
Abstract
We reported previously (±)-2-(5-methylthiophen-2-yl)-3-phenyl-2,3-dihydroquinazolin-4(1H)-one [(±)-Retro-2cycl] as the chemical structure of Retro-2 that showed mouse protection against ricin, a notorious ribosome inactivating protein (RIP). Herein we report our chemical resolution of (±)-Retro-2cycl, analog synthesis and cell-based evaluation showing that the two optically pure enantiomers and their achiral analog have nearly the same degree of cell protection against ricin as (±)-Retro-2cycl. We also report our computational studies explaining the lack of stereo preference and revealing a common pharmacophore of structurally distinct inhibitors of intracellular retrograde trafficking of RIPs. This pharmacophore comprises a central aromatic ring o-substituted by an aromatic ring and a moiety bearing an O or S atom attached to sp2 C atom(s). These results offer new insights into lead identification and optimization for RIP antidote development to minimize the global health threat caused by ribosome-inactivating proteins.
Similar content being viewed by others

Introduction
Ricin produced by the castor bean plant enzymatically depurinates the 28S rRNA and thereby functions as a ribosome-inactivating protein (RIP). This notorious RIP has been used repeatedly for bioterrorism1 as in letters sent to U.S. government officials in 2013. Escherichia coli produces Shiga-like toxins (Stx1 and Stx2; the latter being the most toxic2). Stx1 and Stx2 also function as RIPs and are responsible for food-borne outbreaks, including the deadliest outbreak in 20113, with significant morbidity and mortality4. Presently there are no U.S. Food and Drug Administration approved vaccines or therapeutics to protect against ricin or other RIPs.
In an effort to minimize the global health threat caused by RIPs, we have been working on developing small-molecule RIP inhibitors that target the RIP catalytic domain using our doorstop approach5. In this endeavor we attempted to synthesize Retro-2 (Figure 1) as a benchmark for our in vivo studies, because it was reported that 200 mg/kg Retro-2 protected all inhibitor-treated mice against a dose of ricin that killed 90% of an unprotected control mouse population6. Serendipitously we discovered that the chemical structure of Retro-2 is (±)-2-(5-methylthiophen-2-yl)-3-phenyl-2,3-dihydroquinazolin-4(1H)-one rather than (E)-2-(((5-methylthiophen-2-yl)methylene)amino)-N-phenylbenzamide7. The former is a racemic, cyclic compound abbreviated as (±)-Retro-2cycl (Figure 1) and the latter is an achiral, acyclic chemical (Figure 1). Because (±)-Retro-2cycl is a mixture of two stereoisomers, we set out to separate the mixture, determine the cell protection activities of the two enantiomers and develop analogs of the active enantiomer(s) as a better benchmark for our in vivo studies.

Chemical structures of Retro-2cycl, DA2MT and other RIP transport inhibitors.
Herein we report our chemical resolution of (±)-Retro-2cycl, synthesis of an achiral analog of (±)-Retro-2cycl, computational studies of Retro-2cycl isomers and other known inhibitors of intracellular transport of RIPs and cell-based studies of Retro-2cycl isomers and their achiral analog. The results led to the development of a common pharmacophore of structurally distinct RIP transport inhibitors and offer important insight into RIP antidote development.
