
Abstract
The p63RhoGEF and GEFT proteins are encoded by the same gene and both members of the Dbl family of guanine nucleotide exchange factors. These proteins can be activated by the heterotrimeric G-protein subunit Gαq. We show that p63RhoGEF is located at the plasma membrane, whereas GEFT is confined to the cytoplasm. Live-cell imaging studies yielded quantitative information on diffusion coefficients, association rates and encounter times of GEFT and p63RhoGEF. Calcium signaling was examined as a measure of the signal transmission, revealing more efficient signaling through the membrane-associated p63RhoGEF. A rapamycin dependent recruitment system was used to dynamically alter the subcellular location and concentration of GEFT, showing efficient signaling through GEFT only upon membrane recruitment. Together, our results show efficient signal transmission through membrane located effectors and highlight a role for increased concentration rather than increased encounter times due to membrane localization in the Gαq mediated pathways to p63RhoGEF and PLCβ.
Similar content being viewed by others


Introduction
G-protein coupled receptors (GPCRs) comprise the largest family of receptors at the plasma membrane, with over 800 genes in the human genome encoding a GPCR1. These seven transmembrane proteins can perceive a wide variety of extracellular signals including light, hormones, ions and neurotransmitters2. Activation of the receptor induces a conformational change, which allows for nucleotide exchange factor (GEF) activity acting on the heterotrimeric G-protein complex. The GEF activity catalyzes the release of GDP from the Gα subunit, which is replaced by GTP3. Subsequently, the GTP-bound activated Gα subunit interacts with downstream effectors.
Almost twenty different Gα subunits can be discerned and these are grouped in four classes; Gi/Go, Gs, Gq and G12/G134. The classical effector of the Gq class is phospholipase C-beta (PLCβ)5,6. PLCβ is a 150 kDa enzyme that catalyzes the hydrolysis of phosphoinositide PtdIns(4,5)P2 located in the plasma membrane into diacylglycerol (DAG) and Ins(1,4,5)P3. The latter diffuses into the cytosol and releases calcium from intracellular stores7.
Recently, several studies, using biochemical8,9 and structural10 approaches, have shown that p63RhoGEF (and its homologues, Trio and Kalirin) can physically interact with Gαq. This interaction stimulates the GEF activity of p63RhoGEF, which catalyzes nucleotide exchange on Rho family GTPases, suggesting that p63RhoGEF links Gαq with the actin cytoskeleton.
p63RhoGEF is a protein of 580 amino acids, belonging to the Dbl family of guanine nucleotide exchange factors (GEFs) based on homology11. The members of this family are characterized by a tandem domain structure consisting of a Dbl homology (DH) domain directly followed by a pleckstrin homology (PH) domain12. The DH domain has intrinsic GEF activity, which is regulated by the adjacent PH domain. The PH domain of p63RhoGEF contains a C-terminal extension that was identified to be required for interaction with Gαq9. Deletion of the PH domain increases the GEF activity of the DH domain, indicating an autoinhibitory role for the PH domain9,13. Further studies on the activation mechanism have revealed that Gαq acts allosterically by binding to the C-terminal extension of the PH domain, relieving the DH domain from autoinhibition14. The crystal structure of the Gαq-p63RhoGEF complex supports this mode of activation10.
The gene encoding p63RhoGEF also encodes a splice variant, known as GEFT13. This variant lacks the first 105 amino acids. It was recently reported that the p63RhoGEF has cysteine residues in the N-terminal region that are palmitoylated, conferring plasma membrane localization15. Whereas GEFT lacks the N-terminal lipidation motif, it does contain the full DH/PH region including the complete Gαq interaction surface. It is unclear whether these variants interact similarly with Gαq in living cells, or alternatively, whether one is preferred over the other. To study the effect of palmitoylation of p63RhoGEF and subcellular location of p63RhoGEF and GEFT on the interaction with Gαq, we investigated the interaction in living cells using a variety of fluorescence techniques. We reveal that the plasma membrane located p63RhoGEF interacts efficiently with Gαq, while cytoplasmic GEFT does not.
Results
p63RhoGEF is located at the plasma membrane while GEFT is in the cytoplasm
To address the localization and dynamics of p63RhoGEF and its splice variant GEFT in living cells, fusions with fluorescent proteins were made (Figure 1). The full-length p63RhoGEF fused to the YFP variant mVenus16,17, denoted as YFP-p63, displayed clear plasma membrane localization in living HeLa cells as can be inferred from confocal images depicted in Figure 2A and as was observed previously15. Plasma membrane localization was observed when YFP was fused to the C-terminus of p63RhoGEF (p63-YFP) (Figure 2A), indicating that plasma membrane labeling was independent of the location of the fluorescent protein. In contrast, the splice variant, GEFT, did not localize at the membrane but was located in the cytoplasm and was largely excluded from the nucleus (Figure 2A). To exclude a cell-to-cell variability with respect to the localization, GEFT and p63RhoGEF were co-expressed in the same cell, again showing non-overlapping localization with p63RhoGEF at the plasma membrane and GEFT in the cytoplasm (Figure 2B).

