
Abstract
Here we proposed a novel architectural design of a ternary MnO2-based electrode – a hierarchical Co3O4@Pt@MnO2 core-shell-shell structure, where the complemental features of the three key components (a well-defined Co3O4 nanowire array on the conductive Ti substrate, an ultrathin layer of small Pt nanoparticles and a thin layer of MnO2 nanoflakes) are strategically combined into a single entity to synergize and construct a high-performance electrode for supercapacitors. Owing to the high conductivity of the well-defined Co3O4 nanowire arrays, in which the conductivity was further enhanced by a thin metal (Pt) coating layer, in combination with the large surface area provided by the small MnO2 nanoflakes, the as-fabricated Co3O4@Pt@MnO2 nanowire arrays have exhibited high specific capacitances, good rate capability and excellent cycling stability. The architectural design demonstrated in this study provides a new approach to fabricate high-performance MnO2–based nanowire arrays for constructing next-generation supercapacitors.
Similar content being viewed by others


Introduction
Sustainable and renewable energy sources, such as hydropower, solar energy and wind power, in conjunction with flexible energy storage systems, such as rechargeable batteries and supercapacitors, are one of the key solutions to release the heavy burden on the current energy infrastructure and the environment1,2,3,4,5,6. Recently, supercapacitors have emerged as a new class of promising energy storage devices because of their higher power density, faster charge-discharge rate and longer cycle lifetime than those of rechargeable batteries7,8. Supercapacitors can also store more energy than conventional dielectric capacitors9,10. The central issues in the development of practical supercapacitors are the selection and fabrication of high-performance electrode materials. Among the emerging electrode materials for supercapacitors, redox-active transition-metal oxides such as RuO2, NiO, Fe2O3, SnO2 and MnO2, are the most attractive materials due to their high specific capacitances from the fast and reversible redox reactions on the electrode surface11,12,13,14,15.
In particular, MnO2 has drawn recent interest as a potential electrode material for supercapacitors because of its high specific capacitance (theoretical value of ~1370 F/g), low cost, natural abundance and environmental benignity16,17. However, the practical use of MnO2 as electrode materials is largely weight-down due to its poor electrical conductivity (10−5–10−6 S/cm)18,19,20, which limits the charge-discharge rate of the supercapacitors; as well as a relatively small surface area of bulk MnO2, which constraints the energy that can be stored in a particular electrode. An effective way to improve the utilization of MnO2 (thus increasing the energy density) is to reduce the MnO2 particles to the nanometer range, in which small particles have a characteristic high surface to volume ratio. For instance, ultrathin (<100 nm in thickness) films of MnO2 have shown high specific capacitances21,22. The major strategy to improve the electrical conductivity of the MnO2 electrodes is to combine conductive materials (e.g., carbon-based materials, metals and oxides) to MnO2, forming MnO2 composite electrodes with improved conductivities23,24. Taken together, new designs of high-performance MnO2-based electrodes must feature with a desired combination of small-sized MnO2 nanoparticles and a good electrical conductivity with the help from the conductive additives25,26,27,28.
In this context the recent developments on the conductive metal oxide nanowire arrays (NAs, e.g., SnO2 and Co3O4) look very promising22,29. Metal oxide NAs can be easily fabricated on the conductive substrates (e.g., Ti foil). The well-defined NAs have good conductivity and can work as current collectors, providing effective pathways for ion and electron transport throughout the entire electrode. Moreover, the three-dimensional conductive scaffold could also maximize the loading of thin MnO2 film and increase the area-normalized capacitances. For example, Liu et al. have developed a method to fabricate Co3O4@MnO2 NAs, which showed a good electrochemical performance in an alkali aqueous electrolyte29. The authors attributed its good performance to the MnO2 deposited on the Co3O4 NAs, which can improve the capacitive performance of Co3O4. Co3O4 is a typical electrode material used in the alkali aqueous electrolyte. However, the practical use of the alkali solution as the electrolyte is limited by its corrosive nature to the metal substrate. There is therefore strong interest in developing high-performance MnO2–based electrodes that could be operated in a neutral aqueous electrolyte to fully realize its potential for supercapacitors. In addition, since Co3O4 is still a semicondutor material, the relatively poor conductivity of the Co3O4@MnO2 NAs may further affect the capacitive performance of MnO2 in the neutral aqueous electrolyte.
Herein we report a facile approach to fabricate a novel hierarchical Co3O4@Pt@MnO2 nanostructure. The as-fabricated Co3O4@Pt@MnO2 NA electrode showed very high electrochemical performance for supercapacitors operated in a neutral aqueous electrolyte (1 M Na2SO4 solution). The key to fabricate the well-defined ternary Co3O4@Pt@MnO2 NAs was to introduce an ultrathin layer of Pt nanoparticles on the Co3O4 nanowire surface, which not only enhanced the conductivity of the NAs, but also induced a perfect coating of small MnO2 nanoflakes on the NAs. The as-fabricated Co3O4@Pt@MnO2 electrode showed several desirable electrochemical features for supercapacitors: a high specific capacitance (497 F/g on the basis of MnO2 mass by cyclic voltammetry and 539 F/g by galvanostatic charge-discharge at 1 A/g), good rate performance (39.6 Wh/kg at 40 A/g) and excellent cycling performance (no capacity loss over 5000 cycles).
