
Abstract
We evaluated homologous recombination deficient (HRD) phenotypes in epithelial ovarian cancer (EOC) considering BRCA1, BRCA2 and RAD51C in a large well-annotated patient set. We evaluated EOC patients for germline deleterious mutations (n = 899), somatic mutations (n = 279) and epigenetic alterations (n = 482) in these genes using NGS and genome-wide methylation arrays. Deleterious germline mutations were identified in 32 (3.6%) patients for BRCA1, in 28 (3.1%) for BRCA2 and in 26 (2.9%) for RAD51C. Ten somatically sequenced patients had deleterious alterations, six (2.1%) in BRCA1 and four (1.4%) in BRCA2. Fifty two patients (10.8%) had methylated BRCA1 or RAD51C. HRD patients with germline or somatic alterations in any gene were more likely to be high grade serous, have an earlier diagnosis age and have ovarian and/or breast cancer family history. The HRD phenotype was most common in high grade serous EOC. Identification of EOC patients with an HRD phenotype may help tailor specific therapies.
Similar content being viewed by others


Introduction
Ovarian cancer represents the fifth most common cause of cancer mortality in women due in part to the advanced stage at which patients typically present, with an estimated 14,030 deaths and 22,240 cases in 2013 in the United States1. The most common ovarian cancer is invasive epithelial ovarian cancer, with five common histological subtypes: high grade serous (70%), endometrioid (10%), clear cell (10%), low grade serous (5%) and mucinous (3%). A family history of breast or ovarian cancer in first degree relatives increases the risk for ovarian cancer2; family-based studies have revealed high- and moderate-penetrance genes including BRCA13, BRCA24, DNA mismatch repair genes5, RAD51C6,7, RAD51D8 and BRIP16 and case-control studies have identified eleven common variants associating with modestly increased risks9,10,11,12,13,14,15. Estimates of the contribution of germline BRCA1 and BRCA2 mutations to EOC vary widely from 5% to 20%16; somatic mutations in BRCA1 and BRCA2 occur less frequently (in 2%–8% patients)17,18 and, as has BRCA1 methylation19,20,21,22 have been reported in ovarian cancer patients with no family history23.
Ovarian cancer patients who are BRCA1 and BRCA2 germline mutation carriers have been reported to have an improved outcome compared to non-carriers and the mechanism underlying this benefit has been hypothesized as a high response rate to platinum agents, particularly among patients with high grade serous histology24,25,26,27. This is because patients with germline BRCA1 or BRCA2 mutations have an impaired ability to repair double stranded DNA breaks through homologous recombination28. More recently, favorable responses to poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors have been noted in ovarian cancer patients carrying deleterious germline BRCA1 and BRCA2 mutations29. However, some ovarian cancer patients without germline BRCA1 and BRCA2 mutations also respond to PARP inhibitors30, suggesting that broader dysfunction of homologous recombination than BRCA1 and BRCA2, a homologous recombination deficient (HRD) phenotype may be important. HRD may be the result of deleterious germline mutation in other homologous recombination genes such as RAD51B, RAD51C, or RAD51D or somatic mutation or silencing of the same homologous recombination genes31,32
We evaluated the prevalence of HRD in a large EOC case series and correlated this with clinical outcomes. HRD was determined by sequencing BRCA1, BRCA2 and RAD51C in germline and tumor DNA and assessing DNA methylation of cytosines at CpG sites surrounding these genes.
Results
Table 1 shows the characteristics of patients and Figure 1 shows the number of samples with each data type, after QC exclusions, included in this analysis.

Venn diagram representing data availability for N = 1063 invasive EOC patients.
