h). b AhR mRNA expression in NRK-52E cells after treatment with increasing concentrations of MP for 24 h and 10 nM MP for different times. c Protein expression of AhR in the nucleus and cytoplasm of NRK-52E cells induced by different concentrations of MP. d Quantitative analysis of AhR expression in NRK-52E cells induced by different concentrations of MP. e Protein expression of AhR in the nucleus and cytoplasm of NRK-52E cells induced by MP for different times. f Quantitative analysis of AhR expression in NRK-52E cells induced by MP for different times. g AhR mRNA expression in RAW 264.7 cells after treatment with increasing concentrations of MP for 24 h and 10 nM MP for different times. h Protein expression of AhR in the nucleus and cytoplasm of RAW 264.7 cells induced by different concentrations of MP. i Quantitative analysis of AhR expression in RAW 264.7 cells induced by different concentrations of MP. j Protein expression of AhR in the nucleus and cytoplasm of RAW 264.7 cells induced by MP for different times. k Quantitative analysis of AhR expression in RAW 264.7 cells induced by MP for different times. l AhR mRNA expression in NRK-49F cells after treatment with increasing concentrations of MP for 24 h and 10 nM MP for different times. m Protein expression of AhR in the nucleus and cytoplasm of NRK-49F cells induced by different concentrations of MP. n Quantitative analysis of AhR expression in NRK-49F cells induced by different concentrations of MP. o Protein expression of AhR in the nucleus and cytoplasm of NRK-49F cells induced by MP for different times. p Quantitative analysis of AhR expression in NRK-49F cells induced by MP for different times. q Luciferase assay of AhR activation in MP-induced NRK-52E cells. r Luciferase assay of AhR activation in MP-induced RAW 264.7 cells. s Luciferase assay of AhR activation in HP-induced NRK-49F cells. t The mRNA expression levels of AhR target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in MP-induced NRK-52E cells, RAW 264.7 cells and NRK-49F cells. *P < 0.05, **P < 0.01 vs the CTL group.
MP activated the AhR signalling pathway in TIF
We next examined whether MP activates AhR signalling in MP-induced NRK-52E, RAW 264.7 and NRK-49F cells. AhR mRNA expression was significantly increased in NRK-52E cells treated with different concentrations of MP for 24 h or with MP (10 nM) for different times (Fig. 4b). Nuclear AhR protein expression was significantly upregulated in NRK-52E cells treated with different concentrations of MP for 24 h, while cytoplasmic AhR protein expression was significantly downregulated, indicating the activation of AhR signalling (Fig. 4c, d). Similarly, AhR signalling was activated by stimulation with 10 nM MP for 6, 12 and 24 h (Fig. 4e, f). Consistent with those in MP-induced NRK-52E cells, significantly upregulated AhR mRNA expression, significantly upregulated nuclear AhR protein expression and downregulated cytoplasmic AhR protein expression were observed in MP-induced RAW 264.7 cells (Fig. 4g–k) and NRK-49F cells (Fig. 4l–p). The luciferase assay showed that MP significantly enhanced AhR-driven reporter activity in NRK-52E, RAW 264.7 and NRK-49F cells (Fig. 4q–s). Our findings further demonstrate that AhR target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, are significantly upregulated in MP-induced NRK-52E, RAW 264.7 and NRK-49F cells (Fig. 4t). To further investigate whether MP specifically activates the AhR signalling pathway, we then depleted AhR mRNA in NRK-52E cells with small hairpin RNA (shRNA), which significantly downregulated AhR mRNA expression at 24 h (Fig. 5a). The mRNA expression of CYP1A1, CYP1A2, CYP1B1 and COX-2 was significantly reduced in AhR shRNA-treated NRK-52E cells stimulated with MP (Fig. 5b). Similar results were also observed in MP-induced RAW 264.7 and NRK-49F cells (Fig. 5c–f). These results indicated that MP activates the AhR signalling pathway. We hypothesised that MP might be a crucial component of cell–cell and cell-microenvironment interactions.

a The mRNA expression of AhR in NRK-52E cells transfected with CTL shRNA or AhR shRNA. b The mRNA expression of CYP1A1, CYP1A2, CYP1B1 and COX-2 in MP-induced NRK-52E cells treated with AhR shRNA. c The mRNA expression of AhR in RAW 264.7 cells transfected with CTL shRNA or AhR shRNA. d The mRNA expression of CYP1A1, CYP1A2, CYP1B1 and COX-2 in MP-induced RAW 264.7 cells treated with AhR shRNA. e The mRNA expression of AhR in NRK-49F cells transfected with CTL shRNA or AhR shRNA. f The mRNA expression of CYP1A1, CYP1A2, CYP1B1 and COX-2 in MP-induced NRK-49F cells treated with AhR shRNA. **P < 0.01 vs the CTL, scramble shRNA or scramble group; ##P < 0.01 vs the scramble group treated with MP.