Results
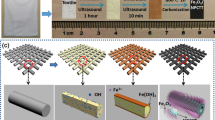
As illustrated in Figure 1, the fabrication of Co3O4@Pt@MnO2 NAs on the Ti foil involved three steps. In the first step, Co3O4 NAs were grown on the Ti foil according to published hydrothermal procedures30. The second step was the coating of small Pt nanoparticles (10–20 nm) on the Co3O4 nanowire surface by using a common sputter-coater. The final step was the deposition of a thin layer of MnO2 on the Pt-coated Co3O4 NAs via a chemical bath deposition method, forming Co3O4@Pt@MnO2 NAs as the final product.

Schematic illustration of the fabrication process of the Co3O4@Pt@MnO2 NAs.
The successful fabrication of Co3O4@Pt@MnO2 NAs on the Ti foil was first confirmed by powder X-ray diffraction (XRD). As shown in Figure 2 (black line), the diffraction patterns of the bare Co3O4 NAs in the 2θ range of 10–80° could be indexed to the cubic phase Co3O4 (JCPDS Card No. 42-1467). The three peaks marked by "#" belong to the Ti substrate. In contrast, besides the peaks from Co3O4 and Ti, the XRD patterns of the Co3O4@Pt@MnO2 NAs (blue line) showed three additional diffraction peaks (marked by "*") in the 2θ range of 10–80°, indexable to the (111), (200) and (220) reflections of face-centered cubic (fcc) Pt (JCPDS, Card No. 4-0802). This data supports the successful coating of Pt nanoparticles on the Co3O4 NAs. It should be mentioned that no peaks pertaining to MnO2 crystals were observed in the XRD patterns of the Co3O4@Pt@MnO2 NAs, which suggests that the deposited MnO2 thin film on the NA surface is most likely amorphous or poorly crystallized, similar to two earlier observations in the MnO2 deposition31,32.

XRD patterns of the Co3O4 NAs (black line) and Co3O4@Pt@MnO2 NAs (blue line).
X-ray photoelectron spectroscopy (XPS) was then used to confirm the chemical composition and metal oxidation states of the Co3O4@Pt@MnO2 NAs. Six elements (Mn, Co, Ti, Pt, O and C) were identified from the full-survey-scan spectrum of the NAs (Figure S1). Panels A, B, C and D of Figure 3 show the Mn 2p, Co 2p, O 1s and Pt 4f core level spectra of the Co3O4@Pt@MnO2 NAs, respectively. Two distinct peaks at binding energies of 642.1 and 653.5 eV with the spin-orbital splitting of 11.4 eV were observed in the Mn 2p core level spectrum (Figure 3a), which correspond well to the Mn 2p3/2 and Mn 2p1/2 in MnO2, respectively33,34. Deconvolution of complex Co 2p spectrum (Figure 3b) suggests the presence of two chemically distinct species: Co2+ and Co3+. In particular, two distinct peaks at 779.9 and 795.1 eV could be assigned to 2p3/2 and 2p1/2 of Co2+, respectively; whereas two small peaks at 788.9 and 803.5 eV could be attributed to 2p3/2 and 2p1/2 of Co3+, respectively. This data provide direct evidence for the presence of Co3O4 phase in the as-fabricated NAs35,36,37. The O 1s core level spectrum (Figure 3c) had three distinct components. Besides the O 1s peaks at 531 and 532.8 eV due to oxygen atoms in the hydroxyl groups and absorbed water38,39, respectively, the strong peak at 529.6 eV could be assigned to oxygen atoms in the oxides of MnO2 and Co3O4. The Pt 4f core level spectrum (Figure 3d) showed two distinct peaks at 70.9 and 74.3 eV, which could be attributed to Pt 4f7/2 and Pt 4f5/2, respectively, confirming the presence of metallic Pt in the as-fabricated NAs40.

XPS Mn 2p (a), Co 2p (b), O 1s (c) and Pt 4f (d) core-level spectra of the as-fabricated Co3O4@Pt@MnO2 NAs.
Scanning electron microscopy (SEM) was used to examine the structure of the bare Co3O4 NAs and the as-fabricated Co3O4@Pt and Co3O4@Pt@MnO2 NAs. Freestanding and well-aligned Co3O4 nanowires on the Ti substrate were observed for the bare Co3O4 NAs, as shown in Figure 4a. The nanowires had a uniform diameter of ~100 nm (Figure 4b). After the Pt coating (forming Co3O4@Pt NAs), the diameter of the nanowires did not change much (Figure 4c and 4d). However, a rough surface of the Co3O4@Pt nanowires can be seen in the SEM images (Figure 4c and 3d) as small Pt nanoparticles covered on the nanowire surface. Further deposition of MnO2 on the surface of Co3O4@Pt NAs formed Co3O4@Pt@MnO2 NAs (Figure 4e) and the diameter of the nanowires increased to ~200 nm (Figure 4f). In addition, a thin layer of MnO2 consisting of small nanoflakes can be seen in the SEM image (Figure 4f). It is worth mentioning that the loading amount of MnO2 or the thickness of the thin layer of MnO2 on the Co3O4@Pt@MnO2 NAs can be tailored by varying the deposition time of MnO2 (Figure S2). In particular, Figure 4e and 3f are representative SEM images of the Co3O4@Pt@MnO2 NAs prepared by 6 h MnO2 deposition.