Results
Chemical resolution of (±)-Retro-2cycl
Although asymmetric syntheses8,9,10,11,12 have been reported for substituted 2,3-dihydroquinazolin-4(1H)-ones that are the scaffold of Retro-2cycl, we found that enantioselective reaction of 5-methylthiophene-2-carboxaldehyde with 2-aminobenzanilide using the readily available Sc(III)-inda-pybox as a catalyst11 gave the desired product with <10% yield and ~20% enantiomeric excess. Recognizing the presence of the aniline moiety in Retro-2cycl, we wanted to perform fractional crystallization of (±)-Retro-2cycl salts to be prepared from chiral acids. Unexpectedly, mixing (±)-Retro-2cycl with acids such as (1s)-(+)-10-camphorsulfonic acid caused decomposition of (±)-Retro-2cycl to 5-methylthiophene-2-carboxaldehyde and 2-aminobenzanilide, while weak acids such as (R)-(−)-mandelic acid, (−)-di-p-toluoyl-L-tartaric acid, (−)-dibenzoyl-L-tartaric acid and (D)-(−)-tartaric acid did not form salts with (±)-Retro-2cycl. We eventually resorted to high-performance liquid chromatography (HPLC) separation using a chiral column. Although diastereomeric chiral stationary phases13 was reported for separation of thiazides that possess an aminal stereo center analogous to the one in (±)-Retro-2cycl, we used the Lux™ Amylose-2 chiral column from Phenomenex (Torrance, California) that has the 5-chloro-2-methylphenylcarbamate derivative of amylose as a chiral stationary phase14. Our rationale for using this type of stationary was that the phenyl rings of the column could form pi-pi interactions with the three aromatic rings of (±)-Retro-2cycl and that the carbamate NH group of the column would permit hydrogen bonds with the amide oxygen atom of (±)-Retro-2cycl. Indeed, using the Lux™ Amylose-2 chiral column, we readily separated (±)-Retro-2cycl as evidenced by the HPLC chromatogram showing two well-separated peaks with retention times of 9.350 and 17.517 minutes and areas under the peaks of 49.769% and 50.231%, respectively, under conditions described in Method (Figure 2). The  values of the two enantiomers are +118.7° and −126.5°, respectively. As described below, the cell protection studies showed that the two optically pure isomers demonstrated nearly the same degree of cell protection against ricin as the racemate. Therefore, we did not carry out the X-ray crystallography study to determine whether (+)-Retro-2cycl corresponds to (R)-Retro-2cycl or (S)-Retro-2cycl, but rather synthesized and tested an achiral analog of (±)-Retro-2cycl.
values of the two enantiomers are +118.7° and −126.5°, respectively. As described below, the cell protection studies showed that the two optically pure isomers demonstrated nearly the same degree of cell protection against ricin as the racemate. Therefore, we did not carry out the X-ray crystallography study to determine whether (+)-Retro-2cycl corresponds to (R)-Retro-2cycl or (S)-Retro-2cycl, but rather synthesized and tested an achiral analog of (±)-Retro-2cycl.

Chromatogram of high-performance liquid chromatography separation of (±)-Retro-2cycl using the Lux™ Amylose-2 chiral column.
Design and synthesis of an achiral analog of Retro-2cycl
Energy minimizations at the HF/6-31G(d) level indicate that both enantiomers can adopt conformations at local minima of their potential energy surfaces (local minimum conformations). In those conformations the thiophene group is at the equatorial position of the heterocyclic ring in Retro-2cycl. The superimposition of the local minimum conformations of the two isomers shows nearly identical molecular shapes in terms of both steric and electrostatic properties (Figure 3), which explains the nearly equal potency of the two isomers for cell protection against ricin. This overlay also prompted us to synthesize and test 2-(5-methylthiophen-2-yl)-3-phenylquinazolin-4(3H)-one (DA2MT, Figure 1) as an achiral analog of (±)-Retro-2cycl. As apparent from Figure 3, the local minimum conformation of DA2MT derived at the HF/6-31G(d) level is nearly identical to those of the two isomers. Furthermore, DA2MT does not carry an aminal carbon, so it is chemically more stable than (±)-Retro-2cycl. The latter decomposes in the presence of (1s)-(+)-10-camphorsulfonic acid as described above. Accordingly, we synthesized DA2MT in quantitative yield through oxidation of (±)-Retro-2cycl using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone rather than following a published procedure15 that uses potassium permanganate in acetone and has a solubility issue for (±)-Retro-2cycl.

Local minimum conformations of structurally distinct RIP inhibitors derived at the HF/6-31G(d) level and their superimpositions.
A (upper left): (S)-Retro-2cycl and (R)-Retro-2cycl; B (upper middle): (S)-Retro-2cycl, (R)-Retro-2cycl and DA2MT; C (upper right): DA2MT, (S)-Retro-1 and Cpd 75; D (lower panel): local minimum conformations of (S)-Retro-2cycl, (R)-Retro-2cycl, DA2MT, (S)-Retro-1 and Cpd 75 in sphere model. For clarity, all H atoms except for those at the chiral centers are undisplayed in the superimposed structures. The Br, S, O, N and H atoms are colored in dark red, dark yellow, red, blue and white, respectively. The C atoms in (S)-Retro-2cycl, (R)-Retro-2cycl, DA2MT, Cpd 75 and (S)-Retro-1 are in green, magenta, cyan, orange and yellow, respectively.