MP leads to ECM accumulation through EMT and MMT in TIF
To further understand whether MP leads to ECM component accumulation through EMT and MMT, we measured the protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-52E, RAW 264.7 and NRK-49F cells. MP upregulated the protein expression of collagen I, α-SMA, and fibronectin and reduced E-cadherin protein expression in NRK-52E cells (Fig. 6a, b). Similar results were observed in MP-induced RAW 264.7 (Fig. 6c, d) and NRK-49F cells (Fig. 6e, f). To further investigate whether MP leads to ECM component accumulation through EMT and MMT by selectively activating the AhR signalling pathway, cells were transfected with AhR shRNA. Compared to those in MP-induced NRK-52E cells, the upregulated protein expression of collagen I, α-SMA, fibronectin and downregulated E-cadherin protein expression were significantly attenuated in MP-induced NRK-52E cells treated with AhR shRNA (Fig. 6g, h). In addition, similar results were observed in MP-induced NRK-52E cells treated with CH223191, a potent and specific AHR antagonist (Fig. 6g, h). Furthermore, the upregulation of ECM component expression was significantly attenuated in MP-induced RAW 264.7 and NRK-49F cells treated with AhR shRNA or CH223191 compared to that in MP-induced RAW 264.7 or NRK-49F cells (Fig. 6i–l). Taken together, these results indicate that MP mediates renal fibrosis through EMT and MMT via activation of the AhR signalling pathway.

a Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-52E cells. b Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-52E cells. c Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced RAW 264.7 cells. d Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced RAW 264.7 cells. e Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-49F cells. f Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-49F cells. g Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-52E cells treated with AhR shRNA or CH223191. h Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-52E cells treated with AhR shRNA or CH223191. i Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced RAW 264.7 cells treated with AhR shRNA or CH223191. j Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced RAW 264.7 cells treated with AhR shRNA or CH223191. k Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-49F cells treated with AhR shRNA or CH223191. l Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-49F cells treated with AhR shRNA or CH223191. *P < 0.05, **P < 0.01 vs the CTL group; #P < 0.05, ##P < 0.01 vs the MP-induced group.
MP mediated renal fibrosis through EMT and MMT by activating the AhR signalling pathway in mice
To further understand whether MP mediates TIF by activating AhR signalling, we examined AhR signalling and ECM components in MP-induced mice. Increased intrarenal α-SMA mRNA expression levels were observed in kidney tissues of mice treated with different doses of MP at Week 8 (Fig. 7a). Therefore, 300 mg/kg was used for further experiments. Increased intrarenal AhR mRNA expression levels were accompanied by the upregulated expression of AhR target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in MP-induced mice at Weeks 4, 8 and 12 (Fig. 7b). Furthermore, MP significantly upregulated nuclear AhR protein levels and downregulated cytoplasmic AhR protein levels at Weeks 4, 8 and 12 (Fig. 7c, d). Immunohistochemical staining further demonstrated significantly increased nuclear AhR protein expression in intrarenal cells, such as renal tubular epithelial cells, macrophages, myofibroblasts and fibroblasts, confirming increased nuclear translocation of AhR in MP-induced mice at Week 12 (Fig. 7e, f). Collectively, these results indicated that MP activated the AhR signalling pathway in vivo.

a α-SMA mRNA expression levels in the kidney tissues of control mice and mice treated with different doses of MP at Week 8. b The mRNA expression levels of AhR and its target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in the kidney tissues of control and MP-induced mice at Weeks 4, 8, and 12. c AhR protein expression in the nucleus and cytoplasm of kidney tissues from control and MP-induced mice at Weeks 4, 8, and 12. d Quantitative analysis of AhR expression in the nucleus and cytoplasm of kidney tissues from control and MP-induced mice at Weeks 4, 8, and 12. e Immunohistochemical analysis with anti-AhR of kidney tissues from control and MP-induced mice at Week 12. f Quantitative immunohistochemical analysis with anti-AhR of kidney tissues from control and MP-induced mice at Week 12. g The levels of serum creatinine and urea in control and MP-induced mice at Week 12. h Images of Masson’s trichrome staining of kidney tissues from control and MP-induced mice at Week 12. i Quantitative analysis of renal fibrosis as shown by Masson’s trichrome staining of kidney tissues in control and MP-induced mice at Week 12. j Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in kidney tissues from control and MP-induced mice at Week 12. k Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in kidney tissues from control and MP-induced mice at Week 12. l AhR mRNA expression levels in AhR shRNA-treated mice. m The mRNA expression levels of AhR target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in AhR shRNA-treated mice induced with MP. n Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced mice treated with AhR shRNA. o Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced mice treated with AhR shRNA. *P < 0.05, **P < 0.01 vs the CTL or CTL Scramble group; ##P < 0.01 vs the MP or MP Scramble group.
