
Abstract
Since the first proposal that fullerenes are capable of hosting atoms, ions, or clusters by the late Smalley in 1985, tremendous examples of endohedral metallofullerenes (EMFs) have been reported. Breaking the dogma that monometallofullerenes (mono-EMFs) always exist in the form of M@C2n while clusterfullerenes always require multiple (two to four) metal cations to stabilize a cluster that is unstable as a single moiety, here we show an unprecedented monometallic endohedral clusterfullerene entrapping an yttrium cyanide cluster inside a popular C82 cage—YCN@Cs(6)-C82. X-ray crystallography and 13C NMR characterization unambiguously determine the cage symmetry and the endohedal cyanide structure, unexpectedly revealing that the entrapped YCN cluster is triangular. The unprecedented monometallic clusterfullerene structure unveiled by YCN@Cs(6)-C82 opens up a new avenue for stabilizing a cluster by a single metal cation within a carbon cage and will surely stimulate further studies on the stability and formation mechanism of EMFs.
Similar content being viewed by others

Introduction
The spherical empty interior of fullerene triggers intuitively the idea of entrapping atoms into the cage so as to modulate the molecular and electronic properties of fullerenes1,2. Endohedral metallofullerenes (EMFs) thus formed have now been revealed to host a variety of species inside the carbon cage, including atoms, ions, or clusters2,3,4,5,6,7,8. EMFs exhibit unique electronic properties as a consequence of charge transfer and electrostatic interactions between the entrapped species and the carbon cage, enabling their potential applications in electronics and biomedicine and so on2,3,4,5,6,7,8. As the first and representative conventional EMFs, monometallofullerenes (mono-EMFs) had been extensively investigated in the early study of EMFs and nowadays it has been commonly accepted that mono-EMFs always exist in the form of M@C2n1,2,3,4,5. Among them, La@C82 is the first stable monometallofullerenes isolated by Smalley et al in 19912, featuring a three-electron transfer from La to the C82 cage1,2,3. Later on, a wealth of other mono-EMFs M@C2n (M = Sc, Y, La-Lu, Ca, Mg) have been successfully isolated, featuring the transfer of two or three electrons from the entrapped metal ion to the carbon cage, which is sufficient for the mutual stabilization of the entrapped metal cation and carbon cage that are not even available in their empty form1,2,3,4,5,6. Thus, it seems improbable to entrap a single metal cation in the form other than M@C2n.
During the past decade the family of EMFs was significantly enlarged by the clusterfullerenes especially metal nitride clusterfullerenes8,9,10,11. With the isolation and identification of Sc3N@C80 as the first metal nitride clusterfullerene in 1999 (ref. 9), the concept of stabilizing multiple metal ions by cluster formation with nonmetal atoms has brought about the consequent discovery of other five types of clusterfullerenes8, including metal carbide clusterfullerenes (MxC2@C2n)12,13, metal hydrocarbon clusterfullerenes (Sc3CH@C80)14, metal oxide clusterfullerenes (e.g., Sc4O2@C80)15, metal sulfide clusterfullerenes (e.g., Sc2S@C82)16 and metal carbonitride clusterfullerene (Sc3NC@C80)17. Interestingly, upon extending the number of entrapped metal ions within endohedral fullerenes, usually the supposed multimetallofullerenes (Mx@C2n+2) turn out to be metal carbide clusterfullerenes (MxC2@C2n) instead12,13. Noteworthy, for all the reported six types of clusterfullerenes, multiple (two to four) metal cations are always required for the metal clusters, which are highly charged and thus unstable as a single molecule. As a consequence, an intriguing question is whether a single metal cation is able to stabilize a cluster in the carbon cage or not.
Herein, we report the discovery of an unprecedented stable monometallic clusterfullerene entrapping an yttrium cyanide cluster inside a popular C82 cage – YCN@Cs(6)-C82. The synthesis, isolation and unambiguous structural determination by single crystal X-ray crystallography and 13C NMR characterization of YCN@Cs(6)-C82 are elaborately presented. The electronic structures of YCN@Cs(6)-C82 have been systematically studied by various spectroscopies, revealing the intriguing feature of this brand-new entrant of EMF family.