Schematic overview of the constructs used in this study.
p63RhoGEF is a 63 kDa protein, comprised of 580 amino acids. It is a member of the Dbl family of Rho GTPase guanine nucleotide exchange factors. The Dbl homology (DH) and pleckstrin homology (PH) domain, identified based on their homology with other proteins, are indicated. Also the C-terminal extension of the PH domain is indicated (XT). The splice variant, termed GEFT, comprises amino acids 106–580. The yellow fluorescent protein variant that is used is mVenus, drawn to scale (238 amino acids) and indicated by yellow rectangles. A mCherry-FKBP12-GEFT was constructed as well as a mCherry-FKBP12 (drawn to scale), which can form a ternary complex with FRB and rapamycin.

Confocal images of living HeLa cells expressing p63RhoGEF and GEFT tagged with YFP.
(A) Both p63-YFP and YFP-p63 are located at the plasma membrane, whereas YFP-GEFT is confined to the cytoplasm. (B) Co-expression of GEFT and p63RhoGEF shows the absence of co-localization. In the overlay, p63RhoGEF is shown in green and GEFT in magenta. (C) The N-terminal 154, 105 or 29 amino acids of p63RhoGEF fused to YFP, all show plasma membrane localization as can also be inferred from the profile plot (D) which shows the fluorescence intensity in arbitray units along the lines indicated in C. The width of the images corresponds to 98 μm.
The clear difference in localization between GEFT and p63RhoGEF is due to the lack of the N-terminal 105 amino acids in case of GEFT. To examine the minimal stretch of amino acids that is sufficient for membrane localization, several variants comprising the first 154, the first 106 or the first 29 amino acids of p63RhoGEF were made, which all localized at the plasma membrane of living cells (Figure 2C and 2D). Together, these results show that the first 29 amino acids at the N-terminus of p63RhoGEF are required and sufficient to confer its plasma membrane localization.
Membrane binding of p63RhoGEF requires cysteines in the N-terminus
It has been shown that cysteines in the N-terminus of p63RhoGEF are palmitoylated15 and that cysteine mutants do not localize to the plasma membrane. To verify this, we mutated several of the cysteines into serines. Three constructs were made as depicted in Figure 3A. The distribution of these constructs in living cells is shown in figure 3B. Only the p63-C2S construct, with mutations C10S and C12S, displayed membrane binding. The other two mutants showed cytoplasmic and nuclear localization. These results suggest that cysteine residues 10 and 12 are dispensible for membrane localization of p63RhoGEF. Our results are in line with previous observations in a different cell type15.

Membrane association requires the second cysteine-stretch of p63RhoGEF.
(A) The amino-acid sequence of the N-terminus of p63RhoGEF, in which cysteines are indicated with boxes and basic residues are indicated with asterisks. The amino acid sequences of the constructs in which the cysteines were mutated to serines are shown, with the mutated cysteine underlined. (B) Confocal images of cells expressing the cysteine mutants of full-length p63RhoGEF tagged with YFP (p63-YFP). The width of the images corresponds to 98 μm. (C) FRAP curve of cells expressing p63-YFP, showing the recovery of fluorescence normalized to unity. The black line indicates the fit yielding D = 0.095 μm2/s. (D) FRAP on HeLa cells expressing cysteine mutants, p63-C2S (circles) and p63-C3S (squares). The black line shows the fit of p63-C2S recovery, yielding D = 0.24 μm2/s. All shown recovery curves are the average of at least 10 cells and the gray bars indicate the s.e.m.
The diffusion time of membrane associated proteins is strongly reduced when compared to the diffusion time of cytosolic proteins18. Fluorescence Recovery After Photobleaching (FRAP) can be used to quantify the diffusion of fluorescent biomolecules in cells19,20. Previously, we have used FRAP to detect the lipidation-dependent interaction of the plant Gα subunit GPα1 with the plasma membrane21. In order to examine membrane interaction of p63RhoGEF, FRAP was performed on the basal membrane of cells expressing p63RhoGEF. The recovery of p63RhoGEF was characterized by a diffusion time of 0.095 μm2/s (Figure 3C), corresponding to values previously reported for palmitoylated proteins22. The recovery of p63(1–29)-YFP was similar with a diffusion time of 0.13 μm2/s (data not shown). These results are in agreement with the observation that the first 29 amino acids are required and sufficient for membrane binding. Next, the recoveries of the cysteine mutants were analyzed. The p63-C3S mutant showed a fast recovery (Figure 3D) which did not allow accurate fitting of the recovery curve. The diffusion time was estimated to be >1 μm/s2. The FRAP recovery curve of p63-C5S was identical to that of p63-C3S (data not shown). However, the mutant p63-C2S, carrying Cys10Ser and Cys12Ser, showed a slow recovery with a diffusion time of 0.24 μm2/s. Hence, the diffusion of this mutant is approximately two-fold increased compared to the diffusion time of wild-type p63RhoGEF. These results agree with the observed localization of the cysteine mutants (Figure 3B) and previous observations15 and indicate that the stretch comprising cysteine residues 23, 25 and 26 is required for membrane localization of p63RhoGEF, but that Cys10 and Cys12, while not sufficient for membrane binding, confer affinity for the membrane.