Frequency of methylation, germline and somatic mutations
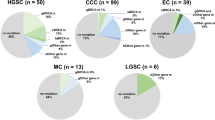
Of the 899 germline-tested patients, deleterious mutations were seen in 83 (9%): 32 (3.5%) in BRCA1, 28 in BRCA2 (3%) and 26 in RAD51C (2.9%; Table 2). Three cases carried mutations in more than one gene: two patients carried deleterious mutations in BRCA1 and RAD51C and one carried deleterious mutations in BRCA2 and RAD51C. For the 619 HGS cases, deleterious mutations were seen in 69 (11%), 27 (4%) in BRCA1, 24 (4%) in BRCA2 and 19 (3%) in RAD51C. One HGS patient carried a mutation in both BRCA2 and RAD51C. Of the six Ashkenazi Jewish patients, one carried a germline BRCA1 mutation (C61G) and none carried either somatic mutations or methylation. Table S1 and S2 provides a complete mutation listing, after exclusions (see Methods section). Another 187 women carried germline missense mutations, none of which had evidence for pathogenicity in the key clinical databases, 65 of these alterations were in BRCA1, 102 in BRCA2 and 32 in RAD51C. Twelve patients had a missense mutation in multiple genes. These variants were considered variants of unknown significance (VUS). In the patients with deleterious mutations, 13 patients also had a missense VUS that was identified. The remaining 629 patients had no deleterious or missense VUS mutations in BRCA1, BRCA2 or RAD51C.
Two hundred and seventy nine patients with available tumor tissue were screened for somatic mutations. Ten tumors (4%) were found to have deleterious somatic mutations, six in BRCA1 and four in BRCA2 (Table 2). Table S2 provides a complete listing for the somatic mutations. Missense VUS were also detected with one in BRCA1 and one in BRCA2. Two hundred and sixty seven patients had no deleterious or missense VUS mutations detected. No deleterious somatic variations were identified in RAD51C.
Forty five of 482 tumors (9%) exhibited a methylated BRCA1 phenotype and seven tumors (1%) showed RAD51C methylation (Table 2). BRCA1 methylation was not detected in any individual carrying a germline BRCA1 deleterious mutation; similarly, RAD51C methylation was exclusive of germline RAD51C mutation carrier status. However, there were two patients with a mutation and methylation of another gene; one patient had a somatic mutation in BRCA2 and BRCA1 methylation and another carried a germline RAD51C mutation and had methylated BRCA1.
Clinical and pathological analyses
In combination, 143 patients exhibited a HRD phenotype (82 due to germline mutation only, nine due to somatic mutation only, 50 due to tumor methylation only, one due to germline mutation and methylation and one additional due to somatic mutation and methylation, Table 2). A total of 79 HRD patients showed alterations in BRCA1, 30 in BRCA2, 29 in RAD51C and five patients showed alterations in more than one gene.
We examined the clinical features of HRD compared to patients with no abnormality detected (NAD) for those with both germline and somatic data (Tables 3, S3). HRD patients had younger age at diagnosis (P = 0.0001) and were more likely to be high grade serous (118/264, P = 0.0004) and have a family history of breast or ovarian cancer (P = 0.002) than patients with NAD. Interestingly, patients with high grade (10/27) endometrioid EOC were HRD, compared to 2/23 patients with low grade endometrioid EOC. One of the most common pathology challenges is whether high grade endometrioid are in fact high grade serous EOC33. Other covariates (debulking, peritoneal cytology, the presence of ascites and smoking and alcohol use history) were not significantly different between the two groups. We also examined clinical features within the HRD patients and found only family history of breast or ovarian cancer was more common in mutated than methylated patients (P = 0.0001, data not shown).