Results
Synthesis, isolation and structural elucidation of YCN@C82
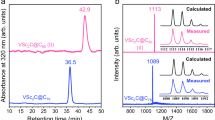
Soot containing YCN@C82 was synthesized by the modified Krätschmer-Huffman DC-arc discharging method using a 1:1 mixture of Y2O3 and TiO2 as the raw material under 400 mbar He and 10 mbar N2 (refs. 19,20) and the introduction of TiO2 in the raw mixture is found to be essential for the formation of YCN@C82. Isolation of YCN@C82 (~10 mg) was accomplished by five-step HPLC and the high purity of the isolated YCN@C82 (≥99.5%) is assured by both the recycling HPLC and laser desorption time-of-flight (LD-TOF) mass spectroscopic (MS) analysis (Supplementary Information S1–S2).
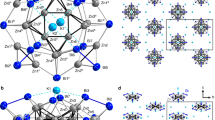
The molecular structure of YCN@Cs(6)-C82 is unambiguously determined by single crystal X-ray diffraction crystallography performed on a high-quality cocrystal of YCN@C82 with NiII(OEP) (OEP = octaethylporphyrin), which was obtained by diffusing a benzene solution of YCN@C82 into a chloroform solution of NiII(OEP)1,2,3,4,5,6,7,8,9,21. The asymmetric unit cell of the cocrystal YCN@C82·NiII(OEP)·1.73C6H6·1.27CHCl3 contains an entire molecule of both YCN@C82 and NiII(OEP), while the cavities are filled with disordered solvent molecules. Although NiII(OEP) is fully ordered, two disordered cage orientations with respective occupancies of 0.55 and 0.45 are present and as many as five metal positions are distinghuishable (occupancy: 0.50, 0.25, 0.19, 0.03 and 0.03 for Y1, Y2, Y3, Y4 and Y5, respectively). In contrast, the CN unit is fully ordered which is located near the center of the fullerene cage (Supplementary Information S3). Fig. 1 shows the X-ray structure of YCN@C82·NiII(OEP)·1.73C6H6·1.27CHCl3 involving only the major cage (0.55 occupancy), the major yttrium (0.50 occupancy) and the CN unit (1.0 occupancy) and its relation to the NiII(OEP) molecule and all solvent molecules were omitted for clarity. It clearly reveals the presence of an 82-atom carbon cage with Cs(6) symmetry instead of an 84-atom cage, thus ruling out the possibility of the endohedral azafullerene structure Y@C83N.

Drawing of the crystallographically determined structure of YCN@Cs(6)-C82 with the major C82 cage (0.55 occupancy) and the most abundant yttrium location (0.50 occupancy) and its relation to the NiII(OEP) molecule.
Thermal ellipsoids are set at 30% probability level. Solvent molecules, the minor cage and minor metal positions are omitted for clarity. Inset shows the triangular configuration of the internal YCN cluster to ensure a clear visualization. Red: Y; Blue: N; Grey: C.