SEM images of (a, b) Co3O4 NAs, (c, d) Co3O4@Pt NAs and (e, f) Co3O4@Pt@MnO2 NAs.
More detailed structural information of the Co3O4@Pt@MnO2 NAs was provided by transmission electron microscopy (TEM). The TEM samples were prepared by scratching the Co3O4@Pt@MnO2 NAs from the Ti substrate, followed by dispersing them in ethanol and casting them on the TEM copper grids. Figure 5a and 5b show the typical TEM images of the Co3O4@Pt@MnO2 NAs taken at different magnifications, indicating that porous Co3O4 nanowires were uniformly covered with small MnO2 nanoflakes. Figure 5c and 5d are enlarged TEM images of the square area 1 and 2 in Figure 5b, respectively. A core-shell-shell structure was observed for a typical Co3O4@Pt@MnO2 nanowire (Figure 5c). The thickness of the deposited Pt nanoparticles and MnO2 nanoflakes was in the range of 10–20 and 50–100 nm, respectively. The primary MnO2 nanoflakes were ultrathin and had a thickness of ~5 nm, as shown in Figure 5c and 5d. The inset of Figure 5d is the high-resolution TEM image of the square area 3, showing single crystallinity and the <111> direction of Pt nanoparticles. The lattice spacing between the (111) planes, 0.23 nm, is the same as that of bulk Pt41,42. In contrast, no clear lattice fringes could be observed for the MnO2 nanoflakes (Figure S3), corroborating the amorphous nature of MnO2, which is similar to the observation in its XRD patterns. Elemental mapping by energy-dispersive X-ray spectroscopy (EDS) (Figure 6) confirmed the core-shell hierarchical structure of the Co3O4@Pt@MnO2 nanowires. The EDS spectrum (Figure 6f) also supports the incorporation of Pt between the MnO2 shell and Co3O4 core. Cu and C signals in Figure 6f are from the TEM copper grid.

TEM images of the Co3O4@Pt@MnO2 nanowires taken at different magnifications: (a, b) low-magnification images; (c, d) high-magnification images of the square area 1 and 2 in (b), respectively.
The inset in (d) shows a HRTEM image of the square area 3.

(a) TEM image of the Co3O4@Pt@MnO2 nanowires and EDS element maps of (b) Co, (c) Pt, (d) Mn and (e) O. (f) EDS spectrum of the Co3O4@Pt@MnO2 nanowires.
The electrochemical performance of the Co3O4@Pt@MnO2 NAs was first examined by cyclic voltammetry (CV) by using an aqueous solution of Na2SO4 (1.0 M) as the electrolyte. The CV curves of the Co3O4@Pt@MnO2 NA electrode were recorded at scan rates of 10, 25, 50 and 100 mV/s. As shown in Figure 7a, all CV curves displayed a quasi-rectangular shape with a perfect mirror-image feature, suggesting that the Co3O4@Pt@MnO2 NAs have an ideal capacitive behavior and high rate capability26,43. In contrast, the rectangularity of CV curves of the bare MnO2 thin film electrode (Figure 7b) was remarkably distorted, especially at a high scan rate of 100 mV/s. The current densities of the CV curves of the Co3O4@Pt@MnO2 NA electrode were much higher than those of the bare MnO2 thin film electrode. Moreover, the CV curve of the Co3O4@Pt@MnO2 NA electrode at the scan rate of 50 mV/s (Figure 7c) had a much larger integrated area compared to that of the bare MnO2 thin film electrode, which suggests that a much higher specific capacitance was achieved in the Co3O4@Pt@MnO2 NA electrode. Since the capacitance contribution of Co3O4 and Pt was small and can be neglected, the specific capacitance of the Co3O4@Pt@MnO2 NA electrode was calculated on the MnO2 mass basis. As shown in Figure 7d, the Co3O4@Pt@MnO2 NA electrode had a specific capacitance of 497, 435, 393 and 354 F/g at scan rates of 10, 25, 50 and 100 mV/s, respectively, which were about 3-fold of increase than those of the bare MnO2 thin film electrode. For example, at the scan rate of 10 mV/s, the Co3O4@Pt@MnO2 NA electrode delivered a specific capacitance of 497 F/g. By comparison, the bare MnO2 thin film electrode can only provide the specific capacitance of 133 F/g. Among all Co3O4@Pt@MnO2 NAs prepared by different MnO2 deposition times, the one prepared by 6 h MnO2 deposition had the best electrochemical performance (Table S1), combining the merits of both high specific capacitance and good MnO2 loading. For example, further increasing the MnO2 deposition time to 10 h, the specific capacitance per gram of the Co3O4@Pt@MnO2 NA electrode decreased to 210.3 F/g (Figure S5 and Table S1).

