
Abstract
The liverwort Marchantia polymorpha is an emerging model organism on account of its ideal characteristics for molecular genetics in addition to occupying a crucial position in the evolution of land plants. Here we describe a method for gene targeting by applying a positive/negative selection system for reduction of non-homologous random integration to an efficient Agrobacterium-mediated transformation system using M. polymorpha sporelings. The targeting efficiency was evaluated by knocking out the NOP1 gene, which impaired air-chamber formation. Homologous recombination was observed in about 2% of the thalli that passed the positive/negative selection. With the advantage of utilizing the haploid gametophytic generation, this strategy should facilitate further molecular genetic analysis of M. polymorpha, in which many of the mechanisms found in land plants are conserved, yet in a less complex form.
Similar content being viewed by others


Introduction
The emergence of land plants from an aquatic ancestor was one of the most critical evolutionary events for life on Earth. The most recent molecular phylogenetic analyses strongly support the sister relationship of the charophycean green algae to land plants and liverworts to all other extant land plants1,2. Therefore, liverworts are considered to be a key group to understand the genetic basis of the critical innovations that allowed green plants to evolve from an aquatic ancestor and to adapt to the terrestrial environment3.
Marchantia polymorpha is a dioecious liverwort species. The haploid generation is dominant in the life cycle, which provides advantages over diploid vascular plants for genetic analysis. M. polymorpha reproduces not only sexually but also asexually by means of bud-like structures called gemmae, which allows rapid propagation of isogenic biomass for molecular and biochemical experiments. Furthermore, an ongoing M. polymorpha genome sequencing project under the Community Sequencing Program at the Joint Genome Institute indicates that many of the biological mechanisms found in other land plants are conserved in a less complex form (http://www.jgi.doe.gov/sequencing/why/CSP2008/mpolymorpha.html)4,5. Thus, M. polymorpha is an emerging model organism, occupying a critical evolutionary position, with which to study specific molecular and cellular developmental processes in detail4,5,6,7,8.
Gene targeting mediated by homologous recombination (HR) is a powerful tool for functional analysis by reverse genetics. This technique is well established not only in bacteria and unicellular eukaryotes, but also in some multicellular model organisms such as fly9 and mouse10. However, gene targeting by HR is still difficult in most plants11,12, with the exception of the moss Physcomitrella patens, in which gene targeting by HR13 is highly efficient. In many other plants, integration of a transgene by HR occurs in the order of 10−3 to 10−6 compared with random integration (RI)14,15,16,17,18 and the overwhelming occurrence of RI of transgenes by non-homologous end-joining relative to targeted HR is a major obstacle for efficient gene targeting in plants. To overcome this problem, a reproducible gene-targeting procedure was developed in the monocot rice using a positive (the hygromycin-resistance gene, hpt)/negative (the diphtheria toxin A fragment gene, DT-A) selection system12,19. DT-A exerts cellular toxicity by ADP-ribosylation of elongation factor 2 upon protein synthesis20. A large number of stable transformants (104 to 105) must be generated to obtain gene-targeted lines because of the high background of the positive/negative selection system and a highly efficient transformation system is a critical factor for the success of such a gene targeting system21.
We recently developed a simple and rapid Agrobacterium-mediated transformation system for M. polymorpha22. Hundreds of stable transformants per sporangium can be obtained using sporelings (immature thalli developed from spores). The large number of transformants obtained per experiment using M. polymorpha is advantageous for gene targeting based on a positive/negative selection system. We selected NOPPERABO1 (NOP1) as a model target gene for evaluation of HR efficiency, because a visible but non-selective phenotype is shown when the gene is mutated. Twenty lines of 930 obtained hygromycin-resistant (Hmr) transformants showed the expected phenotype and their NOP1 loci were successfully disrupted by HR. This gene-targeting strategy opens the door for systematic functional genomics in the basal land plant M. polymorpha, which has a haploid-dominant life cycle.