p63RhoGEF and GEFT interact with Gαq-GTP in living cells
The interaction between Gαq and p63RhoGEF has been thoroughly studied using purified proteins9,10,14 or proteins in cell extracts8, demonstrating a direct interaction between GTP-bound Gαq and p63RhoGEF in vitro. However, interaction data from intact living cells is lacking. It has been shown that overexpressed, wild-type Gαq is able to recruit a palmitoylation-deficient p63RhoGEF to the plasma membrane15. This would suggest an interaction between wild-type Gαq and p63RhoGEF. Since wild-type Gαq is predominantly GDP bound due to intrinsic GTPase activity23 this observation contrasts previous findings from in vitro studies in which only active Gαq (Gαq-GTP) activates p63RhoGEF9. To examine this in more detail, we co-expressed a CFP-Gαq that is fully functional24 with a wild-type version and a palmitoylation-deficient p63RhoGEF, i.e. p63-C5S. Control samples in which p63RhoGEF and Gαq were co-expressed, show that both proteins are located at the plasma membrane (figure 4A). When the palmitoylation-deficient p63-C5S is co-expressed with Gαq, the RhoGEF is confined to the cytoplasm and the Gαq is located at the plasma membrane (67/72 cells show plasma membrane localization of Gq). Similar results were obtained when GEFT is co-expressed with Gαq, although we observed in cells highly expressing GEFT that Gαq shows a more cytoplasmic localization (28 out of 124 cells show cytoplasmic localization of Gq). Together, these results suggest that there is little or no interaction between over-expressed wild-type Gαq and GEFT or p63RhoGEF in HeLa cells.

p63RhoGEF and GEFT interact with Gαq-GTP and not with Gαq-GDP in cells.
(A) Co-expression of CFP tagged Gαq and YFP tagged p63RhoGEF, p63-C5S and GEFT shows that Gαq localizes at the plasma membrane and is independent of the localization of the GEF. Width of the images is 380 μm. (B) Constitutive active Gαq interacts with p63RhoGEF at the plasma membrane. The YFP-Gαq-Q209L is predominantly cytoplasmic, but is recruited to the plasma in those cells that express p63-mCherry. In contrast to full-length, wild-type p63-YFP, the mutant p63(L475)-YFP was not able to recruit CFP-Gαq-Q209L to the plasma membrane. The width of the images corresponds to 146 μm (left) or 73 μm (right). (C) Rapamycin induced recruitment of Gαq or Gαq-Q209L to the plasma membrane shows that GEFT interacts with GTP-bound Gαq. While GEFT is co-recruited to the plasma membrane by Gαq-Q209L, it does not interact with wild type GDP-bound Gαq at the plasma membrane. Recruitment is induced by 100 nM rapamycin (+rapamycin) and the images are acquired after 150s. Width of the images is 100 μm. (D) Quantification of GEFT relocalization induced by recruiting Gαq to the plasma membrane. The relocalization of FKBP12-mRFP-tagged Gαq (induced by addition of 100 nM rapamycin) and YFP-tagged GEFT was determined by measuring the average fluorescence in the cytoplasm. A decrease in cytoplasmic fluorescence indicates relocalization from the cytoplasm to membranes. The intensity measurements were normalized and the traces depict averages (±s.e.m.). Relocalization of the GTP-bound state, Gαq-Q209L (n = 15), resulted in recruitment of GEFT, while recruiting the wild-type GDP-bound Gαq (n = 27) did not result in YFP-GEFT relocalization.
To examine whether and where an interaction occurs between Gαq and p63RhoGEF in living cells, we made use of YFP-Gαq-Q209L, which is a YFP labeled Gαq made constitutively active by a single point mutation (Q209L). This mutant is GTPase deficient and therefore is locked in the active GTP-bound form. The Gαq-Q209L mutant is predominantly located in the cytoplasm (figure 4B) which might be due to diminished interaction with the Gβγ dimer, thereby reducing plasma membrane affinity. Remarkably, upon co-expression of p63-mCherry, the YFP-Gαq-Q209L is located at the plasma membrane (figure 4B), suggesting that the two proteins interact at the plasma membrane. It was previously shown that the L475A point mutation in the extension of the PH domain of p63RhoGEF abolishes the interaction in vitro9. The localization of p63(L475A)-YFP is identical to that of p63-YFP. However, the co-expressed constitutively active Gαq remains cytoplasmic in the presence of p63(L475A)-YFP (figure 4B), confirming in living cells that the interaction between p63 and Gαq is strictly dependent on the L475A mutation in p63RhoGEF.
Since GEFT and the constitutive active Gαq are both located in the cytoplasm, it is unclear from their steady-state localization whether these two proteins interact. To examine whether these proteins interact in intact living cells we performed a co-recruitment assay. Co-recruitment is a strong indicator of a physical interaction or complex formation with the advantage that it can be performed in single, intact, living cells25,26. We have recently used this method to demonstrate the interaction between Gαq-Q209L and PLCβ3 in cells25. To this end, GEFT and FKBP12-Gαq were co-expressed. The FKBP12-Gαq is cytoplasmic due to removal of the lipidation motif. The FKBP12 domain strongly binds a FRB domain after addition of the cell-permeant heterodimerizer rapamycin27,28. By targeting the FRB domain to the plasma membrane, the FKBP12-Gαq can be recruited to the plasma membrane. We performed the co-recruitment with both the wild-type Gαq and the GTPase deficient Gαq. Recruitment of YFP-GEFT was only observed when active, GTP-bound, Gαq was relocalized (figure 4C). Quantification of the translocation of YFP-GEFT observed in multiple cells induced by recruitment of mRFP-Gαq-Q209L or mRFP-Gαq is shown in figure 4D. Together our data suggest that GEFT and p63RhoGEF both interact with Gαq-Q209L, while GDP-bound Gαq does not have affinity for p63RhoGEF and GEFT.