Survival analyses were carried out on those patients with HRD and those with NAD for which all data (germline, somatic mutation and methylation) were available (n = 356). Forty percent (143/356) of all the patients were HRD, while 44.7% (118/264) of HGS patients were HRD. Overall survival for all patients was associated with the HRD phenotype (adjusted HR 0.73, 95% CI 0.54–0.98, P = 0.04, Table 4), but not within each of the defective classes, genes that were mutated at the germline or somatic level or were somatically methylated (respectively, adjusted HR 0.81, 95% CI 0.57–1.15; adjusted HR 0.56, 95% CI 0.20–1.54, adjusted HR 0.65, 95% CI 0.42–1.02, P = 0.18, Table 4), or when outcomes were examined by germline mutation carrier status (P = 0.09, Table 4). The hazard ratios data are similar to those previously reported in a large germline study26. Results were similar whether VUS were included in the NAD category or excluded (data not shown). Analysis of time to recurrence showed no association with any of the HRD phenotypes (Table S3). Survival analyses were also conducted restricted to high grade serous (n = 618 for germline carrier analysis, 264 for HRD analysis) and showed an overall survival differences between HRD and NAD (adjusted HR 0.70, 95% CI 0.51–0.97, P = 0.03, Table 4, Figure 2), but not between germline mutation carriers and-non carriers (adjusted HR 1.07, 95% CI 0.65–1.76; adjusted HR 0.76, 95% CI 0.45–1.30, adjusted HR 0.66, 95% CI 0.37–1.18, P = 0.40, Table 4, Figure 2). Survival at 5 years was similar (adjusted HR 0.68 95% CI 0.48, 0.97, P = 0.04, data not shown). Analysis of time to recurrence was null (Table S4, Figure S3). Finally, we examined HRD phenotypes by debulking status in germline mutation carriers, as Alsop et al27 reported that improved survival was not evident in patients that were sub-optimally debulked. However, we observed no difference in germline mutation carriers stratified by debulking status (data not shown).

Overall survival for high grade serous EOC by HRD and type of alteration.
(A). Red: HRD; Black: NAD. (B). Above is a depiction of samples with type of alteration in each gene; below are the Kaplan Meier curves. Red: germline deleterious mutation; Green: somatic deleterious mutation; Blue: somatic methylation; Black: no germline mutation, somatic mutation or methylation.
Discussion
Using a large homogeneous collection of ovarian cancer patients with clinical follow up unselected for family history, we evaluated the clinical and pathological characteristics associated with a variety of HRD phenotypes. Deleterious germline mutations in BRCA1, BRCA2 or RAD51C were seen 9% of the subjects, BRCA1 or BRCA2 in 6.7% which is within the ranges of those previously reported (5.8%-13.2%)16. Somatic mutations were less common (3.6%), but 11% of the tumors analyzed exhibited BRCA1 or RAD51C methylation, comparable to the prevalence observed in 319 high grade serous ovarian cancers analyzed by the Cancer Genome Atlas19. In total, 143 of the patients had an HRD phenotype. A common phenotype has been reported for sporadic cancers that share the characteristics of tumors associated with germline BRCA1 and BRCA2 mutations34,35 and includes RAD51C in which germline mutations have been identified in patients of familial breast and ovarian cancer6,36,37, as has methylation38,39. Additionally, Abkevich et al40 report an LOH-based score for HRD which included promoter methylation of RAD51C. Here, we analyzed patients regardless of family history, included germline as well as tumor testing for more comprehensive analysis and defined patients as HRD if any deleterious alterations in BRCA1, BRCA2 and RAD51C genes were found.
An HRD phenotype was seen in 45% of high grade serous EOC (69% of patients had HGS EOC), with a smaller proportion observed in low grade serous and the other histologies. This is consistent with observations of others19,26,27. While we did observe a survival advantage for those patients with an HRD phenotype, analysis did not reveal any overall survival differences between BRCA1 or BRCA2 mutation carriers and non-carriers, unlike prior studies. This was unexpected given the literature suggesting longer progression free survival or overall survival for EOC in mutation carriers; however, this advantage appears to be short term. Most studies were out to 60 months only and McLaughlin et al41 showed that an early (up to five years) survival benefit was not evident beyond eight years41. Similarly we do not show a long term survival advantage, thus follow up time may explain the apparent discordance. It may also be that the number of germline BRCA1 and BRCA2 mutations carriers was too small and thus underpowered. Additionally, while care was taken to classify the mutations according to the best available evidence, the power of our analysis will have been reduced by any misclassification of HRD. While the TCGA concluded that methylated patients more closely match survival in wild type BRCA patients, we observed that methylated cases may have a survival benefit similar to that of the germline mutation patients. All categories, while not reaching statistical significance, showed a trend towards a survival benefit; had methylation not been thus, then combining with mutation positive patients would have biased the testing procedure towards the null. Biologically, BRCA1 inactivated by methylation would result in a similar defect as a deleteriously mutated allele. Our study involves patients at one site and all data were similarly processed while the TCGA study involves samples from multiple centers with multiple data sets, which may also account for the differences.