Results
Effectiveness of the positive/negative selection
To efficiently detect rare events of HR in M. polymorpha, we employed a strong positive/negative selection system developed for rice12. We modified the rice targeting vector pINA134 to construct pJHY-TMp1, in which the hygromycin phosphotransferase (hpt) gene was driven by the promoter of the M. polymorpha elongation factor 1 alpha (EF1a) gene instead of the rice Actin1 promoter (Fig. 1a). A control vector, pJHY-CMp1, carried the same hpt cassette as pJHY-TMp1 in the T-DNA region but lacked the DT-A genes (Fig. 1a). To evaluate the efficiency of the negative selection by the DT-A gene in M. polymorpha, we compared the transformation efficiency of pJHY-CMp1 and pJHY-TMp1. Each sporangium contained 2–3 × 105 spores in M. polymorpha and 10–20% of the spores germinated after imbibition in our laboratory conditions. With pJHY-TMp1, only a few dozen Hmr transformants per sporangium survived the positive/negative selection, whereas over 1,000 Hmr transformants per sporangium were obtained using pJHY-CMp1 (Table 1). These results indicated that positive/negative selection with the hpt and DT-A genes was applicable to M. polymorpha.

Strategy for targeted disruption of the NOP1 locus and analysis of homologous recombination events.
(a) Structure of pJHY-TMp1 and pJHY-CMp1. Only the structure between the left and right borders (LB and RB, respectively) of each vector is shown. DT-A, Diphtheria toxin gene; hpt, hygromycin phosphotransferase gene; ΔEn, 3′ part of the maize En element; gus, β-glucuronidase gene. (b) Schematic representation of the genomic structure of the wild-type NOP1 gene region. (c) Structure of pKI406. (d) Structure of the NOP1 locus disrupted by HR. The exons and introns of the NOP1 gene are indicated by blue and open boxes, respectively. The green bar represents the NOP1 flanking region carried by pKI406. Primers are indicated by triangles. Recognition sites of EcoRI and HindIII are indicated by vertical lines with the letters E and H, respectively.
Targeted gene knockout by homologous recombination
As a model gene to target we chose NOP1, which is predicted to encode a plant U-box (PUB) type E3 ligase and acts as a critical positive regulator for air-chamber development in M. polymorpha (Ishizaki et al. in preparation). We selected NOP1 for the first trial of gene targeting in M. polymorpha because the defective gene confers an obvious morphological phenotype that can easily be assessed by visual observation but does not offer any selective disadvantages. The NOP1 gene comprises four exons, of which exon 2 carries the ATG initiation codon. Our targeting strategy was to disrupt NOP1 by replacing a 116 bp portion of exon 2 with an expression cassette for antibiotics resistance. The targeting vector, pKI406, was a pJHY-TMp1 derivative in which hpt-ΔEn is flanked by the 3.6 kb NOP1 upstream region containing the promoter region, first intron and 5′ untranslated region and the 3.5 kb NOP1 coding regions including two intron regions (Fig. 1b and c). HR between the NOP1 sequences was expected to result in substitution of the 116 bp sequence in the NOP1 exon 1 with hpt-ΔEn, without integrating the flanking DT-A genes (Fig. 1d).
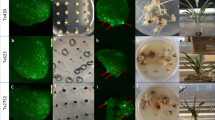
When pKI406 was used for transformation, an average of 186 Hmr thalli were obtained from each of five independent transformation experiments using four sporangia (Table 1). Of 930 Hmr transformants in total, 20 independent lines (#1 to #20) showed impaired air-chamber formation (Fig. 2) in the T1 generation, which was indistinguishable from that of the original NOP1 loss-of-function mutant (Ishizaki et al. in preparation). The loss-of-air-chamber phenotype in line #1 (Fig. 2b) was rescued by introduction of the NOP1 gene (Fig. 2c), which indicated that the phenotype was caused by targeted disruption of NOP1. To examine if the expected HR had occurred in all of the 20 lines, we employed genomic PCR with the three pairs of primers shown in Figure 1D. In all of the lines, but not in the wild type, fragments of the size expected for the HR sites (3.9 kb and 4.3 kb) were amplified by the primer pairs N1F/P1R and H1F/N1R, respectively (Figs. 1d and 2d), suggesting the occurrence of correct crossovers at both sides and eliminating the possibility of one-sided invasion. Furthermore, only a 4.2 kb fragment containing hpt-ΔEn was amplified in the 20 lines, whereas a 0.5 kb fragment was amplified from the wild-type NOP1 locus (Fig. 2d), indicating successful modification of the targeted NOP1 locus.

Phenotype and genotype of targeted transformants.
(a–c) The dorsal surface of the thallus of the wild type (a), transformant #1 (b) and the complemented transformant #1 in which the NOP1 genomic fragment was introduced. (c). Nineteen additional lines (#2 to #20) showed the same phenotype as #1. (d) Genotyping of the nop1 lines. The positions of the primers used for PCR analysis are shown in Figure 1.