Cyclic voltammograms of (a) Co3O4@Pt@MnO2 NA electrode and (b) MnO2 thin film electrode at different scan rates in an aqueous solution of Na2SO4 (1 M). (c) Comparison of the CV curves of Co3O4@Pt@MnO2 NA electrode (red) and MnO2 thin film electrode (black) at a scan rate of 50 mV/s. (d) Specific capacitances of Co3O4@Pt@MnO2 NA electrode (red) and MnO2 thin film electrode (black) at different scan rates.
The Co3O4@Pt@MnO2 NA electrode showed an impressive rate capability (Figure 8). A good symmetry and fairly linear slopes were observed in the galvanostatic charge-discharge curves of the Co3O4@Pt@MnO2 NA electrode at current densities of 1–40 A/g, indicating a good electrochemical capacitive characteristic and superior reversible redox reaction. Figure 8b compares the charge-discharge curves of the Co3O4@Pt@MnO2 NA and MnO2 thin film electrodes at the current density of 1 A/g. The charge storage capacity of the Co3O4@Pt@MnO2 NA electrode was significantly higher than that of the MnO2 thin film electrode with a 235% increase in the discharge time. In addition, the internal resistance (IR) of the Co3O4@Pt@MnO2 NA electrode (the IR drop is 0.0016 V) was significantly reduced compared to that of the MnO2 thin film electrode (the drop is 0.065 V). Figure 8c compares the specific capacitances of the two electrodes derived from the discharging curves at different current densities. The Co3O4@Pt@MnO2 NA electrode delivered a specific capacitance of 539 F/g at the current density of 1 A/g. This value was much higher than that of the MnO2 thin film electrode (171 F/g). In addition, the specific capacitance of our electrode is comparable to the reported values in the high-performance MnO2-based hybrid electrodes, such as graphene/MnO2 (with a specific capacitance of ~380 F/g) and Zn2SnO4@MnO2 (with a specific capacitance of ~621 F/g) electrodes44,26. In addition, at a high current density of 40 A/g, the Co3O4@Pt@MnO2 NA electrode still delivered a high specific capacitance of 298.1 F/g, or ~55.3% of the specific capacitance at 1 A/g, indicating its superior rate capability.

(a) Charge-discharge curves of Co3O4@Pt@MnO2 NA electrode at different current densities. (b) Comparison of charge-discharge curves of Co3O4@Pt@MnO2 NA electrode (red) and MnO2 thin film electrode (black) at a current density of 1 A/g. (c) Specific capacitances of Co3O4@Pt@MnO2 NA electrode (red) and MnO2 thin film electrode (black) at different scan rates. (d) Ragone plots of Co3O4@Pt@MnO2 NA electrode (red) and MnO2 thin film electrode (black).
Power density and energy density are two key performance indicators of supercapacitors. Figure 8d compares the Ragone plots of the Co3O4@Pt@MnO2 NA and MnO2 thin film electrodes, showing that the power and energy densities of the Co3O4@Pt@MnO2 NAs were much higher than that of the MnO2 thin film. In addition, as the current density increased from 1 to 40 A/g, the energy density of the Co3O4@Pt@MnO2 NA electrode decreased from 74.6 to 39.6 Wh/kg and its power density, on the contrary, increased from 0.5 to 19.6 kW/kg.
Cycling stability is another key performance indicator of supercapacitors. Figure 9a compares the cycling performance of the Co3O4@Pt@MnO2 NA and MnO2 thin film electrodes at the current density of 5 A/g over 5000 cycles. The specific capacitance of the Co3O4@Pt@MnO2 NA electrode increased slightly for the initial several hundreds of cycles and was relatively constant from the cycles onwards, indicating an excellent stability for the Co3O4@Pt@MnO2 NA electrode. After 5000 cycles, the specific capacitance was still slightly larger than the value of the first cycle, corresponding to a capacitance retention of ~105.6%. In addition, no obvious dissolution of MnO2 nanoflakes in the electrolyte was observed during cycling, as suggested by the unchanged transparent electrolyte solution during cycling21,22,45. A slight increase of the specific capacitance of the Co3O4@Pt@MnO2 NA electrode after the first cycle was due to the activation of the electrode via increasing the contact area between the electrode and the electrolyte during cycling. In contrast, only 89.8% of the initial capacitance was retained for the MnO2 thin film electrode. The current density dependence of the cycling performance of the Co3O4@Pt@MnO2 NA electrode was also evaluated from 5 to 20 A/g. Figure 9b shows a very good cycling behavior of the Co3O4@Pt@MnO2 NA electrode at these current densities. In addition, the specific capacitance was fully recovered at 5 A/g after cycling at those higher rates and no noticeable decrease was observed after another 300 cycles.

(a) Cycling performance of Co3O4@Pt@MnO2 NA electrode (red) and MnO2 thin film electrode (black) at a current density of 5 A/g. (b) Cycling stability of Co3O4@Pt@MnO2 NA electrode at different current densities.