To investigate whether MP induces TIF in vivo, renal function and ECM components were examined in MP-induced mice at Week 12. MP significantly increased the levels of serum creatinine and urea in mice (Fig. 7g). Pathological analysis showed that MP mediated renal fibrosis (Fig. 7h, i). Further analysis showed that MP significantly upregulated the protein expression of collagen I, α-SMA and fibronectin and preserved E-cadherin protein expression in mice (Fig. 7j, k). These results suggest that ECM deposition and fibrosis determine the MP-mediated decline in renal function. To further investigate whether MP mediates TIF via the AhR signalling pathway, mice were transfected with AhR shRNA. Compared to control shRNA, AhR shRNA significantly decreased AhR mRNA expression in mice (Fig. 7l). The mRNA expression levels of CYP1A1, CYP1A2, CYP1B1 and COX-2 were significantly decreased in AhR shRNA-treated mice induced by MP compared to mice induced with MP alone (Fig. 7m). This effect was accompanied by significant downregulation of the protein expression of collagen I, α-SMA and fibronectin and the maintenance of protein expression in mice (Fig. 7n, o). These results indicate that MP induces ECM component accumulation that leads to TIF. Overall, we conclude that MP mediates ECM component accumulation by activating the AhR signalling pathway in vivo. An in vivo study indicated that MP might be a crucial component of the cell–cell and cell-microenvironment interactions in TIF. We successfully established a mouse model of CKD by using MP, which provides new insights into the pathogenesis and molecular mechanisms of renal fibrosis.
The screening and identification of the natural AhR antagonist from Plantaginis semen
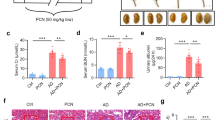
Natural products have been extensively used to treat CKD and have been demonstrated to protect against renal fibrosis [36,37,38]. Increasing evidence has demonstrated the antagonistic or agonistic effects of many natural products on AhR signalling [23]. To verify and evaluate whether the MP-induced CKD model could be used to screen new antifibrotic drugs, 33 isolated compounds from Plantaginis semen, which is used to treat patients with oedema, oliguria and urinary dysfunction, were examined in vitro and in vivo. Recent studies have shown that Plantaginis semen can attenuate hyperuricaemia, gouty nephropathy and diabetic retinal injury and enhance immune responses [39,40,41,42]. GFA (Fig. 8a) was identified as an AhR antagonist and could exhibit antioxidant and antibacterial activities [43]. To assess the effect of GFA on cell viability, NRK-52E cells were exposed to different concentrations of GFA for different times. The results showed that exposure to GFA did not alter proliferation or have cytotoxic effects on NRK-52E cells (Fig. 8b). Therefore, we used 10 μM for further in vitro experiments. Different doses of GFA were orally administered to MP-induced mice. GFA significantly inhibited the upregulation of α-SMA mRNA expression in the kidney tissues of MP-induced mice at Week 12 (Fig. 8c). The 20 mg/kg dose showed a strong inhibitory effect on α-SMA mRNA expression compared to 10 mg/kg. However, the effect of 20 mg/kg was similar to that of 40 mg/kg. Therefore, 20 mg/kg was used for further in vivo experiments.

a Structure of GFA. b The viability of NRK-52E cells after treatment with increasing concentrations of GFA (0–160 µM) for 24 h and 20 µM for different times (0–72 h). c The effects of four doses of GFA on the mRNA expression of α-SMA in MP-induced mice at Week 12. d The mRNA expression levels of AhR target genes, including CYP1A1, CYP1A2, CYP1B1 and COX-2, in the kidney tissues in the different groups as indicated at Week 12. e AhR protein expression in the nucleus and cytoplasm of kidney tissues from the different groups at Week 12. f Quantitative analysis of AhR expression in the nucleus and cytoplasm of kidney tissues from the different groups at Week 12. g The levels of serum creatinine and urea in the different groups at Week 12. h Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in the kidney tissues in the different groups at Week 12. i Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin in the kidney tissues in the different groups at Week 12. j Luciferase assay showing AhR activation in MP-induced NRK-52E cells treated with GFA or CH223191. k Representative immunofluorescent staining of MP-induced NRK-52E cells treated with GFA or CH223191. l Protein expression of collagen I, α-SMA, fibronectin and E-cadherin in MP-induced NRK-52E cells treated with GFA. m Quantitative analysis of collagen I, α-SMA, fibronectin and E-cadherin MP-induced NRK-52E cells treated with GFA. *P < 0.05, **P < 0.01 vs the CTL group (n = 6); #P < 0.05, ##P < 0.01 vs the MP group.