For the endohedral YCN moiety, both N and C atoms are located near the cage center, wheareas the metal is located between the CN-unit and one side of the cage. As a result, the entrapped YCN cluster is triangular, but not linear (see inset of Fig. 1). For the major yttrium atom site with a 0.50 occupancy (Y1), the Y1-N and Y1-C bond distances are 2.383 Å and 2.484 Å, respectively. The shorter bond distance of Y1-N compared to Y1-C suggests a relatively stronger covalent bond between Y and N atoms, which is due to the larger electronegativity of N atom compared to C atom. Note that the determined N-C bond length, 0.935 Å, is smaller than that reported for Sc3NC@C80 (1.193 Å)17 and those of the C-N triplet bonds in traditional inorganic metal cyanide compounds or cyano coordination complexes (e.g. 1.14 – 1.17 Å for AuCN and [Au(CN)2]−)22 and nitrile compounds (e.g. 1.157 Å for CH3CN)23. The unusually short N–C bond indicates an extraordinary bonding nature in the YCN cluster upon entrapped in carbon cage. Interestingly, the shrinking of the C–N bond found in the present study is similar to that found for the C–C bond in the metal carbide clusterfullerenes such as Gd2C2@D3(85)-C92, Sc2C2@C3v(8)-C82(Ad) and Sc3C2@Ih-C80, in which the X-ray crystallographically determined C-C bond lengths (1.04, 1.107, 1.11 Å respectively13,24,25) are shorter than those predicted by theoretical computations and the C-C triplet bonds in alkyne compounds (1.21 Å). The stronger bonding between Y and C/N atoms due to the space confinement of the interior of the carbon cage might be responsible for such a shrinking phenomenon. Besides, the triangular structure of YCN cluster is rather intriguing since it is different from the linear structures commonly found in traditional metal cyanide complexes22,23. Recently, Dorn et al. revealed that the Y2C2 cluster tends to adopt a linear structure if the interior of the fullerene cage is sufficiently spacious whereas a constrained “butterfly” structure is adopted upon decreasing the size of a fullerene cage26. However, in our present case of YCN@Cs(6)-C82, a linear structure is less favoured than the triangular conformation although the space is obviously enough for both and this is presumably due to the conjunct effects of the strong coordination ability of the yttrium metal and the confining effect of the cage. Such an unprecedented triangular cyanide cluster configuration provides a new concept into modern coordination chemistry, specifically with the charged monometallic complexes inside a confined nanospace.
13C NMR spectroscopy further corroborates the determined cyanide cluster structure and the cage symmetry of YCN@Cs(6)-C82 as well (Fig. 2). The apparent 44 signals in the aromatic region (120–155 ppm), in which 38 lines are in full intensity and 6 lines are in half intensity, point to either a Cs-symmetric C82 cage instead of any C84-isomer or the isomeric carbide cluster azafullerene structure YC2@C81N (Supplementary Information S4)27. More convincingly, the 13C NMR signal of the internal CN-unit is observed at δ = 292.37 ppm with the intensity being comparable to those of the 6 cage carbon atoms on the mirror plane. This value is obviously downfield shifted as compared with those of the analogues carbide clusterfullerenes with the same cage (e.g. 244.4 ppm for Sc2C2@Cs(6)-C8228, 256.2 ppm for Y2C2@Cs(6)-C82)26. Besides, compared to the entrapped NC moiety of the reported metal carbonitride clusterfullerenes Sc3NC@C80, for which the experimental 13C NMR signal was unknown17 while the simulated chemical shift is 306.6 ppm18, the chemical shift of the internal CN-unit of YCN@Cs(6)-C82 might be different since the formal charge state of the internal CN-unit of YCN@Cs(6)-C82 is different to that of the entrapped NC moiety of Sc3NC@C80 as discussed below.

13C NMR (125 MHz) spectrum of YCN@C82 showing a 38 × 2C (labeled by blue numbers), 6 × 1C (labeled by red numbers) pattern for the sp2 carbon atoms of the cage.
The filled triangle marks the 13C nuclei signal of the internal YCN cluster and the asterisk labels an unidentified impurity.
A more detailed analysis of the 13C NMR results of YCN@C82 can further exclude the possibility of the isomeric carbide cluster structure with an azafullerene cage, namely YC2@C81N, for which two substitution sites of nitrogen atom in the C82 cage are possible: 1) if the nitrogen atom substitutes any of the 6 carbon atoms on the mirror plane, 43 lines for the sp2 carbon atoms (38 lines with full intensity + 5 lines with 1/2 intensity) plus 1 or 2 line(s) in the much lower field contributed from the encaged C2 moiety would be observed; 2) if the nitrogen atom substitutes any of the 76 carbon atoms apart from the mirror plane, the whole molecule would exhibit as many as 81 lines of the carbon cage because of the lowered symmetry. Therefore, the observed 13C NMR spectrum of our new endohedral compound rules out the possible structure of YC2@C81N. In addition, according to our DFT computations, the alternative most stable structure of YC2@C81N is remarkably energy-unfavorable. Among all the considered YC2@C81N isomers, those with the nitrogen atom substituting any of the 6 carbon atoms on the mirror plane of the Cs-C82 cage (i.e., the C81N cages remain the Cs symmetry) are ~80 kcal/mol higher in energy than the lowest-energy YCN@C82 structure, while others are at least 20.2 kcal/mol higher in energy.