To confirm that HR had occurred as expected, Southern blot analysis was performed using genomic DNA of the wild type and two lines, #5 and #9, randomly selected from among the 20 lines. In the EcoRI digests of the two selected lines, only 5.8 and 6.1 kb fragments corresponding to the disrupted NOP1 locus (Fig. 1d), but not the 8.4 kb wild-type fragment (Fig. 1b), were detected (Fig. 3a). A similar result was obtained for the HindIII digests (Fig. 3b). These results indicated that hpt-ΔEn was integrated only into the NOP1 locus in the haploid genome of the two lines examined.

Southern blot analysis of the NOP1 locus and the integrated hpt sequence in the wild type and the two independent Hmr lines that showed impaired air-chamber formation.
EcoRI (a) and HindIII (b) digests. The probes used and the restriction map of this locus are illustrated in Figure 1.
Assuming the NOP1 locus was disrupted also in the remaining 18 lines, the frequency of HR between pKI406 and the NOP1 locus was approximately 2% of the Hmr transformants that survived the positive/negative selection (Table 1).
Evaluation of gene targeting efficiency using vectors containing homologous arms of various lengths and positions
In order to optimize the design of targeting constructs, a series of vectors containing homologous arms of various lengths and inter-arm distances were constructed for comparison. The targeting vectors, pKI406-1kb and pKI406-2kb, contained shorter homologous arms, 1 and 2 kb, respectively, than those of the original vector, pKI406 (Fig. 4). The distances between the homologous arms of pKI406-d1kb and pKI406-d3kb, 1 and 3 kb, respectively, were larger than 0.1 kb of pKI406 (Fig. 4). When pKI406-1kb and pKI406-2kb were used for transformation, less than 0.015% and 0.7%, respectively, of resulting Hmr transformants showed the nop1 phenotype, whereas 3.2% showed the nop1 phenotype with pKI406 (Table 2). Similarly, 1.0% and 0.1% of resulting Hmr transformants showed the nop1 phenotype with pKI406-d1kb and pKI406-d3kb, respectively (Table 2). These results suggest that, in practice, the length of homologous arms in the targeting vector should be larger than 2 kb and the distance between homologous arms should not exceed 1 kb for gene targeting.

Constructs for optimization of gene-targeting in M. polymorpha.
The exons and introns of the wild-type NOP1 gene are represented by blue and open boxes, respectively. Positions and length of homologous arms cloned in the binary vectors are indicated as orange bars.
Discussion
We established a reproducible procedure for gene targeting in the liverwort M. polymorpha by application of a strong positive/negative selection system developed for rice12. The observed targeting frequency in M. polymorpha, as determined from the ratio of the number of targeted transformants resulting from HR to the number of transformants resulting from RI of the Hmr marker of pJHY-CMp1, was estimated to be 7.7 × 10−4 (Table 1), which is comparable to the frequencies previously reported (10−3 to 10−6)14,15,16. Therefore, high-yield Agrobacterium-mediated transformation by which thousands of transformants can be generated makes it feasible to isolate transformants resulting from HR. In M. polymorpha, it is possible to introduce a fragment of DNA, with a frequency sufficient for isolation of rare HR transformants, by the use of only a few sporangia. We recommend, in practice, to start with 10–20 sporangia to obtain a sufficient number of candidates for gene-targeting constructs. The whole procedure from initiation of sporeling culture to identification of targeted transformants took only 5–6 weeks. In practice, further culture for several weeks is required to establish isogenic lines (the G1 generation) that originate from single gemma-initial cells.
The positive/negative selection reduced the number of false positives and up to 3% of the selected transformants turned out to be correctly targeted as examined by genomic PCR and Southern blot analysis (Figs. 2 and 3). In the similar gene targeting system established in rice, the majority of the surviving calli with the positive/negative selection contained truncated T-DNA molecules mediated by border-independent RI, whereas the most efficient RI of T-DNA mediated by border-associated RI would result in the death of transformed calli caused by the intact DT-A gene19. There is also a possibility of the event called ectopic gene-targeting (EGT), which is also generated by HR-promoted crossover between the transgene and the target sequence and subsequent RI of the resulting recombinant molecule19. The target gene remains intact in both RI and EGT. In the case of the present study on M. polymorpha, the NOP1 locus is expected to be intact in the remainder of the Hmr transformants because they did not show the expected phenotype and the hpt cassette would be integrated either by border-independent RI or EGT.