GFA treatment significantly inhibited the upregulated mRNA expression of AhR and its four target genes in the kidney tissues of MP-induced mice at Week 12 (Fig. 8d). Furthermore, GFA treatment significantly inhibited the upregulated nuclear expression of AhR protein and downregulated cytoplasmic protein expression of AhR in the kidney tissues of MP-induced mice (Fig. 8e, f). GFA significantly lowered the levels of serum creatinine and urea and significantly increased blood haematocrit levels in MP-induced mice at Week 12 (Fig. 8g), indicating that GFA treatment improved renal function. Further analysis showed that GFA inhibited ECM component accumulation, ameliorating TIF (Fig. 8h, i). Mechanistically, MP significantly enhanced AhR-driven reporter activity, and this effect was suppressed by GFA, indicating that GFA directly targets AhR activity (Fig. 8j). Immunofluorescence staining further showed that GFA significantly inhibited the increased nuclear translocation of AhR in MP-induced NRK-52E cells (Fig. 8k). In vitro experiments confirmed that GFA inhibited the accumulation of ECM components, including collagen I, α-SMA and fibronectin, in MP-induced NRK-52E cells and improved TIF damage (Fig. 8l, m). Taken together, these findings demonstrate that GFA attenuates TIF by antagonising MP-induced activation of the AhR signalling pathway.
Discussion
TIF is the final manifestation of the progression of CKD to end-stage renal disease and requires renal replacement treatments such as dialysis and transplantation [44,45,46,47]. In the current study, MP was successfully identified as a crucial component in the contribution of TIF to renal fibrosis via the regulation of cell–cell and cell-microenvironment interactions (Fig. 9). We established a new and simple mouse model of CKD by using MP, which provided new insights into the pathogenesis of CKD and the molecular mechanisms of renal fibrosis in the clinical setting. To validate this model, we examined the natural AhR antagonist GFA, which attenuates renal fibrosis by regulating the AhR signalling pathway. This model could provide a promising therapeutic strategy for the screening and development of new antifibrotic drugs in animal research.

MP activated the AhR signalling pathway in kidney proximal tubular epithelial cells, macrophages and kidney interstitial fibroblasts, as well as in mice. MP mediated renal fibrosis through EMT and MMT by activating the AhR signalling pathway.
ECM components are secreted into the tubulointerstitial matrix that supports and surrounds the different types of cells [48]. ECM components are the major providers of structural scaffolding for certain tissues and are composed of collagen I and fibronectin, providing mechanical strength to tissues [49,50,51]. Increasing evidence indicates that intrarenal ECM components possess many biological functions in addition to providing structural support [52, 53]. An early study suggested that the ECM microenvironment regulated the migratory behaviour of activated renal tubular epithelial cells [11]. The biophysical features of the matrix regulate various profibrotic responses. In addition, ECM components also sequester and act as a reservoir for a wide variety of cytokines, chemokines and growth factors [50, 54, 55] that participate in TIF. Although a number of experiments have been performed on the intrarenal ECM microenvironment, these studies are still in their infancy compared with studies of the tumour microenvironment [56]. Several proteins or enzymes have been shown to be crucial molecules in the microenvironment that contribute to renal injury and TIF [57,58,59]. For example, a recent study demonstrated that glomerular testican-2 was expressed in human kidneys and that testican-2 increased glomerular endothelial tube formation and motility, raising the possibility that its secretion has a functional role in the glomerulus [58]. In addition, intrarenal interleukin-15 acts through interleukin-15 receptor subunit α (CD132) as an epithelial survival factor. A previous study showed that interleukin-15 was a main regulator of cell-microenvironment interactions in human renal homeostasis [4]. Interleukin-15 preserved E-cadherin expression and suppressed EMT in renal epithelial tubular cells with interleukin-15 receptor subunit γ+/+ [4]. A recent study suggested that matrix metalloproteinase-9-positive neutrophils were essential for the profibrotic microenvironment in the obstructed kidney in UUO mice [5]. In contrast, perivascular CD73+ cells ameliorated inflammation and TIF by modulating the renal microenvironment [60]. Taken together, these findings suggest that the renal microenvironment contributes to renal injury in both acute and CKDs.
CKD is associated with an increased risk of death, especially in CKD patients with CVD. Classic risk factors, such as high blood pressure, diabetes mellitus, obesity, hypercholesterolemia and tobacco use, cannot explain the increased rate of cardiovascular events in CKD [61, 62]. Recent studies suggest that chronic and persistent fibrosis in the renal interstitium is more complicated and dynamic than previously thought [5]. Although the specific pathophysiological mechanisms of TIF are not yet well known, the tubulointerstitial microenvironment has become a focus for understanding progressive TIF, similar to the understanding of the effects of the tumour microenvironment on tumour development and progression. Bidirectional communication between a variety of genes, proteins and endogenous metabolites in the kidney orchestrates cellular responses to both normal physiological and pathological conditions [63]. In the last several decades, considerable research has focused on understanding the aberrant expression of many genes and proteins in TIF [64]. In contrast, few studies have demonstrated that the dysregulation of endogenous metabolites plays a critical role in progressive TIF. Purinergic signalling by 5′-ectonucleotidase (CD73), which can convert adenosine monophosphate to adenosine on the extracellular surface, inhibits inflammation. Recently, a study demonstrated that CD73 in renal perivascular cells inhibited myofibroblast transformation and influenced macrophages to promote wound healing [60]. The purinergic signalling pathway in the renal microenvironment orchestrates perivascular cells and macrophages to inhibit inflammation and prevent progressive TIF [60].