Discussion
It is worth mentioning that the intermediate Pt nanoparticle layer is crucial for the construction of the core-shell-shell Co3O4@Pt@MnO2 nanowires. The Pt nanoparticle layer on the Co3O4 nanowires was used to confine the growth of MnO2 nanoflakes on the nanowire surface during the chemical bath deposition. Without the help from the Pt layer, no MnO2 layer can be deposited on the surface of Co3O4 nanowires and only agglomerated MnO2 spheres attaching on the top of the Co3O4 nanowires were obtained (Figure S4). A similar observation was reported by Liu et al. They deposited a carbon layer on the Co3O4 NWs, which could subsequently react with KMnO4, leading to the formation of MnO2 on the Co3O4 NWs29. In the present study, the confinement role of the Pt nanoparticle layer for MnO2 deposition can be attributed to the preferred nucleation of MnO2 on the Pt nanoparticle surface compared to the homogeneous nucleation in solution during the chemical bath deposition, similar to the findings reported in a previous study43. Moreover, Co3O4 is a p-type semiconductor with an electrical conductivity in the range of 10−4 to 10−2 S/cm46. This value is much lower than that of a noble metal (e.g., the electrical conductivity of Pt is ~106 S/cm)47. Therefore, although the incorporation of Co3O4 NWs in our hybrid electrode can improve the loading efficiency of the nanosturctured MnO2, it may still suffer with the poor conductivity between the MnO2 and the current collector. This constraint has been successfully addressed by the smart incorporation of an intermediate Pt nanoparticle layer in our hybrid electrode, which provides a good electrical conductivity, thus leading to a good electrical performance including a large specific capacitance and a good rate capability.
The high specific capacitance and superior rate capability had much to be credited to the hierarchical architecture and the synergy of the three key components in the Co3O4@Pt@MnO2 NAs. First, a highly porous structure was created by growing small MnO2 nanoflakes on the Co3O4@Pt scaffold, which not only increased the utilization of active MnO2 owing to the small size feature of the MnO2 nanoflakes, but also benefited the penetration of the electrolyte due to the highly porous structure. These two effects are essential to the high specific capacitance of the Co3O4@Pt@MnO2 NAs considering the charge storage of MnO2 is a surface or near-surface process. Second, the thin and well-separated MnO2 nanoflakes could shorten the electron and ion diffusion paths and make possible fast and reversible redox reactions, thus enhancing the electrochemical kinetics. Third, a thin layer of Pt nanoparticles between the Co3O4 and MnO2 layer could further improve the transport and collection of electrons, leading to a fast charge-discharge rate. Furthermore, the NAs directly grown on the Ti substrate had excellent mechanical adhesion and electrical connection to the current collector and the polymer binders or conductive additives were not required. In general, the use of the polymer binders or conductive additives might increase the "dead volume" in the electrode materials29,48.
The electrochemical impedance spectra (EIS) of the Co3O4@Pt@MnO2 NAs and MnO2 thin film electrodes were also measured to provide further insights. The Nyquist plots in Figure 10 for the Co3O4@Pt@MnO2 NA and MnO2 thin film electrodes show a straight line in the low frequency region, a depressed semicircle in the high-to-medium frequency region and a high frequency intercept in the real Z′ axis. Almost the same high frequency intercepts were observed for the two electrodes, indicating that they had the same overall ohmic resistance (the combination of the ionic resistance of the electrolyte, the intrinsic resistance of the active materials and the contact resistance between the active materials and the current collector). The semicircle in the high-to-medium frequency region represents the parallel combination of the charge-transfer resistance (Rct) and double-layer capacitance. The size of the semicircle, which equals to Rct, was clearly smaller for the Co3O4@Pt@MnO2 NA electrode (~3 Ω) compared to that of the MnO2 thin film electrode (~300 Ω). The large surface area and improved electrical conductivity of the Co3O4@Pt@MnO2 NA electrode could be the main reason for the reduction of the charge-transfer resistance. The straight line in the low frequency region corresponds to the diffusion-controlled region, where a finite slope represents the diffusive resistance of the electrolyte in the electrode pores and the cation diffusion in the host materials49,50. The slope of the straight line was obviously larger for the Co3O4@Pt@MnO2 NA electrode compared to that of the MnO2 thin film electrode, indicating a lower diffusive resistance for the Co3O4@Pt@MnO2 NA electrode.

Nyquist plots of Co3O4@Pt@MnO2 NA electrode (black) and MnO2 thin film electrode (red).
The inset is the enlarged impedance spectrum of Co3O4@Pt@MnO2 NA electrode at high frequencies.
In summary, a novel hierarchical Co3O4@Pt@MnO2 nanostructure was fabricated by a facile strategy in this study. The complemental features of the three key components: a well-defined Co3O4 NA on the conductive Ti substrate (good conductivity), an ultrathin layer of small Pt nanoparticles (further enhancing the conductivity) and a thin layer of MnO2 nanoflakes (large surface area), have been combined to construct a high-performance electrode for supercapacitors. Hence high specific capacitances [e.g., 539 F/g at 1 A/g in an aqueous Na2SO4 solution (1 M)], good rate capability (a specific energy and power density of 39.6 Wh/kg and 19.6 kW/kg at 40 A/g, respectively) and excellent cycling stability (no capacity loss over 5000 cycles) were achieved in the as-fabricated Co3O4@Pt@MnO2 NA electrode. The architectural design developed herein therefore provides an efficient way to improve the electrochemical performance of MnO2–based electrodes for supercapacitors.