The high frequency of HRD defects in high grade serous EOC suggest screening for this phenotype should be considered in clinical care, particularly for new therapies, such as the PARP inhibitors that may be most efficacious within this phenotype.
Strengths of this report include detailed methylation phenotyping of a subset of patients and sequencing of tumor DNA. Limitations include lack of methylation and somatic mutation data on up to 67% of patients, not confirming the detected mutations in every patient and inclusion of only three of more than ten genes now considered to contribute to the HRD phenotype42. It is also critical to note that most of these patients were treated with the current standard of care at time of diagnosis (carboplatin and/or taxane); upcoming studies will focus on those treated with PARP inhibitors. Other future work includes sequencing of additional DNA repair genes in order to more comprehensively identify those patients with defective homologous recombination pathway abnormalities.
Methods
Patient population and biospecimens
Women with EOC (n = 1063) provided written informed consent between 1992 and 2011 using IRB approved processes at the Mayo Clinic in Rochester, MN. Use of patient derived samples in this study was also approved and the work described was carried out in accordance with these approved guidelines. Eligible participants were women aged 20 years or older with diagnosis of pathologically confirmed invasive primary epithelial ovarian, fallopian tube or primary peritoneal cancer. Patients were followed for vital status using electronic medical records and linkage to vital statistics sources; the median time of follow up was 4.5 years, with a range of 0.1–10 years. Most patients were treated with a platinum agent (carboplatin or cisplatin) and taxane, the standard of care at time of diagnosis. Diagnosis (histology and grade) were reviewed by a gynecologic pathologist. Peripheral blood (n = 899) was used a source of germline DNA43. Pre-chemotherapy tumor tissue (up to n = 482) was snap frozen immediately following surgery and stored at −80°C; tumor DNA was prepared from cryostat sections using Qiagen PureGene chemistry (Qiagen Inc, Valencia, CA, USA) and quantitated using Trinean (Trinean, Gentbrugge, Belgium). Tumor RNA was isolated from fresh frozen samples using the Qiagen RNEasy protocol (Qiagen Inc, Valencia, CA, USA) and quantitated using a Nanodrop Spectrophotomer (Agilent Technologies, Santa Clara, CA, USA). Somatic methylation analyses preceded the mutation screening, thus samples with insufficient DNA for both analyses were not sequenced.
DNA sequencing and bioinformatics
Germline DNA (n = 899, 1.25 μl DNA at a concentration 75 ng/μl) and tumor DNA (n = 279) were sequenced at the University of Cambridge, Cambridge, UK at BRCA1, BRCA2 and RAD51C. The procedure entailed design of PCR primers, testing and optimization using Fluidigm Access Array (Fluidigm Corp., San Francisco, CA, USA). Briefly, the method used a 4-primer amplicon-tagging scheme. Tagged, target-specific primer pairs were combined with sample-specific primer pairs that contained barcoding sequences and the adaptor sequences used by the Illumina sequencing system. Using sample-specific barcodes, all PCR products generated in the 48.48 Access Array Integrated Fluidic Circuit were unique and pooled together to run in a single sequencing experiment. Sequencing reactions were carried out on an Illumina GAIIX next-generation sequencer (Illumina, Inc., San Diego, CA, USA).
GENOME_GPS 1.0 was used for analysis of DNA sequence data including read alignment, variant detection and annotation (Figure S1). Reads were aligned to genome build hg19 using Novoalign and realignment, recalibration used the Genome Analysis Toolkit (GATK) v.1.6.744. The percent of reads mapped on target was used to exclude seven germline samples with <80% of amplicons covered at 20× and twenty one tumor samples with <80% of the amplicons covered at 20×. To avoid false-negative deleterious carrier status assignments for the somatic mutation data, non-carrier patients were excluded if either germline or tumor sequencing data exhibited <80% on-target sequence coverage at 20× or greater [mean coverage, 1st and 3rd quartiles: RAD51C 138.76 (121.68, 150.94); BRCA1 164.81 (142.59, 179.47); BRCA2 157.65 (133.68, 174.13); all three genes, 158.20 (136.06, 173.04)].