In recent decades, metabolomic techniques have been extensively used to identify the dysregulation of metabolites and uncover the biochemical mechanism in various diseases, including renal disease [65]. Based on biological samples such as serum, urine, tissues and faeces, as well as cells, an increasing number of studies have identified many metabolites that are implicated in CKD [66]. However, the normal biological functions and pathogenesis of these metabolites remain poorly understood, and there is no effective therapy. In the current study, we identified that the metabolite MP is associated with progressive TIF (weeks 1, 2 and 3) in mice with UUO, a typical model of renal interstitial fibrosis, based on UPLC-HDMS-based untargeted metabolomics. Our study demonstrates that MP mediates ECM component accumulation through EMT and MMT via activation of the AhR signalling pathway in MP-induced NRK-52E, RAW 264.7 and NRK-49F cells. Consistent with this conclusion, MP leads to ECM component accumulation and TIF via activation of the AhR signalling pathway in mice. These findings of disease-associated metabolite patterns suggest that MP may be a critical component in the microenvironment that contributes to TIF. The dysregulation of MP holds the promise for uncovering novel targets to prevent renal fibrosis in the clinical setting.
Pyrene-derived compounds such as MP, 1-aminopyrene, 1-nitropyrene and 1-hydroxypyrene, which are PAHs, are some of the most important metabolites. A previous study demonstrated that MP induced CYP1B1 mRNA expression in the kidney tissues of rats [67], which was also verified in vitro [68]. In addition, it has been reported that 1-aminopyrene leads to extensive oxidative damage and enhances the expression of proinflammatory cytokines [69], which is consistent with the increased COX-2 mRNA expression observed in our study. Our previous study identified that the increased 1-aminopyrene levels and activated AhR signalling pathway were involved in progressive CKD by using a 5/6 nephrectomized rat model and CKD patients [18]. Furthermore, we demonstrated that 1-aminopyrene upregulated the mRNA expression of AhR and its target genes, including CYP1A1, CYP1A1 and CYP1B1, which were accompanied by the upregulated nuclear expression of AhR protein and downregulated cytoplasmic expression of AhR protein in both NRK-52E cells and mice [18]. Recent studies revealed that 1-hydroxypyrene levels were positively associated with urinary 8-hydroxy-20-deoxyguanosine (a biomarker of oxidative damage) levels and blood AhR expression [70, 71]. In addition, it has been reported that urinary 1-hydroxypyrene levels are related to the levels of catalase, malondialdehyde and β-carotene in postinfarct patients [72]. Thus, the current work and that of others suggests that pyrene-derived metabolites are important mediators of AhR signalling that lead to inflammation and renal injury in kidney disease. These data expand the commonly known and prominent role of pyrene-derived metabolites in the renal microenvironment and homeostasis.
Many known ligands of AhR are man-made, and natural ligands of AhR have remained elusive. Earlier studies have identified the plant-derived dietary compounds curcumin and indolo [2, 3] carbazole as AhR ligands [19, 21, 73]. Furthermore, a number of studies have demonstrated that natural products, such as resveratrol, epigallocatechin gallate, luteolin and myricetin, are AhR antagonists [23]. We demonstrated that GFA treatment attenuated renal fibrosis by regulating the AhR signalling pathway after MP stimulation in vitro and in vivo. This result further confirms that an established MP-induced mouse model may be used to screen new antifibrotic drugs. Overall, this proof-of-principle study provides novel insights into the pathogenesis of CKD and metabolic mechanisms underlying TIF by correlating phenotype and direct metabolite-protein (small molecule-macromolecule) interaction data.
References
Grisaru-Tal S, Itan M, Klion AD, Munitz A. A new dawn for eosinophils in the tumour microenvironment. Nat Rev Cancer. 2020;20:594–607.
Cruz NM, Song X, Czerniecki SM, Gulieva RE, Churchill AJ, Kim YK, et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater. 2017;16:1112–9.
Späth MR, Bartram MP, Palacio-Escat N, Hoyer KJR, Debes C, Demir F, et al. The proteome microenvironment determines the protective effect of preconditioning in cisplatin-induced acute kidney injury. Kidney Int. 2019;95:333–49.
Giron-Michel J, Azzi S, Ferrini S, Chouaib S, Camussi G, Eid P, et al. Interleukin-15 is a major regulator of the cell-microenvironment interactions in human renal homeostasis. Cytokine Growth Factor Rev. 2013;24:13–22.
Wang H, Gao M, Li J, Sun J, Wu R, Han D, et al. MMP-9-positive neutrophils are essential for establishing profibrotic microenvironment in the obstructed kidney of UUO mice. Acta Physiol (Oxf). 2019;227:e13317.
Yu XY, Sun Q, Zhang YM, Zou L, Zhao YY. TGF-β/Smad signaling pathway in tubulointerstitial fibrosis [Systematic Review]. Front Pharmacol. 2022;13:860588.