The length of homologous arms and the inter-arm distances were critical factors for successful gene-targeting in M. polymorpha (Fig. 4 and Table 2). Similarly, in mammalian cells and the moss P. patens, an increase in the length of homologous arms resulted in a dramatic increase in targeting efficiency23,24. The gene targeting is regarded to occur via double crossovers in the hpt-flanking homologous regions on the vector19. A longer homologous arm would be more effective to search for the target locus and form a crossover structure. A Short inter-arm region would enhance the occurrence of crossovers at both homologous arms. As the distance between homologous arms should not exceed 1 kb for gene targeting in practice, it is generally difficult to design a knockout construct that will eliminate the entire protein coding region in a target gene. Therefore, in M. polymorpha the recommended design of a construct for targeted gene knockout is to interrupt gene structure with the hpt cassette. The position of the short inter-arm region should be located at an ATG codon to cancel the translation, or at a domain critical for the function of the target gene.
In M. polymorpha, gene-targeted or knockout transformants can be established immediately after transformation of gametophytes owing to their haploid status. Gene-targeted transformants can be readily screened by PCR or visual examination of their phenotypes in the T1 generation. The method described here is, in principle, applicable to any gene because it does not involve gene-specific selection. Indeed, we have succeeded with application of this strategy to disrupt the other endogenous genes (unpublished data). However, disruption of some endogenous genes would lead to lethality in a haploid system, thus generating no targeted plants. In this respect, development of a conditional knockout strategy based on an inducible site-specific recombination system, such as Cre/lox25 with inducibility, will expand the potential of this gene-targeting strategy in M. polymorpha.
Gene targeting mediated by HR should be useful not only for generation of knockouts but also for modification of targeted sequences and fusion of tags by knock-in. The liverwort M. polymorpha has substantial potential as a model system for plant biology on account of its critical phylogenetic position in the evolution of land plants, its conserved yet less complex developmental pattern and responses to plant growth factors and environmental stimuli and its gametophyte-dominant life cycle, which renders genetic analyses less complicated. HR-mediated gene targeting together with the ongoing genome sequencing project should accelerate functional analysis of genes in M. polymorpha and shed light on the evolution and diversity of regulatory systems in land plants.
Methods
Plant material and growth conditions
Male and female accessions of M. polymorpha, Takaragaike-1 and Takaragaike-2, respectively, were maintained asexually22. F1 spores generated by crossing Takaragaike-1 and Takaragaike-2 were used for transformation. Formation of sexual organs was induced by far-red irradiation as described previously26. Mature sporangia were collected 3–4 weeks after crossing, air-dried for 7–10 d and stored at −80°C until use.
Plants were cultured using half-strength Gamborg's B5 medium27 containing 1% agar under 50–60 μmol photons m−2 s−1 continuous white fluorescent light at 22°C unless otherwise defined.
Vector construction for gene targeting
The vectors pJHY-CMp1 and pJHY-TMp1 were derivatives of pRIT1 and pINA13412, respectively. To generate pJHY-CMp1 and pJHY-TMp1, the rice Actin I promoter driving hpt expression in pRIT1 and pINA134 was replaced with an endogenous promoter of the M. polymorpha elongation factor 1α (EF1a) gene. To generate the NOP1-targeting vector, pKI406, the 5′- and 3′-homologous arms (3.6 kb and 3.5 kb, respectively) were amplified from Takaragaike-1 genomic DNA by PCR using KOD FX Neo (Toyobo Co., Ltd., Osaka, Japan) with primer pairs NOP1-5IF-L/NOP1-5IF-R and NOP1-3IF-L/NOP1-3IF-R, respectively. The PCR-amplified 5′- and 3′-homologous arms were cloned into the PacI and AscI sites, respectively, of pJHY-TMp1 with the In-Fusion HD cloning kit (Clontech, Mountain View, CA).