The interaction of p63RhoGEF with Gαq is dynamic and reversible
Although co-recruitment and co-localization strongly suggest interaction, it is not direct proof. To study the interaction more directly we made use of Förster Resonance Energy Transfer (FRET) which is highly distance dependent and is therefore suited to monitor proximity of molecules in living cells. To examine whether the interaction between Gαq and p63RhoGEF can be observed in living cells, a Gαq subunit tagged with mTurquoise, CFP-Gαq and p63RhoGEF tagged with YFP were co-expressed. Both proteins are located at the plasma membrane (figure 5A). In case of interaction, it is expected that the YFP over CFP ratio increases. Upon addition of histamine, a significant increase in the YFP/CFP ratio was observed (figure 5B). When an H1R antagonist, pyrilamine, was added, the ratio decreased and returned to the baseline value. Kinetic analysis of the interaction yielded a kon of 0.23 s−1 and a koff of 0.045 s−1, corresponding to half-times of 3 and 15 seconds, respectively. To examine the specificity of the interaction, the mutant p63(L475A)-YFP was used. We hardly observed any change in YFP/CFP ratio when histamine receptors were activated (fig. 5B), indicating that the interaction between Gαq and the L475A mutant is strongly reduced.

The dynamics of the interaction between Gαq and p63RhoGEF.
(A) mTurquoise-tagged Gαq was co-expressed with p63RhoGEF or p63RhoGEF-L475A tagged with YFP. (B) FRET ratio imaging reveals a substantial increase in YFP/CFP ratio for wild-type p63RhoGEF (n = 14 cells), but hardly any increase for the L475A mutant (n = 17 cells) upon addition of histamine. The interaction is readily reversed when the H1R antagonist pyrilamine is added. The graph shows the average YFP/CFP ratio change ±s.e.m. Width of the images corresponds to 230 μm.
p63RhoGEF rather than GEFT inhibits PLCβ-dependent calcium signaling
The different location of the Gαq effectors, i.e. membrane associated p63RhoGEF versus cytoplasmic GEFT, comprises an excellent model to address the effects of location on interaction efficiency and signal transmission. We took advantage of the well-characterized signaling pathway in which Gαq activation of PLCβ results in intracellular calcium increases5,6. First, we examined whether the Gαq-p63RhoGEF interaction competes significantly with PLCβ activation. In HeLa cells, the Gαq-PLCβ pathway is activated upon addition of histamine, leading to an increase in the intracellular calcium concentration. Calcium imaging was performed on a population of cells in which the cells expressing YFP-tagged p63RhoGEF could be easily identified based on their YFP fluorescence. The non-transfected cells were used as a control for timing of the onset of the calcium response. When the calcium increase in cells expressing p63RhoGEF was analyzed, a marked delay was observed (Figure 6A), suggesting that p63RhoGEF is capable of inhibiting calcium signaling by competing with PLCβ for interaction with Gαq. A similar experiment with the L475A mutant demonstrated no significant delay in calcium signaling (Figure 6B).

Overexpressed p63RhoGEF inhibits Gαq mediated calcium signaling.
(A) Single cell traces of calcium release measured with Fura Red in HeLa cells stimulated with histamine. The time delay in the calcium release between control HeLa cells (dashed line) and cells expressing p63RhoGEF-YFP (solid line) was quantified. (B) Bar graph that displays the delay in calcium signaling in cells expressing YFP fused to p63RhoGEF, p63RhoGEF-L475A, GEFT or the indicated cysteine mutants. Average values (±s.e.m.) of at least three independent experiments with a total of at least 10 cells are shown. The P-values were calculated with an unpaired two-tailed Student's t-test: P < 0.0002 (**), P < 0.004 (*) and P < 0.05 (#).
Gαq is part of a peripheral membrane protein complex and is itself palmitoylated. Therefore, we examined whether membrane localization of p63RhoGEF is necessary for efficient competition with PLCβ for Gαq interaction, by quantifying the delay in calcium signaling.
When the cysteine mutants were analyzed, only p63-C2S-YFP showed calcium delays similar to wild-type p63-YFP, corresponding with its plasma membrane binding. On the other hand, the cytoplasmic localized p63-C3S and p63-C5S showed only a modest delay in calcium signaling, which was comparable to that of YFP-GEFT (Figure 6B). These results correlate membrane binding with the efficiency of delaying calcium signaling, the latter suggesting competition with PLCβ.
Attenuation of calcium signals upon recruitment of GEFT
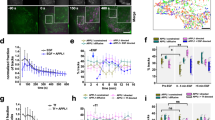
Our data highlights the importance of the location of the effector for its interaction efficiency with Gαq. To obtain direct control over the subcellular location of GEFT, we fused it to FKBP12. When co-expressed with plasma membrane-located FRB, the mCherry-FKBP12-GEFT fusion can be recruited to the plasma membrane by adding rapamycin, inducing dimerization of FKBP12 and FRB. Next, we examined whether relocalization of GEFT to the plasma membrane would interfere with PLCβ signaling (Figure 7A) since this interaction predominantly takes place at the plasma membrane24,25,29. Calcium oscillations, measured using the biosensor yellow cameleon 3.6030, were induced in HeLa cells by stimulating with histamine. After about 90s, when several calcium spikes were observed, mCherry-FKBP12-GEFT was recruited to the plasma membrane by addition of rapamycin. A clear reduction in the calcium response was noted, seconds after addition of rapamycin (Figure 7B). In contrast, no attenuation of the calcium signal was observed in control experiments, in which mCherry-FKBP12 was recruited (Figure 7C). Average traces of multiple experiments are included as supplemental information (figure S1). These results show that recruitment of GEFT to the membrane attenuates calcium signals, suggesting that GEFT competes dynamically with PLCβ for Gq binding.