Bhargava V, Singh K, Meena P, Sanyal R. Nephrogenic systemic fibrosis: A frivolous entity. World J Nephrol. 2021;10:29–36.
Mantovani A, Chiara Z. PNPLA3 gene and kidney disease. Explor Med. 2020;1:42–50.
Hansrivijit P, Chen YJ, Lnu K, Trongtorsak A, Puthenpura MM, Thongprayoon C, et al. Prediction of mortality among patients with chronic kidney disease: A systematic review. World J Nephrol. 2021;10:59–75.
Nakamura J, Sato Y, Kitai Y, Wajima S, Yamamoto S, Oguchi A, et al. Myofibroblasts acquire retinoic acid-producing ability during fibroblast-to-myofibroblast transition following kidney injury. Kidney Int. 2019;95:526–39.
Zeisberg M, Maeshima Y, Mosterman B, Kalluri R. Extracellular matrix microenvironment regulates migratory behavior of activated tubular epithelial cells. Am J Pathol. 2002;160:2001–8.
Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol. 2020;16:269–88.
Feng YL, Chen DQ, Vaziri ND, Guo Y, Zhao YY. Small molecule inhibitors of epithelial-mesenchymal transition for the treatment of cancer and fibrosis. Med Res Rev. 2020;40:54–78.
Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93:568–79.
Tang PM, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019;15:144–58.
Tang PM, Zhou S, Li CJ, Liao J, Xiao J, Wang QM, et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018;93:173–87.
Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Disco. 2020;19:57–75.
Miao H, Cao G, Wu XQ, Chen YY, Chen DQ, Chen L, et al. Identification of endogenous 1-aminopyrene as a novel mediator of progressive chronic kidney disease via aryl hydrocarbon receptor activation. Br J Pharmacol. 2020;177:3415–35.
Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. 2020;117:19376–87.
Zhao H, Chen L, Yang T, Feng YL, Vaziri ND, Liu BL, et al. Aryl hydrocarbon receptor activation mediates kidney disease and renal cell carcinoma. J Transl Med. 2019;17:302.
Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019;19:184–97.
Baker JR, Sakoff JA, McCluskey A. The aryl hydrocarbon receptor (AhR) as a breast cancer drug target. Med Res Rev. 2020;40:972–1001.
Xue Z, Li D, Yu W, Zhang Q, Hou X, He Y, et al. Mechanisms and therapeutic prospects of polyphenols as modulators of the aryl hydrocarbon receptor. Food Funct. 2017;8:1414–37.
Murray IA, Patterson AD, Perdew GH. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat Rev Cancer. 2014;14:801–14.
Miao H, Wu XQ, Wang YN, Chen DQ, Chen L, Vaziri ND, et al. 1-Hydroxypyrene mediates renal fibrosis through aryl hydrocarbon receptor signalling pathway. Br J Pharmacol. 2022;179:103–24.
Wang M, Chen DQ, Chen L, Cao G, Zhao H, Liu D, et al. Novel inhibitors of the cellular renin-angiotensin system components, poricoic acids, target Smad3 phosphorylation and Wnt/β-catenin pathway against renal fibrosis. Br J Pharmacol. 2018;175:2689–708.
Zhao YY, Cheng XL, Wei F, Bai X, Tan XJ, Lin RC, et al. Intrarenal metabolomic investigation of chronic kidney disease and its TGF-β1 mechanism in induced-adenine rats using UPLC Q-TOF/HSMS/MSE. J Proteome Res. 2013;12:2692–703.
Chen DQ, Cao G, Chen H, Argyopoulos CP, Yu H, Su W, et al. Identification of serum metabolites associating with chronic kidney disease progression and anti-fibrotic effect of 5-methoxytryptophan. Nat Commun. 2019;10:1476.
Chen H, Yang T, Wang MC, Chen DQ, Yang Y, Zhao YY. Novel RAS inhibitor 25-O-methylalisol F attenuates epithelial-to-mesenchymal transition and tubulo-interstitial fibrosis by selectively inhibiting TGF-β-mediated Smad3 phosphorylation. Phytomedicine. 2018;42:207–18.
Bisson WH, Koch DC, O’Donnell EF, Khalil SM, Kerkvliet NI, Tanguay RL, et al. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem. 2009;52:5635–41.
Ginalski K. Comparative modeling for protein structure prediction. Curr Opin Struct Biol. 2006;16:172–7.
Zhao YY, Liu J, Cheng XL, Bai X, Lin RC. Urinary metabonomics study on biochemical changes in an experimental model of chronic renal failure by adenine based on UPLC Q-TOF/MS. Clin Chim Acta. 2012;413:642–49.
Zhao YY, Cheng XL, Cui JH, Yan XR, Wei F, Bai X, et al. Effect of ergosta-4,6,8(14),22-tetraen-3-one (ergone) on adenine-induced chronic renal failure rat: a serum metabonomic study based on ultra performance liquid chromatography/high-sensitivity mass spectrometry coupled with MassLynx i-FIT algorithm. Clin Chim Acta. 2012;413:1438–45.