Cell-based assays of Retro-2cycl Enantiomers and DA2MT
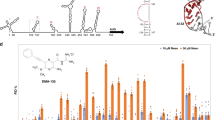
To determine the relative potencies of (±)-Retro-2cycl, (+)-Retro-2cycl, (−)-Retro-2cycl and DA2MT in protecting cells against ricin, we examined protein synthesis inhibition by ricin in Vero cells by measuring [35S]-Met-incorporation. The concentrations used were based on the reported dose-response curves for ricin7. As shown in Figure 4, the presence of 2.5 and 5.0 μM each of the four inhibitors restored ~7 and ~12% of protein synthesis in the presence of 50 ng/mL ricin. Interestingly, all four inhibitors showed nearly equal potency of cell protection against ricin. This observation offers important insight into generalizing the structurally distinct chemicals that inhibit the intracellular transport of RIPs to be discussed below.

Cell protection against 50 ng/mL ricin by (±)-Retro-2cycl, (+)-Retro-2cycl, (−)-Retro-2cycl and DA2MT in Vero cells.
Discussion
Substituted 2,3-dihydroquinazolin-4(1H)-ones have been well studied for various biological properties8,16,17,18,19,20,21,22,23. Racemization of the aminal stereo center in the substituted 2,3-dihydroquinazolin-4(1H)-ones has been reported8. One would suspect that the aminal stereo center in Retro-2cycl could be sensitive to racemization and possibly cause the lack of the stereo preference for cell protection that we observed. However, the Retro-2cycl structure has the substitution by a phenyl ring at its amide nitrogen atom. Theoretically this substitution lowers the propensity of the aminal stereo center for racemization. Indeed, through HPLC separation using a chiral column, we were able to separate (±)-Retro-2cycl. The optical activities and purities of the two enantiomers are demonstrated by the specific optical rotations and the HPLC chromatogram shown in Figure 2. In addition, we measured the rotations of the two enantiomers 14 months after the HPLC separation and found no racemization of the two isomers over the 14-month period. Furthermore, DA2MT, an achiral analog of (±)-Retro-2cycl, showed similar cell protection against ricin as the two isomers. These results preclude the possibility that racemization of the enantiomers led to the paucity of stereo preference for the cell protection against ricin.
DA2MT has been briefly reported to show no protection of HeLa cells against Shiga-like toxin 1, while a procedure for preparing DA2MT and the standard structure characterization have not been reported24. In this study, we found that DA2MT is as active as Retro-2cycl in protecting Vero cells against ricin. In view of its chemical stability and the paucity of chirality, DA2MT is a chemically improved analog of Retro-2cycl for cell protection against ricin and a useful lead for RIP antidote development as exemplified below.
As described above, the local minimum conformations of (+)-Retro-2cycl, (−)-Retro-2cycl and DA2MT superimpose well (Figure 3). These conformations also superimpose well with the local minimum conformations of (S)-Retro-16 and Compound 7525, both of which are effective intracellular RIP transport inhibitors (Figure 3). It is now well accepted that small molecules do not necessarily adopt their lowest potential energy conformations but do adopt local minimum conformations when binding to proteins26,27. All the chemical structures shown in Figure 3 are rigid. Their local minimum conformations were derived from ab initio calculations at the HF/6-31G(d) level and confirmed by frequency calculations using the same theory and basis set. Therefore the overlay shown in Figure 3 is reliable and strongly suggests that (+)-Retro-2cycl, (−)-Retro-2cycl and DA2MT, (S)-Retro-1 and Compound 75 (abbreviated as Cpd 75, Figure 1) share a common pharmacophore that targets the same protein critically involved in intracellular RIP transport. Structurally, this pharmacophore comprises a central aromatic ring with two substituents, one carrying an aromatic ring and the other bearing an O or S atom attached to sp2 C atom(s). Detailed structural information is available from the Cartesian coordinates of the superimposed structures at their local-minimum conformations provided in Supplementary Information.