Dynamic inhibition of calcium oscillations by recruitment of GEFT.
(A) Cartoon illustrating the principle of the experiment. Rapamycin recruits the FKBP12-GEFT (F12-GEFT) fusion to the FRB domain located at the plasma membrane, effectively competing with PLCβ for activated Gq, thereby attenuating calcium signals. (B) Single cell calcium imaging data showing oscillations due to GPCR activation by histamine. Subsequent recruitment of mCherry-FKBP12-GEFT strongly attenuates the signal. (C) Single cell calcium imaging data from a control cell expressing mCherry-FKBP12. Recruitment induced by rapamycin does not alter the frequency or amplitude of the calcium spiking.
Discussion
In this study, we used fluorescent protein tagged p63RhoGEF and GEFT to study its subcellular location and interaction with Gαq. The heterotrimeric G-protein complex itself is predominantly located at the plasma membrane by lipid modification of the Gα and Gγ subunits. Both GEFT and p63RhoGEF contain the entire region which is necessary for Gαq interaction10. However, while p63RhoGEF localizes almost exclusively to the plasma membrane, GEFT is confined in the cytoplasm. Therefore, these signaling components comprise an excellent model to address the effects of location on interaction efficiency and signal transmission.
The p63RhoGEF and GEFT do not differ in their Gαq interaction domain, encompassing roughly residues 149–502, which has an affinity for active Gαq (Gαq-AlF4−) of <0.1 μM10 or 1 μM9 depending on the assay. Our data confirm that active GTP-bound Gαq (Gαq-Q209L) is capable of interacting with both GEFT and p63RhoGEF in cells, while the wildtype Gαq, which is predominantly GDP-bound, has hardly any affinity.
To examine the effects on signal transmission, we used calcium imaging as an output signal for GPCR activation. Interaction of Gαq with its RhoGEF effector leads to a reduction in the calcium signal. The overexpression of membrane located p63RhoGEF strongly inhibited calcium signaling. In contrast, the effect of overexpressed cytoplasmic cysteine mutants and GEFT are far less efficient in inhibiting calcium signaling. This indicates that the interaction between Gαq and membrane located p63RhoGEF is stronger. Our data fit with our previous observations that Gαq activation and interaction with PLCβ occurs predominantly at the plasma membrane24,25,29. Since p63RhoGEF with L475A did not interfere with calcium signaling, the interaction between Gαq and p63RhoGEF is required to delay calcium signaling. Still, it is possible that some other mechanism than competition between p63RhoGEF and PLCβ inhibits the calcium signal, e.g. downregulation of PLCβ activity by p63RhoGEF or modulation of calcium channels by p63RhoGEF. Further experiments are required to observe directly the competition between PLCβ and p63RhoGEF for Gαq, for instance by multiplex FRET measurements.
Since our experiments employ overexpressed proteins, it is necessary to understand whether p63RhoGEF competes with PLCβ under physiological conditions. Recently, Momotami et al. detected relative high expression of p63RhoGEF in vascular smooth muscle cells and especially portal veins31. Previous studies have revealed a role for Gq-PLCβ in portal vein myocytes32. Therefore further studies using knock-down approaches may shed light on the balance between the PLCβ and p63RhoGEF pathway in smooth muscle cells.
How does membrane localization of the RhoGEF result in a more efficient interaction with Gαq? Two effects have been brought forward which are (i) a decrease in time necessary for a productive interaction and (ii) an increased concentration of the interactors33. Theoretical analysis of interacting membrane components favors the second idea34.
The encounter time between Gαq and p63RhoGEF, calculated based on the p63RhoGEF diffusion time of 0.1 μm/s2 that we measured, is about 0.3s for a cell expressing 10,000 copies of each molecule34. The encounter time probably reflects a minimum as we can easily obtain concentrations around 1 μM35,36 in transient expression systems, which would correspond to 1 million copies and hence a shorter encounter time. The actual half time of association between p63RhoGEF and Gαq as judged from dynamic FRET data, is 3s, which is at least an order of magnitude slower than the encounter time. Since non-membrane bound p63RhoGEF moves with a diffusion coefficient that is an order of magnitude faster (and GEFT diffuses with 2–5 μm2/s, M.A. Hink personal communication) the encounter rate with Gαq will be even faster. Still, our experiments show that Gαq and p63RhoGEF interact more efficiently. These considerations support the idea that concentration rather than encounter time is the factor that determines signaling efficiency. This hypothesis can be tested by controlling the local concentration in a cell and compare the situation before and after the manipulation. Experiments with the recruitment of GEFT under influence of rapamycin increase the effective concentration of the protein at the plasma membrane27,33 and have a strong effect on the signal transmission. Therefore, our observations support the concept that the increased concentration is the dominant factor.
Together, our data imply that, in our system, GEFT is under hardly any or no control of Gαq and suggest that p63RhoGEF mediates Gαq dependent remodeling of the cytoskeleton37,38. Still, it is possible that the location of GEFT is under control of other, unknown, factors and therefore GEFT may play a role in signaling through Gαq under conditions where it is recruited to the plasma membrane39.