Zhao YY, Tang DD, Chen H, Mao JR, Bai X, Cheng XH, et al. Urinary metabolomics and biomarkers of aristolochic acid nephrotoxicity by UPLC-QTOF/HDMS. Bioanalysis. 2015;7:685–700.
Shinde R, McGaha TL. The aryl hydrocarbon receptor: connecting immunity to the microenvironment. Trends Immunol. 2018;39:1005–20.
Luo LP, Suo P, Ren LL, Liu HJ, Zhang Y, Zhao YY. Shenkang injection and its three anthraquinones ameliorates renal fibrosis by simultaneous targeting IƙB/NF-ƙB and Keap1/Nrf2 signaling pathways. Front Pharmacol. 2021;12:800522.
Yang Y, Wu C. Traditional chinese medicine in ameliorating diabetic kidney disease via modulating gut microbiota. Integr Med Nephrol Androl. 2021;8:8.
Zhou XF, Wang Y, Luo MJ, Zhao TTPL. Tangshen formula attenuates renal fibrosis by downregulating transforming growth factor β1/Smad3 and LncRNA-MEG3 in rats with diabetic kidney disease. Integr Med Nephrol Androl. 2021;8:1.
Zhao H, Xu J, Wang R, Tang W, Kong L, Wang W, et al. Plantaginis Semen polysaccharides ameliorate renal damage through regulating NLRP3 inflammasome in gouty nephropathy rats. Food Funct. 2021;12:2543–53.
Yang F, Shi W, Wang L, Qin N, Wang C, Guo Y, et al. Lipidomics study of the therapeutic mechanism of Plantaginis Semen in potassium oxonate-induced hyperuricemia rat. BMC Complement Med Ther. 2021;21:175.
Tzeng TF, Liu WY, Liou SS, Hong TY, Liu IM. Antioxidant-rich extract from Plantaginis semen ameliorates diabetic retinal injury in a streptozotocin-induced diabetic rat model. Nutrients 2016;8:572.
Ren Z, Yu R, Meng Z, Sun M, Huang Y, Xu T, et al. Spiky titanium dioxide nanoparticles-loaded Plantaginis Semen polysaccharide as an adjuvant to enhance immune responses. Int J Biol Macromol. 2021;191:1096–104.
Sharief Nohammad N, Srinivasulu A, Chittibabu B, Umamaheswara Rao V. Isolation and purification of antibacterial principle from Avicennia marina L in methanol. Int J Pharm Pharm Sci. 2014;7:38–41.
Sawhney H, Gill SS. Renal transplant recipient seizure practical management. World J Nephrol. 2020;9:1–8.
Carta P, Lorenzo DM, Luciano M, Aida L, Caroti L, Cirami L. Malignancies after renal transplantation: A single center retrospective study. J Nephrol Hypertens. 2020;3:1009.
Bacharaki D, Chrysanthopoulou E, Grigoropoulou S, Giannakopoulos P, Simitsis P, Frantzeskaki F, et al. Siblings with coronavirus disease 2019 infection and opposite outcome-the hemodialysis’s better outcome paradox: Two case reports. World J Nephrol. 2021;10:21–8.
Chuengsaman P, Narenpitak S, Sritippayawan S. Efficacy and safety of recombinant human erythropoietin (Hema-Plus(®)) for management of anemia in Thai patients on peritoneal dialysis. World J Nephrol. 2021;10:109–21.
Tamura H. Trends in pediatric nephrotic syndrome. World J Nephrol. 2021;10:88–100.
Chaudhuri O, Cooper-White J, Janmey PA, Mooney DJ, Shenoy VB. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature. 2020;584:535–46.
Romani P, Brian I, Santinon G, Pocaterra A, Audano M, Pedretti S, et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat Cell Biol. 2019;21:338–47.
Li SS, Sun Q, Hua MR, Suo P, Chen JR, Yu XY, et al. Targeting the Wnt/β-catenin signaling pathway as a potential therapeutic strategy in renal tubulointerstitial fibrosis. Front Pharmacol. 2021;12:719880.
Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, et al. Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell. 2020;182:388–403.e15.
Piperigkou Z, Karamanos NK. Dynamic interplay between mirnas and the extracellular matrix influences the tumor microenvironment. Trends Biochem Sci. 2019;44:1076–88.
Deng M, Lin J, Nowsheen S, Liu T, Zhao Y, Villalta PW, et al. Extracellular matrix stiffness determines DNA repair efficiency and cellular sensitivity to genotoxic agents. Sci Adv. 2020;6:eabb2630.
Merchant ML, Barati MT, Caster DJ, Hata JL, Hobeika L, Coventry S, et al. Proteomic analysis identifies distinct glomerular extracellular matrix in collapsing focal segmental glomerulosclerosis. J Am Soc Nephrol. 2020;31:1883–904.