Electronic structure of YCN@C82
The electronic structure of YCN@C82 was studied by UV-vis-NIR, X-ray photoemission spectroscopy (XPS), electron spin resonance (ESR) spectroscopy and electrochemistry (Supplementary Information S5-S7). The XPS result shows that the Y atom takes a valency of +3 in YCN@C82 (Supplementary Information S6). Room-temperature ESR spectroscopic measurement reveals that YCN@C82 is ESR-silent. This is dramatically different to the analogous C82-based mono-EMFs entrapping a trivalent Group-III metal M@C82 (M = Sc, Y, La etc.), which are typically ESR-active due to their open-shell electronic structures3,4,5,6,7. Instead, divalent metal-based C82 mono-EMFs, such as Yb@C82, Ca@C82 and Sm@C82, are usually ESR-silent owing to their closed-shell structures5,27. The similarity of the ESR behaviour of YCN@C82 to those of divalent metal-based C82 mono-EMFs suggests a similar two-electron transfer. Thus, an electronic configuration of [Y3+(CN)−]2+@[C82]2− can be proposed, for which the (CN)− moiety typically existing in the traditional metal cyanide inorganic compounds22 is obviously different to the [NC]3− unit within the reported [Sc3+]3[NC]3−@[C80]6− (refs. 17, 18). Indeed, the proposed electronic configuration of [Y3+(CN)−]2+@[C82]2− is confirmed by the cyclic voltammetric study. YCN@C82 exhibits one reversible oxidation step at +0.56 V and four reversible reduction steps at −0.59, −0.84, −1.76 and −1.92 V versus Fc/Fc+, respectively (Fig. 3). Interestingly, the first two reduction steps as well as the last two reduction steps are mutually close, whereas there is an abrupt large separation (0.92 V) between the second and third reduction steps (see also Supplementary Information S8). Such a reduction behaviour of YCN@C82 highly resembles that of the reported Yb@Cs(6)-C82, for which one (quasi-) reversible oxidation step and four reversible reduction steps specifically with a comparable large separation (0.89 V) between the second and third reduction steps were observed27. Again, such a high resemblance of the reduction behaviour of YCN@Cs(6)-C82 to that of Yb2+@[Cs(6)-C82]2− confirms the proposed electronic configuration of [Y3+(CN)−]2+@[C82]2− featuring a formal two-electron transfer from the entrapped YCN moiety to C82 cage.

Cyclic voltammogram of YCN@C82 in o-dichlorobenzene (o-DCB) solution with ferrocene (Fc) as the internal standard and tetrabutylamonium hexafluorophosphate (TBAPF6) as supporting electrolyte.
Scan rate: 100 mV/s. Each redox step is marked with a number and a solid dot to aid comparison. The small peak at around −1.50 V is due to an unknown impurity. The asterisk labels the oxidation peak of ferrocene.
Discussion
It has been commonly believed that monometallofullerenes (mono-EMFs) always exist as a simple form of M@C2n and clusterfullerenes always require multiple (two to four) metal cations to stabilize a cluster that is unstable as a single moiety. Now we break these dogmas by presenting the first example of monometallic clusterfullerene entrapping an improbable yttrium cyanide cluster — YCN@Cs(6)-C82. X-ray crystallography and 13C NMR spectroscopic study assure the elucidation of its cage isomeric structure of Cs(6)-C82, which entraps an unprecedented triangular YCN cluster. A closed-shell electronic configuration of [Y3+(CN)−]2+@[C82]2− is established on the basis of XPS, ESR and cyclic voltammetric studies. The existence of such an improbable monometallic cyanide clusterfullerene portends the possibility of creating novel EMFs entrapping other types of clusters. Our successful synthesis and structural elucidation of the first monometallic clusterfullerene mark a breakthrough for fullerene research and will stimulate further studies on the stability and formation mechanism of EMFs, both theoretically and experimentally.