The observation that signal transmission is dominated by membrane-located proteins has important implications for subsequent signaling steps. Both Gαq and p63RhoGEF are palmitoylated, a post-translational modification that is dynamically and spatially regulated40. Since p63RhoGEF and Gαq are implicated as oncoproteins41,42, it will be relevant to study whether the malignant activity of these proteins can be reduced by small molecules that interfere with palmitoylation43.
We show directly that the subcellular location is a key determinant for the interaction between Gαq and its effector, with membrane located p63RhoGEF interacting strongly with Gαq. For several other RhoGEFs, it was shown that they translocate to the plasma membrane upon stimulation of cells44,45. These data suggest that the plasma membrane is an important platform for RhoGEF activity, possibly by activating a specific subset of RhoGTPases. Hence, it will be key to study where the RhoGTPases and regulatory proteins, such as mixed lineage kinase 3 (MLK3)46 are located to understand signal transduction further down the cascade.
Methods
Materials
Fura Red/AM and lipofectamine 2000 were from Invitrogen (Bleiswijk, The Netherlands). Rapamycin was from LC Laboratories (Woburn, USA).
Construction of fluorescent protein fusions
Mammalian expression vectors were based on the pEGFP-N1 and pEGFP-C1 plasmids in which EGFP was replaced by the YFP variant mVenus16 or mCherry47. PCR products were inserted into pYFP-N1 using KpnI and AgeI restriction enzymes and into pYFP-C1 using BsrGI and KpnI restriction enzymes. Restriction sites are underlined in the primer sequences. Full length human p63RhoGEF9 was used as a template for PCR. To construct p63RhoGEF-YFP, p63RhoGEF was amplified using forward primer 5′-TAGGTACCACCATGCGGGGGGGGCACAAA-3′ and reverse primer 5′- CGACCGGTGACAGCTCATCTTCATCCAG-3′. To construct YFP-p63RhoGEF, template was amplified with forward primer 5′- GCTGTACAAGTCCCGGGGGGGGCACAAAGGG-3′ and reverse primer 5′- ACGGTACCTTACAGCTCATCTTCATCC-3′. To construct YFP-GEFT, template was amplified with forward primer 5′- GCTGTACAAGATGTTGGAGCCAGCTCTA-3′ and reverse primer 5′-ACGGTACCTTACAGCTCATCTTCATCC-3′. Deletion constructs p63(1–154)-YFP and p63(1–106)-YFP were made similar to p63RhoGEF-YFP, but instead using the reverse primer 5′-CGACCGGTGACTTCTGTTCCTCCTCACTC-3′ and 5′-CGACCGGTGACCAGTTCTCATATGGTCC-3′ respectively. The p63(1–29)-YFP was made by using the unique SmaI restriction site in p63RhoGEF. The L475A mutant was made using site-directed mutagenesis with the mutagenesis primers 5′- GACTTCCTCAACGCAGCGCAGTCACCCATTGAG-3′ and 5′- CTCAATGGGTGACTGCGCTGCGTTGAGGAAGTC-3′.
p63RhoGEF-YFP was cut with KpnI and SmaI to replace a fragment comprising aa 1–29 with phosphorylated oligonucleotide linkers that contained the Cys10Ser and Cys12Ser mutation or Cys23Ser, Cys25Ser and Cys26Ser mutations or all five Cys to Ser mutations. These constructs are indicated as p63-C2S-YFP, p63-C3S-YFP and p63-C5S-YFP respectively and their N-terminal amino acid sequence is shown in Figure 3A. A fully functional fluorescently tagged Gαq was made by inserting mVenus (YFP-Gαq) or mTurquoise (CFP-Gαq) in between residues 127 and 128 of Gαq24. A constitutively active VFP-tagged Gαq was made by introducing the Q209L mutation. Details of all constructs are available upon request. All constructs were verified by sequencing. The plasmids encoding mRFP-FKBP12-Gαq (wt and Q209L) and Lck-FRB-ECFP(W66A), were a kind gift of Mateusz Putyrski28.
To simplify the construction of FKBP12-tagged domains, we amplified the FKBP12 by PCR (fw 5′-GGTGTACAGAGCGGCGATGGGAGTGC-3′, rv 5′- CCAGATCTCCGGTACCCTGCTCCTCCTGCTCCC-3′) and inserted it into the mCherry-C1 plasmid using BsrGI and BglII to obtain mCherry-FKBP12-C1, retaining the reading frame and the restriction sites that are present in the Clontech-like C1 plasmids. Subsequently we obtained GEFT from YFP-GEFT using BsrGI and MfeI restriction sites and inserted it into mCherry-FKBP12-C1 cut with Acc65I and MfeI to obtain mCherry-FKBP12-GEFT. Yellow cameleon 3.60 was a kind gift of A. Miyawaki.
Cell culture and sample preparation
HeLa cells were cultured and prepared for microscopy as described48.
Microscopy
Confocal laser scanning microscopy was performed with a Zeiss LSM510 microscope equipped with a 63x oil immersion objective (Plan-Apochromat, NA 1.4) and the pinhole set to 1 airy unit to obtain optical slices (<0.8 μm). For YFP fluorescence, excitation was at 514 nm and the emission light was filtered through a 530–600 nm bandpass filter. For co-imaging of green and red fluorophores, excitation wavelengths of 488 nm and 543 nm were used and emission filters were 505–530 nm and LP560 nm or LP585 nm. For co-imaging of cyan and yellow fluorophores, excitation wavelengths of 458 nm and 514 nm were used and emission filters were 470–500 nm and 520–555 nm. To avoid bleed-through, images were acquired in multitrack mode. Calcium imaging with Fura Red (figure 6) was performed as described29. All images are representative of multiple experiments, i.e. multiple transfections performed on different days.