Mayeux PR, MacMillan-Crow LA. Pharmacological targets in the renal peritubular microenvironment: implications for therapy for sepsis-induced acute kidney injury. Pharmacol Ther. 2012;134:139–55.
Rinschen MM, Saez-Rodriguez J. The tissue proteome in the multi-omic landscape of kidney disease. Nat Rev Nephrol. 2021;17:205–19.
Ngo D, Wen D, Gao Y, Keyes MJ, Drury ER, Katz DH, et al. Circulating testican-2 is a podocyte-derived marker of kidney health. Proc Natl Acad Sci USA. 2020;117:25026–35.
Schunk SJ, Floege J, Fliser D, Speer T. WNT-β-catenin signalling - a versatile player in kidney injury and repair. Nat Rev Nephrol. 2020;17:172–84.
Perry HM, Görldt N, Sung SJ, Huang L, Rudnicka KP, Encarnacion IM, et al. Perivascular CD73+ cells attenuate inflammation and interstitial fibrosis in the kidney microenvironment. Am J Physiol Ren Physiol. 2019;317:F658–F69.
Sallee M, Dou L, Cerini C, Poitevin S, Brunet P, Burtey S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: a new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014;6:934–49.
Dehlin M, Jacobsson L, Roddy E. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol. 2020;16:380–90.
Fang Y, Gong AY, Haller ST, Dworkin LD, Liu Z, Gong R. The ageing kidney: Molecular mechanisms and clinical implications. Ageing Res Rev. 2020;63:101151.
Edeling M, Ragi G, Huang S, Pavenstadt H, Susztak K. Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nat Rev Nephrol. 2016;12:426–39.
Rinschen MM, Ivanisevic J, Giera M, Siuzdak G. Identification of bioactive metabolites using activity metabolomics. Nat Rev Mol Cell Biol. 2019;20:353–67.
Hocher B, Adamski J. Metabolomics for clinical use and research in chronic kidney disease. Nat Rev Nephrol. 2017;13:269–84.
Hatanaka N, Yamazaki H, Kizu R, Hayakawa K, Aoki Y, Iwanari M, et al. Induction of cytochrome P450 1B1 in lung, liver and kidney of rats exposed to diesel exhaust. Carcinogenesis. 2001;22:2033–8.
Iwanari M, Nakajima M, Kizu R, Hayakawa K, Yokoi T. Induction of CYP1A1, CYP1A2, and CYP1B1 mRNAs by nitropolycyclic aromatic hydrocarbons in various human tissue-derived cells: chemical-, cytochrome P450 isoform-, and cell-specific differences. Arch Toxicol. 2002;76:287–98.
Wu M, Jiang Y, Liu M, Shang Y, An J. Amino-PAHs activated Nrf2/ARE anti-oxidative defense system and promoted inflammatory responses: the regulation of PI3K/Akt pathway. Toxicol Res (Camb). 2018;7:465–72.
Liu Y, Zhang H, Zhang H, Niu Y, Fu Y, Nie J, et al. Mediation effect of AhR expression between polycyclic aromatic hydrocarbons exposure and oxidative DNA damage among Chinese occupational workers. Environ Pollut. 2018;243:972–77.
Serdar B, Lee D, Dou Z. Biomarkers of exposure to polycyclic aromatic hydrocarbons (PAHs) and DNA damage: a cross-sectional pilot study among roofers in South Florida. BMJ Open. 2012;2:e001318.
Freitas F, Brucker N, Durgante J, Bubols G, Bulcao R, Moro A, et al. Urinary 1-hydroxypyrene is associated with oxidative stress and inflammatory biomarkers in acute myocardial Infarction. Int J Environ Res Public Health. 2016;11:9024–37.
Ciolino HP, Daschner PJ, Wang TT, Yeh GC. Effect of curcumin on the aryl hydrocarbon receptor and cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Biochem Pharmacol. 1998;56:197–206.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 82074002, 81922073, 81872985) and the National Key Research and Development Project of China (No. 2019YFC1709405).
Author information
Authors and Affiliations
Contributions
YYZ designed the study. GC, HM, YNW, DQC, XQW, LC and LZ performed the experiments. YG and YYZ performed the statistical analysis. GC, HM and YYZ prepared the paper. NDV and PL revised the paper. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Cao, G., Miao, H., Wang, Yn. et al. Intrarenal 1-methoxypyrene, an aryl hydrocarbon receptor agonist, mediates progressive tubulointerstitial fibrosis in mice. Acta Pharmacol Sin 43, 2929–2945 (2022). https://doi.org/10.1038/s41401-022-00914-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00914-6
Keywords
This article is cited by
-
Sirtuin 6 protects against podocyte injury by blocking the renin-angiotensin system by inhibiting the Wnt1/β-catenin pathway
Acta Pharmacologica Sinica (2024)
-
Integrative phosphatidylcholine metabolism through phospholipase A2 in rats with chronic kidney disease
Acta Pharmacologica Sinica (2023)