In order to generate a series of NOP1-targeting vectors with homologous arms of different lengths and inter-arm distances, 5′- and 3′-homologous arms were amplified by PCR using the following primer pairs and cloned into the PacI and AscI sites of pJHY-TMp1, respectively, as in the construction of pKI406. For pKI406-1kb, the 5′- and 3′-homologous arms (1.1 and 1.2 kb, respectively) were amplified using primer pairs NOP1-5IF-L-1kb/NOP1-5IF-R and NOP1-3IF-L/NOP1-3IF-R-1kb, respectively. For pKI406-2kb, the 5′- and 3′-homologous arms (2.1 and 2.0 kb, respectively) were amplified using primer pairs NOP1-5IF-L-2kb/NOP1-5IF-R and NOP1-3IF-L/NOP1-3IF-R-2kb, respectively. The 5′-homologous arms of pKI406-d1kb and pKI406-d3kb were the same as that of pKI406 and their 3′-homologous arms (3.6 kb for each) were amplified using primer pairs NOP1-3IF-L-d1kb/NOP1-3IF-R-d1kb and NOP1-3IF-L-d3kb/NOP1-3IF-R-d3kb, respectively.
Sequences of all primers used in this study are given in Supplementary Table S1.
Introduction of targeting constructs into M. polymorpha
Agrobacterium tumefaciens strain C58C1 (GV2260) was used for transformation. Transformation of M. polymorpha was performed as previously described22 with minor modifications. After co-cultivation with A. tumefaciens, sporelings were transferred to half-strength Gamborg's B5 agar medium containing 1% agar, 10 mg l−1 hygromycin (Wako Pure Chemical Industries, Osaka, Japan) and 100 mg l−1 cefotaxime (Claforan, Sanofi-Aventis K. K., Tokyo, Japan). Independent T1 lines were transferred to fresh selection medium, grown for a further 2 weeks and screened for gene-targeted lines by visual examination of phenotypes and also by genotyping as described below. Isogenic lines (the G1 generation) were established from T1 lines using gemmae that arose from single cells as described previously7. Plants grown from gemmae of G1 lines (the G2 generation) were used for further confirmation of targeted disruption of the NOP1 locus.
Complementation of NOP1 targeted lines
For complementation a binary vector, pMpGWB301 (manuscript in preparation), harbouring a mutated ALS gene (mALS) that confers chlorosulfuron resistance was used. For construction of a plasmid containing the NOP1 genomic fragment, the promoter and coding region were amplified by PCR using the primer sets ProL/ProR and gCDSL/gCDSR, respectively. These PCR fragments were cloned into pENTR/D-TOPO (Life Technologies, Carlsbad, CA) to generate pAM101 for the promoter and pAM102 for the coding region of the NOP1 genomic fragment, respectively. The AvrII-AscI DNA fragment of pAM101 was subcloned into the AvrII-AscI site of pAM102 to generate the plasmid pAM103, which contained the NOP1 genomic fragment from 5.7 kb upstream of ATG to 0.2 kb downstream of the 3′UTR. The resultant NOP1 cassette was subcloned into pMpGWB301 using LR clonase (Life Technologies) in accordance with the manufacturer's protocol. The resulting plasmid was transformed into regenerating thalli of the NOP1 targeted line #1 by the method described previously28. Selection was conducted with 0.5 μM chlorosulfuron (DuPont, Wilmington, DE).
Genotyping
Small pieces (3 × 3 mm) of thalli were taken from individual plants and crushed with a micro-pestle in 100 μl buffer containing 100 mM Tris-HCl, 1 M KCl and 10 mM EDTA (pH 9.5). Sterilized water (400 μl) was added to each tube and 1 μl aliquot of the extract was used as a template for PCR using KOD FX Neo DNA polymerase (Toyobo). The PCR conditions were 96°C for 2 min, followed by 35 cycles of 98°C for 10 s, 68°C for 4 min and final extension at 72°C for 5 min. Primers used are shown in Supplementary Table S1.
Southern blot analysis
Total DNA for Southern blot analysis was extracted from approximately 5 g fresh weight of tissue with a cetyltrimethylammonium bromide (CTAB) method29 with modifications. The tissue was frozen in liquid nitrogen and disrupted with a mortar and pestle. Disrupted cells were extracted with 10 ml CTAB buffer (1.5% CTAB, 75 mM Tris-HCl [pH 8.0], 15 mM EDTA and 1.05 M NaCl) for 20 min at 56°C. The extract was mixed thoroughly with an equal volume of chloroform–isoamylalcohol (24:1, v/v) for 20 min at room temperature. After centrifugation at 4,000 × g for 20 min at 20°C, the aqueous phase was transferred to a fresh plastic tube and mixed with an equal volume of chloroform–isoamylalcohol (24:1, v/v) for 20 min at room temperature. After centrifugation at 4,000 × g for 20 min at 20°C, the aqueous phase was mixed slowly with 1.5 volume of CTAB precipitation buffer (1% CTAB, 50 mM Tris-HCl [pH 8.0] and 10 mM EDTA). After centrifugation at 10,000 × g for 30 min at 20°C, the precipitate was dissolved in 1 M sodium chloride supplemented with RNaseA (final 10 μg ml−1) and incubated for 30 min at 37°C. DNA was precipitated with ethanol and dissolved in TE buffer.