Germline variants were called through GATK Unified Genotyper. Germline filtering used genotype quality greater than 20 and read depth greater than 20. Somatic single nucleotide variants were called using SomaticSniper45, whereas insertions and deletions were called by GATK Somatic Indel Detector44. Somatic variants were considered with a depth of at least 20× coverage for the germline and tumor DNA at the given site as well as a quality score ≥ 20 and excluded if close to indel calls, aligned to multiple positions and/or occurred in a known repeat region. Indel calls required ≥ 10 supporting tumor sequencing reads and no reads supporting the indel in the germline.
Each variant was annotated using the Targeted RE-sequencing Annotation Tool (TREAT)46. Missense coding variants were functionally annotated by snpEFF (http://snpeff.sourceforge.net/) and PolyPhen-2 to predict biological effects. Somatic variants were considered deleterious if they were stop-gains (nonsense) or occurred at splice sites junctions, along with frameshifts and insertion/deletions as deleterious. In addition, missense mutations were hand-curated using LOVD (www.LOVD.nl), IARC (www.iarc.fr), HGMD (www.hgmd.cf.ac.uk) and Alamut (http://www.interactive-biosoftware.com/software/alamut/features) databases. Variants for which deleteriousness could not be determined with confidence, or which were known polymorphisms, were considered VUS.
DNA methylation
Tumor methylation status for 482 patients was examined using the Illumina Infinium HumanMethylation450 BeadChip as previously described47. Positive bisulfite modified (BSM) controls (SssI treated DNA, CpGenome Universal Methylated DNA, Millipore, Billerica, MA, USA and placental DNA), negative BSM controls (unmodified SssI treated DNA and WGA DNA) and a CEPH DNA (Coriell Institute), were included in each 96 well plate. Validation of nine CpG loci, including one at BRCA1 (cg25288140), was accomplished by Pyrosequencing, as previously described47 and suggested that BeadChip data were satisfactory; Pearson correlation of methylation values between BeadChip and Pyrosequencing assays ranged from 0.57 to 0.96.
To dichotomize tumor methylation status for each gene, we used expression array data from 171 patients to identify CpG probes which negatively correlated with expression. Expression data were generated using 750 ng of high quality total RNA and Agilent Whole Human Genome 4 × 44K Expression Arrays (using a mixed reference of 107 tumors) as previously described47. We estimated pairwise Spearman correlations between gene expression and CpG methylation probes within 20 kb of each gene to identify CpG probes most associated with expression. For each probe, we confirmed that each CpG probe was close to known promoter and active regulatory elements defined by ENCODE project using the UCSC Genome Browser (Figure S2A). Thresholds for dichotomization of methylation status were defined based upon the median methylation value of the selected probe set. For example, Figure S2B plots BRCA1 expression versus the median of N = 21 BRCA1 CpG probes; based on this, patients with mean BRCA1 CpG methylation > 0.15 were considered BRCA1 methylated. A similar process was used for six CpG probes in RAD51C; however, no BRCA2 CpG was clearly associated with altered gene expression, thus BRCA2 was not part of methylation analyses.
Association analysis
HRD patients were defined as those with deleterious germline mutations, deleterious somatic mutations or methylation of BRCA1, BRCA2, or RAD51C. Clinical characteristics of HRD and patients with NAD were compared using Fisher exact tests. Survival analysis with HRD status was conducted using multivariable Cox proportional hazards modeling and, for adjusted analyses, included age, stage, debulking, menopausal status, grade and ascites, estimating hazard ratios (HRs) and 95% confidence intervals (CIs). Time at risk was defined from the date of diagnosis to date of death allowing for left truncation with right censoring at last date of follow up to 10 years. In addition to overall survival, we considered time-to-recurrence or death from any cause. We analyzed survival using the HRD phenotype defined by germline status only and also by combined germline, somatic and methylation status (using the subset with complete data). Analyses were for all patients and among patients with high grade serous EOC and were conducted both with VUS considered to be non-carriers or excluded. Each analysis used all available data (thus sample sizes vary) and was implemented in SAS, with corresponding Kaplan-Meier curves generated in R.