FRAP
The strip-FRAP method was applied essentially as described35,49 to measure diffusion in the basal membrane of cells. A 1.4 μm wide strip (40 pixels, zoom 8) was bleached by two scans at maximum excitation power (AOTF at 100% transmission) of the 488 nm and 514 nm laser line. Ten pre-bleach and 100 post-bleach images were acquired with a 500 ms interval. The resulting mean fluorescence from the strip was quantified. The recovery was fit to the empirical formula described previously50,51 to yield the effective diffusion time. At least ten cells were measured for each construct.
FRET ratio imaging
The calcium imaging experiments reported in figure 7 were performed using FRET ratio imaging of yellow cameleon 3.60. FRET ratio-imaging was performed as described24,52.
References
Fredriksson, R., Lagerstrom, M. C., Lundin, L. G. & Schioth, H. B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups and fingerprints. Mol Pharmacol 63, 1256–72 (2003).
Pierce, K. L., Premont, R. T. & Lefkowitz, R. J. Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3, 639–50 (2002).
Cabrera-Vera, T. M. et al. Insights into G protein structure, function and regulation. Endocr Rev 24, 765–81 (2003).
Wettschureck, N. & Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol Rev 85, 1159–204 (2005).
Rebecchi, M. J. & Pentyala, S. N. Structure, function and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 80, 1291–335 (2000).
Rhee, S. G. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 70, 281–312 (2001).
Berridge, M. J. Inositol trisphosphate and calcium signalling. Nature 361, 315–25 (1993).
Lutz, S. et al. The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem 280, 11134–9 (2005).
Rojas, R. J. et al. Galphaq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem 282, 29201–29210 (2007).
Lutz, S. et al. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318, 1923–7 (2007).
Souchet, M. et al. Human p63RhoGEF, a novel RhoA-specific guanine nucleotide exchange factor, is localized in cardiac sarcomere. J Cell Sci 115, 629–40 (2002).
Rossman, K. L., Der, C. J. & Sondek, J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6, 167–80 (2005).
Lutz, S. et al. p63RhoGEF and GEFT are Rho-specific guanine nucleotide exchange factors encoded by the same gene. Naunyn Schmiedebergs Arch Pharmacol 369, 540–6 (2004).
Shankaranarayanan, A. et al. Galpha q allosterically activates and relieves autoinhibition of p63RhoGEF. Cell Signal 22, 1114–23 (2010).
Aittaleb, M., Nishimura, A., Linder, M. E. & Tesmer, J. J. Plasma membrane association of p63 Rho guanine nucleotide exchange factor (p63RhoGEF) is mediated by palmitoylation and is required for basal activity in cells. J Biol Chem 286, 34448–56 (2011).
Kremers, G. J., Goedhart, J., van Munster, E. B. & Gadella, T. W., Jr Cyan and yellow super fluorescent proteins with improved brightness, protein folding and FRET Forster radius. Biochemistry 45, 6570–80 (2006).
Nagai, T. et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20, 87–90 (2002).
Teruel, M. N. & Meyer, T. Translocation and reversible localization of signaling proteins: a dynamic future for signal transduction. Cell 103, 181–4 (2000).
Kenworthy, A. K. Fluorescence-based methods to image palmitoylated proteins. Methods 40, 198–205 (2006).
Reits, E. A. & Neefjes, J. J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol 3, E145–7 (2001).
Adjobo-Hermans, M. J., Goedhart, J. & Gadella, T. W., Jr Plant G protein heterotrimers require dual lipidation motifs of Galpha and Ggamma and do not dissociate upon activation. J Cell Sci 119, 5087–97 (2006).
Henis, Y. I., Rotblat, B. & Kloog, Y. FRAP beam-size analysis to measure palmitoylation-dependent membrane association dynamics and microdomain partitioning of Ras proteins. Methods 40, 183–90 (2006).
Gilman, A. G. G proteins: transducers of receptor-generated signals. Annu Rev Biochem 56, 615–49 (1987).
Adjobo-Hermans, M. J. et al. Real-time visualization of heterotrimeric G protein Gq activation in living cells. BMC Biol 9, 32 (2011).
Adjobo-Hermans, M. J. et al. PLCbeta isoforms differ in their subcellular location and their CT-domain dependent interaction with Galphaq. Cell Signal 25, 255–263 (2013).
Piljic, A. & Schultz, C. Analysis of protein complex hierarchy in living cells. ACS Chem Biol 3, 749–55 (2008).
Goedhart, J. & Gadella, T. W., Jr Bypassing GPCRs with chemical dimerizers. Chem Biol 18, 1067–8 (2011).
Putyrski, M. & Schultz, C. Switching heterotrimeric G protein subunits with a chemical dimerizer. Chem Biol 18, 1126–33 (2011).
Adjobo-Hermans, M. J., Goedhart, J. & Gadella, T. W., Jr Regulation of PLCbeta1a membrane anchoring by its substrate phosphatidylinositol (4,5)-bisphosphate. J Cell Sci 121, 3770–7 (2008).