This pharmacophore unifies structurally distinct chemicals identified by different research laboratories calling attention to the possibility that Retro-1, Retro-2cycl, DA2MT and Compound 75 may have the same target site for their inhibition of RIP transport. It also offers new insights into lead identification and optimization for RIP antidote development to minimize the global health threat caused by RIPs. For example, focused combinatorial library synthesis to derivatize DA2MT with the guidance of the pharmacophore holds promise in identifying improved RIP transport inhibitors. Alternatively, in silico database screening for chemicals with the pharmacophore described above may result in new leads for RIP transport inhibitors. Reverse engineering with 20 essential amino acids for proteins with cavities that can accommodate the pharmacophore is now technically feasible with today's computing capability. Such reverse engineered proteins can thus offer guidance in lead optimization of RIP transport inhibitors, which may address the concern that the RIP transport inhibitors are undesirable because of the lack of structural information about the targets of current RIP transport inhibitor leads.
Methods
Separation of (±)-Retro-2cycl
About 10 mg of (±)-Retro-2cycl prepared according to our published procedure7 was separated using the Lux™ Amylose-2 chiral column on a Beckman Coulter HPLC System Gold 125P eluting with n-hexane/2-propanol (60/40) at a flow rate of 1.0 mL/min. The retention times of the plus and minus enantiomers are 9.350 and 17.517 minutes, respectively. The two fractions were collected and the eluent was removed in vacuo. The chemical identities of both isomers were confirmed by proton NMR analysis. The specific optical rotations of the two enantiomers are 118.7° (c = 0.21 in CHCl3) and −126.5° (c = 0.29 in CHCl3), respectively, which were measured using a Jasco DIP-370 Digital Polarimeter at 598 nm at room temperature with L = 0.5 dm.
General description of chemical synthesis
All commercially available reagents were used as received. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Mercury 400 spectrometer from Varian (Palo Alto, CA). Chemical shifts are reported in ppm using the solvent peak as an internal standard. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet and m = multiplet), coupling constant and integration. Low-resolution mass spectra (LRMS) were recorded using either a Hewlett Packard 5973 Mass Spectrometer with SIS Direct Insertion Probe (Palo Alto, CA) or a Waters ZQ/EMD 1000 Mass Spectrometer (Milford, MA). High-resolution mass spectra (HRMS) were obtained on a Bruker BioTOF II ESI. IR spectra were obtained on a ThermoNicolet Avatar 370 FT-IR (Waltham, MA) using a KBr pellet. Melting points were determined by using Mel-Temp II (Holliston, MA). Elemental Analysis was performed at NuMega (San Diego, CA).
Synthesis of DA2MT (2-(5-methylthiophen-2-yl)-3-phenylquinazolin-4(3H)-one)
To a stirred solution of Retro-2cycl (64.0 mg, 0.20 mmol) in chloroform (5 mL) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (48.0 mg, 0.21 mmol) at room temperature. After 15 minutes of stirring, the reaction mixture was washed with saturated NaHCO3 solution (10 mL) to remove the remaining 2,3-dichloro-5,6-dicyano-1,4-benzoquinone and 4,5-dichloro-3,6-dihydroxyphthalonitrile as the by-product. The layers were separated and the organic layer was dried over MgSO4, filtered and concentrated to give 63.1 mg of pure product as a white solid in quantitative yield. Melting point 172–173.5°C; 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 8.0 Hz, 1H), 7.78–7.72 (m, 2H), 7.54–7.51 (m, 3H), 7.44 (ddd, J = 8.0, 6.2, 2.0 Hz, 1H), 7.36–7.33 (m, 2H), 6.42 (d, J = 3.8 Hz, 1H), 6.04 (d, J = 3.8 Hz, 1H) and 2.40 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.82, 148.85, 148.06, 146.34, 138.08, 135.78, 134.92, 131.86, 130.02, 129.79, 129.39, 127.56, 127.34, 126.83, 126.40, 120.46 and 15.56; IR (KBr) ν 3061, 2915, 1678, 1539, 768 and 700 cm−1; LRMS m/z 318 (100%, [M]+), HRMS-ESI m/z 319.0898 ([M + H]+, C19H15N2OS+ requires 319.0905). Anal. calcd for C19H14N2OS: C, 71.67; H, 4.43; N, 8.80. Found: C, 71.70; H, 4.80; N, 8.87.
Computational studies
Different conformations of each inhibitor shown in Figure 3 were systematically generated by alternating the bulky groups at axial and equatorial positions and subsequently energy minimized with the MMX force field using the PCModel 91 program (Serena Software). These resulting conformations were subjected to energy minimization at the HF/6-31G(d) level using the Gaussian 98 program28. All the energy-minimized conformations at the HF/6-31G(d) level were checked for possible imaginary frequencies by subsequent frequency calculations using the same theory and basis set. The energy-minimized conformations with no imaginary frequencies were then manually superimposed using the Pair Fitting tool of the MacPyMOL V1.5.0 (Schrödinger LLC, Portland, OR), which led to the superimposed inhibitor structures shown in Figure 3.