Methods
Synthesis and isolation
YCN@C82 was synthesized in a modified Krätschmer-Huffman generator by vaporizing composite graphite rods (Φ8 × 150 mm) containing a mixture of Y2O3 (99.99%), TiO2 (99.99%) and graphite powder with a molar ratio of 1:1:15 (Y:Ti:C) with the addition of 10 mbar N2 into 400 mbar He as described previously19,20. For comparison, a reference synthesis by using a mixture of Y2O3 and graphite powder (1:15, without involvement of TiO2) was also carried out. The as-produced soot was Soxhlet-extracted by CS2 for 24 h and the resulting brown-yellow solution was distilled to remove CS2 and then immediately redissolved in toluene (~200 ml) and subsequently passed through a 0.2 μm Telflon filter (Sartorius AG, Germany) for HPLC separation. The isolation of YCN@C82 was performed by five-step HPLC as described in details in Supplemental Information S1. The purity of the isolated YCN@C82 was further checked by both the recycling HPLC and laser desorption time-of-flight (LD-TOF) mass spectroscopic (MS) analysis running in both positive and negative ion modes (Biflex III, Bruker Daltonics Inc., Germany).
Spectroscopic characterizations
UV-vis-NIR spectrum of YCN@C82 dissolved in toluene was recorded on a UV-vis-NIR 3600 spectrometer (Shimadzu, Japan) using a quartz cell of 1 mm layer thickness and 1 nm resolution. For XPS measurements, thin films of YCN@C82 drop-coated onto KBr single crystal disks were transferred under ultrahigh vacuum conditions into an ESCALAB 250 spectrometer (Thermo-VG Scientific, England) where they were studied using monochromatic Al Kα radiation (1486.6 eV) with an energy resolution of 0.6 eV. ESR spectrum of YCN@C82 was measured in toluene solution using a JES-FA200 FT-EPR X-band spectrometer (JEOL, Japan). The 13C NMR spectroscopic study was performed at 125 MHz in an Advance 500 spectrometer (Bruker, Germany) at room temperature in carbon disulfide with d6-acetone as an external lock.
Electrochemical study
Electrochemical study of YCN@C82 was performed in o-dichlorobenzene (o-DCB, anhydrous, 99%, Aldrich). The supporting electrolyte was tetrabutylamonium hexafluorophosphate (TBAPF6, puriss. electrochemical grade, Fluka) which was dried under a pressure at 340 K for 24 h and stored in glove box prior to use. Cyclic voltammogram experiments were performed with a CHI 660 potentiostat (CHI Instrument, USA) at room temperature in a glove box. A standard three-electrode arrangement of a platinum (Pt) wire as working electrode, a platinum coil as counter electrode and a silver wire as a pseudo-reference electrode was used. In a comparison experiment, ferrocene (Fc) was added as the internal standard and all potentials are referred to the Fc/Fc+ couple.
X-ray crystallographic study
Crystal growth forYCN@C82 ·NiII(OEP)·1.73C6H6·1.27CHCl3 was accomplished by layering a solution of ca. 1.0 mg YCN@C82 in 1 ml benzene over a solution of 3.0 mg NiII(OEP) in 3 ml chloroform. After the two solutions diffused together over a period of 7 days, small black crystals suitable for X-ray crystallographic study formed upon a slow evaporation of benzene. X-ray data collection for the crystal of YCN@C82·NiII(OEP)·1.73C6H6·1.27CHCl3 (0.37 × 0.30 × 0.30 mm3) was carried out at 107 K on a Gemini S Ultra diffractometer (Oxford diffraction Ltd., UK) with a 92 mm Sapphire CCD image plate detector. YCN@C82·NiII(OEP)·1.73C6H6·1.27CHCl3 crystallizes in the monoclinic space group P21/c; a = 17.685(5) Å, b = 16.933(5) Å, c = 27.340(5) Å, V = 7795(3) Å3, Z = 4. A numerical absorption correction utilizing equivalents was employed. The structure was solved by direct methods and refined using all data (based on F2). Hydrogen atoms were located in a difference map, added geometrically and refined with a riding model. Refinement of 13450 reflections, 1765 parameters and 1922 restraints yielded wR2 = 0.2246 for all data and a conventional R1 of 0.0806 based on 10964 reflections with I > 2σ(I). CCDC 886828 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge on application to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44) 1223-336-033; e-mail: deposit@ccdc.cam.ac.uk).