For Southern blot analyses, 2 μg genomic DNA was digested overnight with EcoRI and HindIII at 37°C. The DNA was fractionated by electrophoresis on a 0.8% (w/v) agarose gel and blotted onto a positively charged nylon membrane Biodyne A (PALL, Port Washington, NY). The probes PN3.6 (3,624 bp), Hm1.0 (1,000 bp) and CN0.7 (700 bp) were amplified by PCR with the primer pairs NOP1-5IF-L/NOP1-5IF-R, Hm1.0-F/Hm1.0-R and CN0.7-F/CN0.7-R, respectively. The blotted membranes were hybridized in Church hybridization buffer30 at 65°C with the probes labelled with [α-32P] dCTP by random priming using the Random Primer Labeling Kit Ver. 2 (Takara, Shiga, Japan). Washing and analysis of the blots was performed as described previously26.
References
Turmel, M., Otis, C. & Lemieux, C. The mitochondrial genome of Chara vulgaris: insights into the mitochondrial DNA architecture of the last common ancestor of green algae and land plants. Plant Cell 15, 1888–1903 (2003).
Qiu, Y. L. et al. The deepest divergences in land plants inferred from phylogenomic evidence. Proc. Natl. Acad. Sci. U. S. A. 103, 15511–15516 (2006).
Bowman, J. L., Floyd, S. K. & Sakakibara, K. Green genes-comparative genomics of the green branch of life. Cell 129, 229–234 (2007).
Zobell, O., Faigl, W., Saedler, H. & Munster, T. MIKC* MADS-box proteins: conserved regulators of the gametophytic generation of land plants. Mol. Biol. Evol. 27, 1201–1211 (2010).
Ueda, M. et al. Composition and physiological function of the chloroplast NADH dehydrogenase-like complex in Marchantia polymorpha. Plant J. 72, 683–693 (2012).
Tougane, K. et al. Evolutionarily conserved regulatory mechanisms of abscisic acid signaling in land plants: characterization of ABSCISIC ACID INSENSITIVE1-like type 2C protein phosphatase in the liverwort Marchantia polymorpha. Plant Physiol. 152, 1529–1543 (2010).
Ishizaki, K., Nonomura, M., Kato, H., Yamato, K. T. & Kohchi, T. Visualization of auxin-mediated transcriptional activation using a common auxin-responsive reporter system in the liverwort Marchantia polymorpha. J. Plant Res. 125, 643–651 (2012).
Okumura, M. et al. Characterization of the plasma membrane H+-ATPase in the liverwort Marchantia polymorpha. Plant Physiol. 159, 826–834 (2012).
Rong, Y. S. & Golic, K. G. Gene targeting by homologous recombination in Drosophila. Science 288, 2013–2018 (2000).
Capecchi, M. R. Altering the genome by homologous recombination. Science 244, 1288–1292 (1989).
Puchta, H. Gene replacement by homologous recombination in plants. Plant Mol. Biol. 48, 173–182 (2002).
Terada, R., Urawa, H., Inagaki, Y., Tsugane, K. & Iida, S. Efficient gene targeting by homologous recombination in rice. Nat. Biotechnol. 20, 1030–1034 (2002).
Schaefer, D. G. & Zryd, J. P. Efficient gene targeting in the moss Physcomitrella patens. Plant J. 11, 1195–1206 (1997).
Paszkowski, J., Baur, M., Bogucki, A. & Potrykus, I. Gene targeting in plants. EMBO J. 7, 4021–4026 (1988).
Risseeuw, E., Offringa, R., Franke-van Dijk, M. E. & Hooykaas, P. J. Targeted recombination in plants using Agrobacterium coincides with additional rearrangements at the target locus. Plant J. 7, 109–119 (1995).
Hanin, M. et al. Gene targeting in Arabidopsis. Plant J. 28, 671–677 (2001).
Hanin, M. & Paszkowski, J. Plant genome modification by homologous recombination. Curr. Opin. Plant Biol. 6, 157–162 (2003).