Nagai, T., Yamada, S., Tominaga, T., Ichikawa, M. & Miyawaki, A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A 101, 10554–9 (2004).
Momotani, K. et al. p63RhoGEF couples Galpha(q/11)-mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ Res 109, 993–1002 (2011).
Macrez-Lepretre, N., Kalkbrenner, F., Schultz, G. & Mironneau, J. Distinct functions of Gq and G11 proteins in coupling alpha1-adrenoreceptors to Ca2+ release and Ca2+ entry in rat portal vein myocytes. J Biol Chem 272, 5261–8 (1997).
Haugh, J. M. & Lauffenburger, D. A. Physical modulation of intracellular signaling processes by locational regulation. Biophys J 72, 2014–31 (1997).
Kholodenko, B. N., Hoek, J. B. & Westerhoff, H. V. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol 10, 173–8 (2000).
Luijsterburg, M. S. et al. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci 120, 2706–16 (2007).
van der Wal, J., Habets, R., Varnai, P., Balla, T. & Jalink, K. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J Biol Chem 276, 15337–44 (2001).
Barnes, W. G. et al. beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem 280, 8041–50 (2005).
Chikumi, H., Vazquez-Prado, J., Servitja, J. M., Miyazaki, H. & Gutkind, J. S. Potent activation of RhoA by Galpha q and Gq-coupled receptors. J Biol Chem 277, 27130–4 (2002).
Guo, X. et al. A Rac/Cdc42-specific exchange factor, GEFT, induces cell proliferation, transformation and migration. J Biol Chem 278, 13207–15 (2003).
Rocks, O. et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141, 458–71 (2010).
Liu, D., Yang, X., Yang, D. & Songyang, Z. Genetic screens in mammalian cells by enhanced retroviral mutagens. Oncogene 19, 5964–72 (2000).
Van Raamsdonk, C. D. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602 (2009).
Dekker, F. J. et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol 6, 449–56 (2010).
Bhattacharyya, R. & Wedegaertner, P. B. Characterization of G alpha 13-dependent plasma membrane recruitment of p115RhoGEF. Biochem J 371, 709–20 (2003).
Michiels, F. et al. Regulated membrane localization of Tiam1, mediated by the NH2-terminal pleckstrin homology domain, is required for Rac-dependent membrane ruffling and C-Jun NH2-terminal kinase activation. J Cell Biol 137, 387–98 (1997).
Swenson-Fields, K. I. et al. MLK3 limits activated Galphaq signaling to Rho by binding to p63RhoGEF. Mol Cell 32, 43–56 (2008).
Shaner, N. C. et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22, 1567–1572 (2004).
Goedhart, J., Vermeer, J. E., Adjobo-Hermans, M. J., van Weeren, L. & Gadella, T. W. Sensitive Detection of p65 Homodimers Using Red-Shifted and Fluorescent Protein-Based FRET Couples. PLoS ONE 2, e1011 (2007).
Hoogstraten, D. et al. Rapid switching of TFIIH between RNA polymerase I and II transcription and DNA repair in vivo. Mol Cell 10, 1163–74 (2002).
Ellenberg, J. & Lippincott-Schwartz, J. Dynamics and mobility of nuclear envelope proteins in interphase and mitotic cells revealed by green fluorescent protein chimeras. Methods 19, 362–72 (1999).
Ellenberg, J. et al. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol 138, 1193–206 (1997).
Verbeek, D. S., Goedhart, J., Bruinsma, L., Sinke, R. J. & Reits, E. A. PKC{gamma} mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J Cell Sci 121, 2339–49 (2008).
Acknowledgements
Part of this work is supported by the EU integrated project on "Molecular Imaging" LSHG-CT-2003-503259. We thank the members of our lab for encouraging discussions and especially Erik Manders for microscopy related support. We thank John Sondek & Rafael Rojas (Department of Pharmacology, Chapel Hill, USA) for supplying us with p63RhoGEF and for sharing data ahead of publication. R.Y. Tsien (UCSD, USA) generously provided the mFruit plasmids and M.Putyrski and C.Schultz (EMBL, Germany) provided the FKBP12-Gq constructs. The biosensor YC3.60 was provided by A. Miyawaki (RIKEN BSI, Japan). We thank Nathalie Reinhard for her contribution to the construction of mCherry-FKBP12-GEFT and Kevin Crosby for stimulating discussions.
Author information
Authors and Affiliations
Contributions
J.G., J.v.U., M.J.W.A. and T.W.J.G. designed experiments; J.G., J.v.U. and M.J.W.A. conducted experiments and analysed data; J.G. aided by the other authors, wrote the paper; All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SUPPLEMENTARY Figure S1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Goedhart, J., van Unen, J., Adjobo-Hermans, M. et al. Signaling efficiency of Gαq through its effectors p63RhoGEF and GEFT depends on their subcellular location. Sci Rep 3, 2284 (2013). https://doi.org/10.1038/srep02284
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02284
This article is cited by
-
The C-terminus of the oncoprotein TGAT is necessary for plasma membrane association and efficient RhoA-mediated signaling
BMC Cell Biology (2018)
-
Kinetics of recruitment and allosteric activation of ARHGEF25 isoforms by the heterotrimeric G-protein Gαq
Scientific Reports (2016)
-
Plasma membrane restricted RhoGEF activity is sufficient for RhoA-mediated actin polymerization
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