References
Heath, J. R. et al. Lanthanum complexes of spheroidal carbon shells. J. Am. Chem. Soc. 107, 7779–7780 (1985).
Chai, Y. et al. Fullerenes with metals inside. J. Phys. Chem. 95, 7564–7568 (1991).
Akasaka, T. & Nagase, S. Endofullerenes: a new family of carbon clusters, Kluwer Academic Publishers, Dordrecht ; Boston, 2002.
Yang, S. F. & Dunsch, L. Endohedral fullerenes. Chapter in “Nanomaterials: Inorganic and Bioinorganic Perspectives”, ed., John Wiley & Sons, Ltd., Chichester, United Kingdom, 2008, pp 189–214.
Shinohara, H. Endohedral metallofullerenes. Rep. Prog. Phys. 63, 843–892 (2000).
Chaur, M. N., Melin, F., Ortiz, A. L. & Echegoyen, L. Chemical, electrochemical and structural properties of endohedral metallofullerenes. Angew. Chem. Int. Ed. 48, 7514–7538 (2009).
Rodríguez-Fortea, A., Balch, A. L. & Poblet, J. M. Endohedral metallofullerenes: a unique host–guest association. Chem. Soc. Rev. 40, 3551–3563 (2011).
Yang, S. F., Liu, F. P., Chen, C. B., Jiao, M. Z. & Wei, T. Fullerenes encaging metal clusters — clusterfullerenes. Chem. Commun. 47, 11822–11839 (2011).
Stevenson, S. et al. Small-bandgap endohedral metallofullerenes in high yield and purity. Nature 401, 55–57 (1999).
Dunsch, L. & Yang, S. F. Metal nitride cluster fullerenes: Their current state and future prospects. Small 3, 1298–1320 (2007).
Dunsch, L. & Yang, S. F. Endohedral clusterfullerenes - playing with cluster and cage sizes. Phys. Chem. Chem. Phys. 9, 3067–3081 (2007).
Wang, C. R. et al. A scandium carbide endohedral metallofullerene: (Sc2C2)@C84 . Angew. Chem. Int. Ed. 40, 397–399 (2001).
Iiduka, Y. et al. Structural determination of metallofuIlerene Sc3C82 revisited: A surprising finding. J. Am. Chem. Soc. 127, 12500–12501 (2005).
Krause, M., Ziegs, F., Popov, A. A. & Dunsch, L. Entrapped bonded hydrogen in a fullerene: The five-atom cluster Sc3CH in C80 . Chem. Phys. Chem. 8, 537–540 (2007).
Stevenson, S. et al. A distorted tetrahedral metal oxide cluster inside an lcosahedral carbon cage. Synthesis, isolation and structural characterization of Sc4(μ3-O)2@Ih-C80 . J. Am. Chem. Soc. 130, 11844–11845 (2008).
Dunsch, L. et al. Metal sulfide in a C82 fullerene cage: A new form of endohedral clusterfullerenes. J. Am. Chem. Soc. 132, 5413–5421 (2010).
Wang, T. S. et al. Planar quinary cluster inside a fullerene cage: synthesis and structural characterizations of Sc3NC@C80-Ih. J. Am. Chem. Soc. 132, 16362–16364 (2010).
Jin, P. et al. NC unit trapped by fullerenes: a density functional theory study on Sc3NC@C2n (2n = 68, 78 and 80). Phys. Chem. Chem. Phys. 12, 12442–12449 (2010).
Yang, S. F. et al. An endohedal titanium (III) in a clusterfullerene: Putting a non-group-III metal nitride into the C80-Ih fullerene cage. Chem. Commun. 6391–6393 (2009).
Chen, C. B. et al. Titanium/yttrium mixed metal nitride clusterfullerene TiY2N@C80: Synthesis, isolation and effect of the group-III metal. Inorg. Chem. 51, 3039–3045 (2012).