[35S]-Methionine incorporation assay
Vero cells were maintained in Dulbecco's modified Eagle medium with 10% fetal calf serum and 1 mM glutamine. The cells were resuspended after trypsin treatment at 4 × 104 cells/mL in the same medium and 0.5 mL of the medium was dispensed into 24-well plates. After 24 hours at 37°C and 5% CO2, the medium was changed to Dulbecco's modified Eagle medium without Met, Gln, or fetal calf serum and equilibrated for 1 hour. An inhibitor solution with a final dimethyl sulfoxide concentration of 0.5% was added to the medium at 25 hours. Ricin was added after 26 hours at varied concentrations. [35S]-Met was added 2 hours after ricin exposure. The [35S]-Met incorporation was terminated 30 minutes after the Met addition via medium removal and addition of 150 μL of 0.2 M aqueous KOH to dissolve cells, as described elsewhere29. Proteins were precipitated with 10% trichloroacetic acid, harvested on glass fiber filters and counted. The control incorporation was determined after treatment with 0.5% dimethyl sulfoxide alone. Ricin was purchased from Vector Laboratories (Burlingame, CA).
References
Audi, J., Belson, M., Patel, M., Schier, J. & Osterloh, J. Ricin poisoning: a comprehensive review. JAMA 294, 2342–2351 (2005).
Liu, Y.-N. et al. Shiga toxin type 2 (Stx2), a potential agent of bioterrorism, has a short distribution and a long elimination half-life and induces kidney and thymus lesions in rats. Arch. Toxicol. 85, 1133–1140 (2011).
Frank, C. et al. Epidemic profile of Shiga-toxin-producing Escherichia coli O104:H4 outbreak in Germany. N. Engl. J. Med. 365, 1771–1780 (2011).
Johannes, L. & Römer, W. Shiga toxins–from cell biology to biomedical applications. Nat. Rev. Microbiol. 8, 105–116 (2010).
Pang, Y.-P. et al. Small-molecule inhibitor leads of ribosome-inactivating proteins developed using the doorstop approach. PLoS One 6, e17883 (2011).
Stechmann, B. et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141, 231–242 (2010).
Park, J. G., Kahn, J. N., Tumer, N. E. & Pang, Y. P. Chemical structure of Retro-2, a compound that protects cells against ribosome-inactivating proteins. Sci. Rep. 2, 631 (2012).
Chinigo, G. M. et al. Asymmetric synthesis of 2,3-dihydro-2-arylquinazolin-4-ones: methodology and application to a potent fluorescent tubulin inhibitor with anticancer activity. J. Med. Chem. 51, 4620–4631 (2008).
Cheng, X., Vellalath, S., Goddard, R. & List, B. Direct catalytic asymmetric synthesis of cyclic aminals from aldehydes. J. Am. Chem. Soc. 130, 15786–15787 (2008).
Rueping, M., Antonchick, A. P., Sugiono, E. & Grenader, K. Asymmetric Brønsted acid catalysis: catalytic enantioselective synthesis of highly biologically active dihydroquinazolinones. Angew. Chem. Int. Ed. 48, 908–910 (2009).
Prakash, M. & Kesavan, V. Highly enantioselective synthesis of 2,3-dihydroquinazolinones through intramolecular amidation of imines. Org. Lett. 14, 1896–1899 (2012).
Cheng, D.-J., Tian, Y. & Tian, S.-K. Catalytic asymmetric synthesis of dihydroquinazolinones from imines and 2-aminobenzamides. Adv. Synth. Catal. 354, 995–999 (2012).
Hyun, M. H. & Pirkle, W. H. Liquid chromatographic separation of the stereoisomers of thiazide diuretics. J. Chromatogr. A 876, 221–227 (2000).
Chankvetadze, B., Yashima, E. & Okamoto, Y. Dimethyl-, dichloro- and chloromethylphenylcarbamates of amylose as chiral stationary phases for high-performance liquid chromatography. J. Chromatogr. A 694, 101–109 (1995).