Iida, S. & Terada, R. A tale of two integrations, transgene and T-DNA: gene targeting by homologous recombination in rice. Curr. Opin. Biotechnol. 15, 132–138 (2004).
Terada, R., Johzuka-Hisatomi, Y., Saitoh, M., Asao, H. & Iida, S. Gene targeting by homologous recombination as a biotechnological tool for rice functional genomics. Plant Physiol. 144, 846–856 (2007).
Pappenheimer, A. M. Jr. Diphtheria toxin. Annu. Rev. Biochem. 46, 69–94 (1977).
Terada, R., Asao, H. & Iida, S. A large-scale Agrobacterium-mediated transformation procedure with a strong positive-negative selection for gene targeting in rice (Oryza sativa L.). Plant Cell Rep. 22, 653–659 (2004).
Ishizaki, K., Chiyoda, S., Yamato, K. T. & Kohchi, T. Agrobacterium-mediated transformation of the haploid liverwort Marchantia polymorpha L., an emerging model for plant biology. Plant Cell Physiol. 49, 1084–1091 (2008).
Hasty, P., Rivera-Perez, J. & Bradley, A. The length of homology required for gene targeting in embryonic stem cells. Mol. Cell. Biol. 11, 5586–5591 (1991).
Kamisugi, Y., Cuming, A. C. & Cove, D. J. Parameters determining the efficiency of gene targeting in the moss Physcomitrella patens. Nucleic Acids Res. 33, e173 (2005).
Sauer, B. Manipulation of transgenes by site-specific recombination: use of Cre recombinase. Methods Enzymol. 225, 890–900 (1993).
Chiyoda, S., Ishizaki, K., Kataoka, H., Yamato, K. T. & Kohchi, T. Direct transformation of the liverwort Marchantia polymorpha L. by particle bombardment using immature thalli developing from spores. Plant Cell Rep. 27, 1467–1473 (2008).
Gamborg, O. L., Miller, R. A. & Ojima, K. Nutrient requirements of suspension cultures of soybean root cells. Exp. Cell Res. 50, 151–158 (1968).
Kubota, A., Ishizaki, K., Hosaka, M. & Kohchi, T. Efficient Agrobacterium-mediated transformation of the liverwort Marchantia polymorpha using regenerating thalli. Biosci. Biotechnol. Biochem. 77, 167–172 (2013).
Murray, M. G. & Thompson, W. F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4325 (1980).
Church, G. M. & Gilbert, W. Genomic sequencing. Proc. Natl. Acad. Sci. U. S. A. 81, 1991–1995 (1984).
Acknowledgements
We thank Akane Kubota and Miya Mizutani for technical assistance and discussions. We thank Katsuyuki T. Yamato and Ryuichi Nishihama for critical reading of the manuscript. This work was supported by a KAKENHI grant-in-aid for Scientific Research on Priority Area (No. 23012025 to T.K.), Scientific Research on Innovative Area (No. 23119510 to K.I.), from the Ministry of Education, Culture, Sports, Science and Technology of Japan and Scientific Research (B) (No. 22370001 for S. Iida), Young Scientists B (No. 22770035 to K.I.) from the Japan Society for the Promotion of Science, the Asahi Glass Foundation and SUNTRY Foundation for Life Sciences (to K.I.).
Author information
Authors and Affiliations
Contributions
K.I., Y.J.-H., S. Iida and T.K. designed the research, interpreted the data and wrote the paper. Binary vectors for gene targeting were designed and constructed by Y.J.-H. and S. Iida. Gene targeting experiments were performed by K.I. and S. Ishida.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Ishizaki, K., Johzuka-Hisatomi, Y., Ishida, S. et al. Homologous recombination-mediated gene targeting in the liverwort Marchantia polymorpha L.. Sci Rep 3, 1532 (2013). https://doi.org/10.1038/srep01532
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01532
This article is cited by
-
A non-canonical BZR/BES transcription factor regulates the development of haploid reproductive organs in Marchantia polymorpha
Nature Plants (2024)
-
Stomatal regulators are co-opted for seta development in the astomatous liverwort Marchantia polymorpha
Nature Plants (2023)
-
Charting the genomic landscape of seed-free plants
Nature Plants (2021)
-
Design principles of a minimal auxin response system
Nature Plants (2020)
-
Physiological function of photoreceptor UVR8 in UV-B tolerance in the liverwort Marchantia polymorpha
Planta (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