Yang, S. F., Troyanov, S. I., Popov, A., Krause, M. & Dunsch, L. Deviation from the planarity – a large Dy3N cluster encapsulated in an Ih-C80 Cage: An X-ray crystallographic and vibrational spectroscopic study. J. Am. Chem. Soc. 128, 16733–16739 (2006).
Harris, K. J. & Wasylishen, R. E. A. 13C and 15N solid-state NMR study of structural disorder and aurophilic bonding in Au-I and Au-III cyanide complexes. Inorg. Chem. 48, 2316–2332 (2009).
Stevens, P. A., Madix, R. J. & Stöhr, J. The bonding of acetonitrile and CH2CN on Ag(110) determined by near edge x-ray absorption fine structure: Evidence for π-donor bonding and azimuthal ordering. J. Chem. Phys. 91, 4338–4345 (1989).
Yang, H. et al. Detection of a family of gadolinium-containing endohedral fullerenes and the isolation and crystallographic characterization of one member as a metal-carbide encapsulated inside a large fullerene cage. J. Am. Chem. Soc. 130, 17296–17300 (2008).
Iiduka, Y. et al. Experimental and theoretical studies of the scandium carbide endohedral metallofullerene Sc2C2@C82 and its carbene derivative. Angew. Chem. Int. Ed. 46, 5562–5564 (2007).
Zhang, J. et al. Nanoscale fullerene compression of an yttrium carbide cluster. J. Am. Chem. Soc. 134, 8487–8493 (2012).
Lu, X. et al. Yb@C2n (n = 40, 41, 42): new fullerene allotropes with unexplored electrochemical properties. J. Am. Chem. Soc. 132, 5896–5905 (2010).
Lu, X. et al. Structural elucidation and regioselective functionalization of an unexplored carbide cluster metallofullerene Sc2C2@Cs(6)-C82 . J. Am. Chem. Soc. 133, 19553–19558 (2011).
Acknowledgements
This work was supported by National Basic Research Program of China (2010CB923300, 2011CB921400), the National Natural Science Foundation of China (Nos. 90921013, 21132007), “100 Talents Programme of CAS” from Chinese Academy of Sciences and Innovation Project of Anhui Municipal Education Commission (2009Z029) [to SFY]. X. L. thanks “The National Thousand Talents Programme”, the National Natural Science Foundation of China (No. 21171061) and HUST for generous financial supports. T. A. is grateful for the financial supports from a Grant-in-Aid for Scientific Research on Innovative Areas (No. 20108001, "pi-Space"), a Grant-in-Aid for Scientific Research (A) (No. 20245006) and (B) (No. 24350019) from MEXT of Japan. Z. C. thanks the financial support by NSF Grant EPS-1010094 and Department of Defense (Grant W911NF-12-1-0083).
Author information
Authors and Affiliations
Contributions
S.F.Y., X. L. and Z. F. C. conceived and designed the experiments, S.F.Y., X. L., Z. F. C. and T. A. co-wrote the paper, C. B. C. and M. Z. J. conducted the synthesis, F. P. L. accomplished the crystal growth and diffraction data collection, X. L. carried out the NMR measurement and analyzed the data, T. W. performed the isolation, C. B. C. and F. P. L. made the spectroscopic measurements, S. W. carried out the electrochemical study, F. Y. L. and Z. F. C. performed the computational work, Y. P. X., M. S., X. L. and T. A. participated in the X-ray structure analysis. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yang, S., Chen, C., Liu, F. et al. An Improbable Monometallic Cluster Entrapped in a Popular Fullerene Cage: YCN@Cs(6)-C82. Sci Rep 3, 1487 (2013). https://doi.org/10.1038/srep01487
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01487
This article is cited by
-
Theoretical study on monometallic cyanide cluster fullerenes MCN@C74 (M=Y, Tb)
Journal of Molecular Modeling (2015)
-
Carbide clusterfullerenes with odd number of carbon atoms: molecular and electronic structures of Sc4C@C80, Sc4C@C82, and Sc4C3@C80
Theoretical Chemistry Accounts (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