Hisano, T., Ichikawa, M., Nakagawa, A. & Tsuji, M. Studies on organosulfur compounds XII: syntheses and pharmacological activities of 2-heterocyclic substituted 4(3H)-quinazolinones. Chem. Pharm. Bull. 23, 1910–1916 (1975).
Cohen, E., Klarberg, B. & Vaughan, J. R. Quinazolinone sulfonamides as diuretic agents. J. Am. Chem. Soc. 81, 5508–5509 (1959).
Yale, H. L. & Kalkstein, M. Substituted 2,3-dihydro-4(1H)-quinazolinones. A new class of inhibitors of cell multiplication. J. Med. Chem. 10, 334–336 (1967).
Bonola, G., DaRe, P., Magistretti, M. J., Massarani, E. & Setnikar, I. l-Aminoacyl-2,3-dihydro-4(1H)-quinazolinoiie derivatives with choleretic and antifibrillatory activity. J. Med. Chem. 11, 1136–1139 (1968).
Okumura, K., Oine, T., Yamada, Y., Hayashi, G. & Nakama, M. 4-Oxo-1,2,3,4-tetrahydroquinazolines. I. Syntheses and pharmacological properties of 2-methyl-3-aryl-4-oxo-1,2,3,4-tetrahydroquinazolines and their 1-acyl derivatives. J. Med. Chem. 11, 348–352 (1968).
Alaimo, R. J. & Russell, H. E. Antibacterial 2,3-Dihydro-2-(5-nitro-Zthieny1)-quinazolin-4(1H)-one. J. Med. Chem. 15, 335–336 (1972).
Neil, G. L., Li, L. H., Buskirk, H. H. & Moxley, T. E. Antitumor effects of the antispermatogenic agent, 2,3-dihydro-2-(1-naphthyl)-4(1H)-quinazolinone (NSC-145669). Cancer Chemother. Rep. 56, 163–173 (1972).
Levin, J. I., Chan, P. S., Bailey, T., Katocs, A. S. & Venkatesan, A. The synthesis of 2,3-dihydro-4(1H)-quinazolinone angiotensin II receptor antagonists. Bioorg. Med. Chem. Lett. 4, 1141–1146 (1994).
Hour, M. J. et al. 6-Alkylamino- and 2,3-dihydro-3′-methoxy-2-phenyl-4-quinazolinones and related compounds: their synthesis, cytotoxicity and inhibition of tubulin polymerization. J. Med. Chem. 43, 4479–4487 (2000).
Noel, R. et al. N-methyldihydroquinazolinone derivatives of Retro-2 with enhanced efficacy against Shiga toxin. J. Med. Chem. 56, 3404–3413 (2013).
Saenz, J. B., Doggett, T. A. & Haslam, D. B. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun. 75, 4552–4561 (2007).
Wang, Q. & Pang, Y.-P. Preference of small molecules for local minimum conformations when binding to proteins. PLoS ONE 2, e820 (2007).
Wang, Q. & Pang, Y.-P. Normal-mode-analysis–monitored energy minimization procedure for generating small–molecule bound conformations. PLoS ONE 2, e1025 (2007).
Frisch, M. J. et al. GAUSSIAN 98, Revision A.7. Gaussian, Inc. Pittsburgh, PA (1999).
Jacewicz, M., Feldman, H. A., Donohue-Rolfe, A., Balasubramanian, K. A. & Keusch, G. T. Pathogenesis of Shigella diarrhea. XIV. Analysis of Shiga toxin receptors on cloned HeLa cells. J. Infect. Dis. 159, 881–889 (1989).
Acknowledgements
This work was supported by the U.S. National Institute of Allergy and Infectious Diseases (5U01 AI082120-04).
Author information
Authors and Affiliations
Contributions
Y.-P.P. and N.E.T. conceived and supervised the project; S.Y. designed and performed the chemical resolution studies; J.G.P. designed and performed chemical synthesis of DA2MT; J.N.K. performed the cell-based assays; Y.-P.P. designed and performed the computational studies; all authors analyzed the data; Y.-P.P. wrote the paper; all authors contributed with revisions.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Yu, S., Park, J., Kahn, J. et al. Common Pharmacophore of Structurally Distinct Small-Molecule Inhibitors of Intracellular Retrograde Trafficking of Ribosome Inactivating Proteins. Sci Rep 3, 3397 (2013). https://doi.org/10.1038/srep03397
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03397
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