References
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2013. CA: A Cancer Journal for Clinicians 63, 11–30 (2013).
Pharoah, P. D. & Ponder, B. A. The genetics of ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 16, 449–468 (2002).
Miki, Y. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71 (1994).
Wooster, R. et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792 (1995).
Aarnio, M., Mecklin, J. P., Aaltonen, L. A., Nystrom-Lahti, M. & Jarvinen, H. J. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 64, 430–433 (1995).
Meindl, A. et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 42, 410–414 (2010).
Meindl, A., Ditsch, N., Kast, K., Rhiem, K. & Schmutzler, R. K. Hereditary breast and ovarian cancer: new genes, new treatments, new concepts. Dtsch Arztebl Int. 108, 323–330 (2011).
Loveday, C. et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 43, 879–882 (2011).
Bojesen, S. E. et al. Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat Genet. 45, 371–384 (2013).
Permuth-Wey, J. et al. Identification and molecular characterization of a new ovarian cancer susceptibility locus at 17q21.31. Nat Commun. 4, 1627 (2013).
Pharoah, P. D. et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 45, 362–370 (2013).
Shen, H. et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat Commun. 4, 1628 (2013).
Bolton, K. L. et al. Common variants at 19p13 are associated with susceptibility to ovarian cancer. Nat Genet. 42, 880–884 (2010).
Goode, E. L. et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 42, 874–879 (2010).
Song, H. et al. A genome-wide association study identifies a new ovarian cancer susceptibility locus on 9p22.2. Nat Genet. 41, 996–1000 (2009).
Ramus, S. J. & Gayther, S. A. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol. 3, 138–150 (2009).
Berchuck, A. et al. Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res. 4, 2433–2437 (1998).
Foster, K. A. et al. Somatic and germline mutations of the BRCA2 gene in sporadic ovarian cancer. Cancer Res. 56, 3622–3625 (1996).
Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011).
Baldwin, R. L. et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. 60, 5329–5333 (2000).
Bol, G. M. et al. Methylation profiles of hereditary and sporadic ovarian cancer. Histopathology 57, 363–370 (2010).
Bianco, T., Chenevix-Trench, G., Walsh, D. C., Cooper, J. E. & Dobrovic, A. Tumour-specific distribution of BRCA1 promoter region methylation supports a pathogenetic role in breast and ovarian cancer. Carcinogenesis 21, 147–151 (2000).
Demsky, R. et al. Keeping it simple: Genetics referrals for all invasive serous ovarian cancers. Gynecol Oncol. 130, 329–333 (2013).
Pharoah, P. D., Easton, D. F., Stockton, D. L., Gayther, S. & Ponder, B. A. Survival in familial, BRCA1-associated and BRCA2-associated epithelial ovarian cancer. United Kingdom Coordinating Committee for Cancer Research (UKCCCR) Familial Ovarian Cancer Study Group. Cancer Res. 59, 868–871 (1999).
Liu, J. et al. Clinical characteristics and outcomes of BRCA-associated ovarian cancer: genotype and survival. Cancer Genet. 205, 34–41 (2012).
Bolton, K. L. et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. Jama 307, 382–390 (2012).
Alsop, K. et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 30, 2654–2663 (2012).
Tutt, A. & Ashworth, A. The relationship between the roles of BRCA genes in DNA repair and cancer predisposition. Trends Mol Med. 8, 571–576 (2002).
Fong, P. C. et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 28, 2512–2519 (2010).
Tinker, A. V. & Gelmon, K. The role of PARP inhibitors in the treatment of ovarian carcinomas. Curr Pharm Des. 18, 3770–3774 (2012).
Walsh, T. et al. Mutations in 12 genes for inherited ovarian, fallopian tube and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U.S.A. 108, 18032–18037 (2011).