Methods
Fabrication of Co3O4@Pt@MnO2 NAs
Co3O4 NAs were grown on the Ti foil by a common hydrothermal method according to a previous publication30. A thin film of Pt nanoparticles was then coated on the Co3O4 NAs by using a sputter-coater JS1600. After that, a thin MnO2 layer was deposited onto the surface of Co3O4@Pt NAs via a facile chemical bath deposition method43. In a typical MnO2 deposition process, the Ti foil grown with Co3O4@Pt NAs was submerged into an aqueous solution (30 mL, 25°C) of MnSO4·H2O, (NH4)2S2O8 and Na2SO4 (molar ratio of 1:1:1 and the concentration was 0.1 mol/L). The mixture was stirring at room temperature for 2 to 10 h, which led to 0.1 to 0.3 mg MnO2 deposited on the nanowire surface on the Ti foil. The as-fabricated Co3O4@Pt@MnO2 NAs were then washed with copious water and subsequently dried at 60°C in an oven. For comparison, a planar MnO2 thin film was also fabricated on the Ti foil by the anodic deposition according to published procedures51.
Materials characterization
The microstructure, surface morphology and chemical composition of the samples were characterized using powder X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). The powder XRD measurements were performed on a Bruker D8 advanced diffractometer with a Cu Kα radiation in the 2θ range of 10–80°. XPS spectra were recorded on a Phi Quantera spectrometer using A1 Kα X-ray as the excitation source. FESEM and TEM images were taken on a Hitachi S-4800 microscope and a JEOL JEM2100 microscope equipped with an Energy Dispersive Spectroscopy (EDS), respectively.
Electrochemical measurements
Electrochemical measurements were performed on an electrochemical workstation (CHI 660D, CH Instruments Inc., Shanghai). The three-electrode cell with Ag/AgCl as the reference electrode, Pt foil as the counter electrode and the as-fabricated samples as the working electrode, was used for the electrochemical measurements. 1 M Na2SO4 was used as the electrolyte. CV measurements were carried out between 0 and 1 V at different scan rates of 10, 25, 50 and 100 mV/s. Galvanostatic charge-discharge measurements were used to evaluate the power and energy densities at different current densities of 1, 5, 10, 20 and 40 A/g. Electrochemical impedance spectroscopy (EIS) was performed by applying an AC voltage of 5 mV in a frequency range of 0.01 Hz to 100 KHz at an open circuit potential.
References
Wang, X. J. et al. An Aqueous Rechargeable Lithium Battery Using Coated Li Metal as Anode. Sci. Rep. 3, 1401 (2013).
Liu, C., Li, F., Ma, L. P. & Cheng, H. M. Advanced Materials for Energy Storage. Adv. Mater. 22, E28–E62 (2010).
Song, Z. P. et al. Polymer-Graphene Nanocomposites as Ultrafast-Charge and -Discharge Cathodes for Rechargeable Lithium Batteries. Nano Lett. 12, 2205–2211 (2012).
Xu, B., Qian, D., Wang, Z. Y. & Meng, Y. S. Recent Progress in Cathode Materials Research for Advanced Lithium Ion Batteries. Mater. Sci. Eng. R. 73, 51–65 (2012).
Lim, H. D. et al. Enhanced Power and Rechargeability of a Li-O2 Battery Based on a Hierarchical-Fibril CNT Electrode. Adv. Mater. 25, 1348–1352 (2013).
Deng, D. & Lee, J. Y. Reversible Storage of Lithium in a Rambutan-Like Tin-Carbon Electrode. Angew. Chem. Int. Ed. 48, 1660–1663 (2009).
Tang, Z., Tang, C. H. & Gong, H. A High Energy Asymmetric Supercapacitor from Nano-architectured Ni(OH)2/Carbon Nanotube Electrodes. Adv. Funct. Mater. 22, 1272–1278 (2012).
Lei, Z. B., Lu, L. & Zhao, X. S. The Electrocapacitive Properties of Graphene Oxide Reduced by Urea. Energy Environ. Sci. 5, 6391–6399 (2012).
Chen, S. et al. Graphene Oxide-MnO2 Nanocomposites for Supercapacitors. ACS Nano. 4, 2822–2830 (2010).
Yuan, C. Z. et al. Flexible Hybrid Paper Made of Monolayer Co3O4 Microsphere Arrays on rGO/CNTs and Their Application in Electrochemical Capacitors. Adv. Funct. Mater. 22, 2560–2566 (2012).
Zhang, Y. J. et al. Crystallization Design of MnO2 Towards Better Supercapacitor. CrystEngComm. 14, 5892–5897 (2012).
Xia, H. et al. A Symmetric RuO2/RuO2 Supercapacitor Operating at 1.6 V by Using a Neutral Aqueous Electrolyte. Electrochem. Solid State Lett. 15, A60–A63 (2012).
Tang, C. H., Tang, Z. & Gong, H. Hierarchically Porous Ni-Co Oxide for High Reversibility Asymmetric Full-Cell Supercapacitors. J. Electrochem. Soc. 159, A651–A656 (2012).