Huang, J. -W., Wang, Y., Dhillon, K. K., Calses, P., Villegas, E., Mitchell, P. S., Tewari, M., Kemp, C. J. & Tanaguchi, T. Sytematic screen identifies miRNAs that target RAD51C and RAD51D to enhance chemotherapy. Mol Cancer Res. 11, 1–10 (2013).
Kobel, M. et al. Biomarker-based ovarian carcinoma typing: a histologic investigation in the ovarian tumor tissue analysis consortium. Cancer Epidemiol Biomarkers Prev. 22, 1677–1686 (2013).
Turner, N., Tutt, A. & Ashworth, A. Hallmarks of 'BRCAness' in sporadic cancers. Nat Rev Cancer 4, 814–819 (2004).
Tan, D. S. et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 26, 5530–5536 (2008).
Clague, J. et al. RAD51C germline mutations in breast and ovarian cancer cases from high-risk families. PLoS One 6, e25632 (2011).
Coulet, F. et al. Germline RAD51C mutations in ovarian cancer susceptibility. Clin Genet. 83, 332–336 (2013).
Hansmann, T. et al. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum Mol Genet. 21, 4669–4679 (2012).
Loveday, C. et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat Genet. 44, 475–476 (2012).
Abkevich, V. et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer 107, 1776–1782 (2012).
McLaughlin, J. R. et al. Long-term ovarian cancer survival associated with mutation in BRCA1 or BRCA2. J Natl Cancer Inst. 105, 141–148 (2013).
Rigakos, G. & Razis, E. BRCAness: Finding the achilles heel in ovarian cancer. Oncologist 17, 956–962 (2012).
Goode, E. L. et al. Assessment of hepatocyte growth factor in ovarian cancer mortality. Cancer Epidemiol Biomarkers Prev. 20, 1638–1648 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Larson, D. E. et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012).
Asmann, Y. W. et al. TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics 28, 277–278 (2012).
Cicek, M. S. et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum Mol Genet. 22, 3038–3047(2013).
Acknowledgements
This work was supported by National Institutes of Health grants R01-CA122443, P50-CA136393, P30-CA15083 and the Fred C. and Katherine B. Andersen Foundation. We thank Gary Kenney, M.D. for pathology review of tumor tissue. We thank Craig Luccarini, Caroline Baynes from University of Cambridge for assisting our sample sequencing.
Author information
Authors and Affiliations
Contributions
E.L.G. and P.P.D.P. designed the study. J.M.C. coordinated the analyses and prepared the manuscript. All authors read and approved the final version of the manuscript. M.C.L., N.B.L., C.W., B.L.F., E.M.D. and J.D. performed statistical and informatics analyses. G.C., H.H. and G.E.K. provided the gene expression data. H.S., P.H. and P.P.D.P. performed the next generation sequencing. J.M.C. performed the methylation analysis. Sample provision and management were coordinated by K.K. Clinical information was provided by B.W., N.M.L., M.W. and M.B. Technical support and advice were provided by M. C., J.C., M.B. and J.-B.F.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary figure Legends and tables
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Cunningham, J., Cicek, M., Larson, N. et al. Clinical Characteristics of Ovarian Cancer Classified by BRCA1, BRCA2 and RAD51C Status. Sci Rep 4, 4026 (2014). https://doi.org/10.1038/srep04026
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04026
This article is cited by
-
BRCA1/2 in non-mucinous epithelial ovarian cancer: tumour with or without germline testing?
British Journal of Cancer (2022)
-
Perspectives on the role of breast cancer susceptibility gene in breast cancer
International Journal of Clinical Oncology (2022)
-
Development of a 3D functional assay and identification of biomarkers, predictive for response of high-grade serous ovarian cancer (HGSOC) patients to poly-ADP ribose polymerase inhibitors (PARPis): targeted therapy
Journal of Translational Medicine (2020)
-
BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases
Journal of Ovarian Research (2020)
-
Genetic and epigenetic profiling of BRCA1/2 in ovarian tumors reveals additive diagnostic yield and evidence of a genomic BRCA1/2 DNA methylation signature
Journal of Human Genetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