Xia, X. F. et al. Nanostructured Ternary Composites of Graphene/Fe2O3/Polyaniline for High-performance Supercapacitors. J. Mater. Chem. 22, 16844–16850 (2012).
Wang, W. J. et al. Graphene/SnO2/Polypyrrole Ternary Nanocomposites as Supercapacitor Electrode Materials. RSC Adv. 2, 10268–10274 (2012).
Tang, W. et al. A Hybrid of MnO2 Nanowires and MWCNTs as Cathode of Excellent Rate Capability for Supercapacitors. J. Power Sources. 197, 330–333 (2012).
Huang, H. J. & Wang, X. Graphene Nanoplate-MnO2 Composites for Supercapacitors: a Controllable Oxidation Approach. Nanoscale 3, 3185–3191 (2011).
Xu, C. J., Kang, F. Y., Li, B. H. & Du, H. D. Recent Progress on Manganese Dioxide Based Supercapacitors. J. Mater. Res. 25, 1421–1432 (2010).
Pang, H. et al. Cu Superstructures Fabricated Using Tree Leaves and Cu-MnO2 Superstructures for High Performance Supercapacitors. J. Mater. Chem. A 1, 5053–5060 (2013).
Sun, Z. P. et al. Hierarchically Structured MnO2 Nanowires Supported on Hollow Ni Dendrites for High-performance Supercapacitors. Nanoscale 5, 4379–4387 (2013).
Pang, S. C. & Anderson, M. A. Novel Electrode Materials for Electrochemical Capacitors: Part II. Material Characterization of Sol-gel-derived and Electrodeposited Manganese Dioxide Thin Films. J. Mater. Res. 15, 2096–2106 (2000).
Yan, J., Khoo, E., Sumboja, A. & Lee, P. S. Facile Coating of Manganese Oxide on Tin Oxide Nanowires with High-Performance Capacitive Behavior. ACS Nano 4, 4247–4255 (2010).
Zhang, H. et al. Growth of Manganese Oxide Nanoflowers on Vertically-Aligned Carbon Nanotube Arrays for High-Rate Electrochemical Capacitive Energy Storage. Nano. Lett. 8, 2664–2668 (2008).
Wang, H. L., Casalongue, H. S., Liang, Y. Y. & Dai, H. J. Ni(OH)2 Nanoplates Grown on Graphene as Advanced Electrochemical Pseudocapacitor Materials. J. Am. Chem. Soc. 132, 7472–7477 (2010).
Hou, Y., Cheng, Y. W., Hobson, T. & Liu, J. Design and Synthesis of Hierarchical MnO2 Nanospheres/Carbon Nanotubes/Conducting Polymer Ternary Composite for High Performance Electrochemical Electrodes. Nano Lett. 10, 2727–2733 (2010).
Bao, L. H., Zang, J. F. & Li, X. D. Flexible Zn2SnO4/MnO2 Core/Shell Nanocable-Carbon Microfiber Hybrid Composites for High-Performance Supercapacitor Electrodes. Nano Lett. 11, 1215–1220 (2011).
Lu, X. H. et al. WO3−x@Au@MnO2 Core-Shell Nanowires on Carbon Fabric for High-Performance Flexible Supercapacitors. Adv. Mater. 24, 938–944 (2012).
He, Y. B. et al. Single-crystal ZnO Nanorod/Amorphous and Nanoporous Metal Oxide Shell Composites: Controllable Electrochemical Synthesis and Enhanced Supercapacitor Performances. Energy Environ. Sci. 4, 1288–1292 (2011).
Liu, J. P. et al. Co3O4 Nanowire@MnO2 Ultrathin Nanosheet Core/Shell Arrays: A New Class of High-Performance Pseudocapacitive Materials. Adv. Mater. 23, 2076–2081 (2011).
Jiang, J. et al. General Synthesis of Large-Scale Arrays of One-Dimensional Nanostructured Co3O4 Directly on Heterogeneous Substrates. Cryst. Growth Des. 10, 70–75 (2010).
Lee, S. W. et al. Carbon Nanotube/Manganese Oxide Ultrathin Film Electrodes for Electrochemical Capacitors. ACS Nano 4, 3889–3896 (2010).
Liu, R. & Lee, S. B. MnO2/Poly (3, 4-ethylenedioxythiophene) Coaxial Nanowires by One Step Coelectrodeposition for Electrochemical Energy Storage. J. Am. Chem. Soc. 130, 2942–2943 (2008).
Sui, N., Duan, Y. Z., Jiao, X. L. & Chen, D. R. Large-Scale Preparation and Catalytic Properties of One-Dimensional α/β–MnO2 Nanostructures. J. Phys. Chem. C 113, 8560–8565 (2009).
Reddy, A. L. M., Shaijumon, M. M., Gowda, S. R. & Ajayan, P. M. Coaxial MnO2/Carbon Nanotube Array Electrodes for High-Performance Lithium Batteries. Nano Lett. 9, 1002–1006 (2009).
Yan, N. et al. Co3O4 Nanocages for High-Performance Anode Material in Lithium-Ion Batteries. J. Phys. Chem. C 116, 7227–7235 (2012).
Li, L., Li, Y., Gao, S. Y. & Koshizaki, N. Ordered Co3O4 Hierarchical Nanorod Arrays: Tunable Superhydrophilicity without UV Irradiation and Transition to Superhydrophobicity. J. Mater. Chem. 19, 8366–8371 (2009).
Huang, H. et al. Nanocrystal-Constructed Mesoporous Single-Crystalline Co3O4 Nanobelts with Superior Rate Capability for Advanced lithium-Ion Batteries. ACS Appl. Mater. Interfaces 4, 5974–5980 (2012).
Wei, W. F., Cui, X. W., Chen, W. X. & Ivey, D. G. Phase-Controlled Synthesis of MnO2 Nanocrystals by Anodic Electrodeposition: Implications for High-Rate Capability Electrochemical Supercapacitors. J. Phys. Chem. C 112, 15075–15083 (2008).
Xia, H. et al. MnO2 Nanotube and Nanowire Arrays by Electrochemical Deposition for Supercapacitors. J. Power Sources 195, 4410–4413 (2010).
Wang, F. C. et al. Effect of Pt Loading Order on Photocatalytic Activity of Pt/TiO2 Nanofiber in Generation of H2 from Neat Ethanol. J. Phys. Chem. C 113, 13832–13840 (2009).
Li, Y. M., Tang, L. H. & Li, J. H. Preparation and Electrochemical Performance for Mrthanol Oxidation of Pt/Graphene Nanocomposites. Electrochem. Commun. 11, 846–849 (2009).
Jiang, L. H. et al. Structure and Chemical Composition of Supported Pt-Sn Electrocatalysts for Ethanol Oxidation. Electrochim. Acta 50, 5384–5389 (2005).
Xia, H., Xiao, W., Lai, M. O. & Lu, L. Facile Synthesis of Novel Nanostructured MnO2 Thin Films and Their Application in Supercapacitors. Nanoscale Res. Lett. 4, 1035–1040 (2009).
Yu, G. H. et al. Enhancing the supercapacitor performance of graphene/MnO2 nanostructured electrodes by conductive wrapping. Nano. Lett. 11, 4438–4442 (2011).
Jiang, H. et al. Ultrafine Manganese Dioxide Nanowire Network for High-Performance Supercapacitor. Chem. Commun. 47, 1264–1266 (2011).
Zhi, M. J. et al. Nanostructured carbon-metal oxide composite electrodes for supercapacitors: a review. Nanoscale 5, 72–88 (2013).
Kim, K. et al. High-frequency characterization of contact resistance and conductivity of platinum nanowires. IEEE Trans. Microwave Theo. Tech. 59, 2647–2654 (2011).
Jiang, J., Li, Y. Y., Liu, J. P. & Huang, X. T. Building One-dimensional Oxide Nanostructure Arrays on Conductive Metal Substrates for Lithium-Ion Battery Anodes. Nanoscale 3, 45–58 (2011).
Era, A., Takehara, Z. & Yoshizawa, S. Discharge Mechanism of the Manganese Dioxide Electrode. Electrochim. Acta 12, 1199–1212 (1967).
Guan, C. et al. Hybrid Structure of Cobalt Monoxide Nanowire @ Nickel Hydroxidenitrate Nanoflake Aligned on Nickel Foam for High-Rate Supercapacitor. Energy Environ. Sci. 4, 4496–4499 (2011).
Xia, H., Lai, M. O. & Lu, L. Nanostructured Manganese Oxide Thin Films as Electrode Material for Supercapacitors. JOM 1, 54–59 (2011).
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 51102134 and 11134004), Natural Science Foundation of Jiangsu Province (No. BK20131349 and No. BK2011709), China Postdoctoral Science Foundation (No. 2013M530258) and Jiangsu Planned Projects for Postdoctoral Research Funds (No. 1202001B).
Author information
Authors and Affiliations
Contributions
H.X., D.Z. and J.X. designed project, carried out data analyses and co-wrote the manuscript. Z.L. and Y.Y. performed the materials characterization. X.S. and G.Y. prepared figures 8–10.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supporting Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Xia, H., Zhu, D., Luo, Z. et al. Hierarchically Structured Co3O4@Pt@MnO2 Nanowire Arrays for High-Performance Supercapacitors. Sci Rep 3, 2978 (2013). https://doi.org/10.1038/srep02978
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02978
This article is cited by
-
Enhanced cycling performance of lithium-ion batteries with V2O5 as cathode by Co-doping for structural stability
Journal of Applied Electrochemistry (2024)
-
Spinel-structured hollow nanospheres prepared by a soft-template solvothermal method for high-performance supercapacitors
Journal of Materials Science: Materials in Electronics (2024)
-
Potentiodynamic electrodeposited MnO2:Co3O4 thin films electrodes for supercapacitor application
Journal of Materials Science: Materials in Electronics (2023)
-
Reduced graphene oxide supported MXene based metal oxide ternary composite electrodes for non-enzymatic glucose sensor applications
Scientific Reports (2022)
-
Sea-Island-Like Morphology of CuNi Bimetallic Nanoparticles Uniformly Anchored on Single Layer Graphene Oxide as a Highly Efficient and Noble-Metal-Free Catalyst for Cyanation of Aryl Halides
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
