
Abstract
Metallic aluminum (Al) is of interest as a reducing agent because of its low standard reduction potential. However, its surface is invariably covered with a dense aluminum oxide film, which prevents its effective use as a reducing agent in wet-chemical synthesis. Pitting corrosion, known as an undesired reaction destroying Al and is enhanced by anions such as F−, Cl− and Br− in aqueous solutions, is applied here for the first time to activate Al as a reducing agent for wet-chemical synthesis of a diverse array of metals and alloys. Specifically, we demonstrate the synthesis of highly dispersed palladium nanoparticles on carbon black with stabilizers and the intermetallic Cu2Sb/C, which are promising candidates, respectively, for fuel cell catalysts and lithium-ion battery anodes. Atomic hydrogen, an intermediate during the pitting corrosion of Al in protonic solvents (e.g., water and ethylene glycol), is validated as the actual reducing agent.
Similar content being viewed by others


Introduction
Metallic aluminum (Al) is a strong reducing agent with a standard reduction potential E°Al3+/Al = −1.662 V. However, Al metal is invariably passivated by a dense aluminum oxide film a few nanometers thick, which can serve as a barrier to accessing Al as a reducing agent; the oxide film can form instantly on Al surface when exposed to air and/or water. The dense oxide film is, however, not a hurdle for employing Al as a reducing agent at high temperatures, such as at 1450°C in melted glass1 and in a high-energy mechanical milling process2,3. In contrast, it is indeed a barrier for employing Al as a reducing agent in wet-chemical synthesis.
Fortunately, Al is susceptible to pitting corrosion which is enhanced by a variety of anions, such as F−, Cl−, Br−, SCN−, ClO3−, ClO4−, gluconate anions, etc., in aqueous solutions at different rates4,5,6,7,8,9,10,11. For instance, the pitting corrosion of Al in an aqueous solution with Cl− follows three steps: adsorption of Cl− on the oxide surface, penetration of Cl− through the oxide film via oxygen vacancies and localized dissolution of Al below the oxide film to release hydrogen gas12,13,14,15,16. This pitting corrosion, known as an undesired reaction destroying Al and Al alloys, makes it possible to use Al as a reducing agent in wet-chemical synthesis. To the best of our knowledge, there are no reports in the literature that pitting corrosion is applied beneficially to activate Al as a reducing agent for wet-chemical synthesis.
Using Al as a reducing agent offers several advantages: strong reducing capability, easy transportation and storage due to its solid form and stability (protected with a dense oxide film) and environmentally benign reactions. In this study, we investigate the mechanism for using Al as a reducing agent with the assistance of pitting corrosion in protonic solvents for wet-chemical synthesis. The discovered mechanism answers a question raised in a classical general chemistry laboratory demonstration of the supposed displacement reactions, i.e., why does Al react with CuCl2 in aqueous solution to produce Cu metal, but not with CuSO4 or Cu(NO3)217,18? We then demonstrate the use of Al as a reducing agent by preparing a wide array of metals and alloys at room temperature; particularly, synthesis of highly dispersed palladium nanoparticles (NPs) on carbon black with the use of stabilizers for use as fuel cell catalysts and intermetallic Cu2Sb/C for use as lithium-ion battery anodes.
Results
Mechanisms: role of pitting corrosion
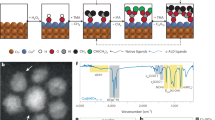
The UV-Vis spectra in Figure 1 show that a 10 mM Na2PdCl4 aqueous solution can be reduced by the addition of Al foil with > 95% conversion at room temperature within 5 min. The precipitated particles were confirmed as metallic Pd by the XRD pattern (right inset of Figure 1). These results imply that the dense aluminum oxide film barrier on Al foil is overcome and Pd ions are reduced without requiring any assistance or special conditions, such as ultrasonication, mechanical force, pH, temperature, atmosphere, etc.

UV-Vis spectra, photos and XRD pattern.
The UV-Vis spectra at 0 min were obtained for the aqueous Na2PdCl4 solutions (10 mM, 0.1 mM) without the addition of Al foil. The UV-Vis spectra at 2 and 5 min were acquired for the supernatants by centrifuging the 10 mM Na2PdCl4 aqueous solutions after the addition of Al foil for 2 and 5 min. The left inset is the images of the supernatants after reacting for 2 and 5 min and the right inset is the XRD pattern of the precipitated particles after reacting for 5 min.
The mechanism for [PdCl4]2− to overcome the dense oxide film on Al foil could be the pitting corrosion via Cl−. The inset in Figure 2 shows that the pH of deionized water sharply decreases from about 6 to < 4 on adding a small amount of Na2PdCl4. This suggests that the [PdCl4]2− complex releases free Cl− by the replacement of the Cl− ligands with OH− from water to form [PdClx(OH)y]2−, leading to an increase in H+ concentration in the solution. It is also supported by a shifting of the peak position of the Pd d–d spin-forbidden transition to a lower value in the UV-Vis spectra in Figure 2 as the concentration of Na2PdCl4 decreases19.

UV-Vis spectra and pH measurement of Na2PdCl4 aqueous solutions with different concentrations.
To confirm the formation of pitting corrosion on the Al foil with the exclusion of stirring effects, two drops (20 μL) of a 10 mM Na2PdCl4 aqueous solution were, respectively, placed on Al foil for 1 min and 3 min and then the Al foil was rinsed with water. The dark spots without Pd NPs (marked with circles) and with Pd NPs (marked with squares) on the Al foil shown in Figure 3a are blisters where pitting corrosion has occurred, causing the oxide film to bubble12. The reactions may occur first at the defect sites on the pristine surface of the Al foil (Supplementary Fig. S1), where the positively charged hydrated oxide surface is easily attacked by Cl− ions12,13,14,15,16. As the contacting time increases, the corrosion below the oxide film at those sites propagates12 and more new sites start pitting. Accordingly, the already formed Pd particles grow and more Pd particles nucleate as shown in Figure 3b. When the pitting corrosion evolves to the stage of blistering, the high pressure hydrogen gas built up in the blisters ruptures the oxide film and causes the Pd particles to detach from the Al foil. New oxide film forms atop the Al instantly because of the fast reaction between Al and water (3 × 10−9 s at 25°C)12. New pitting corrosion thus occurs below the new oxide films and Pd particles are produced nearby as shown in the right inset of Figure 3b.

SEM images of the Al foil surfaces.
The Al foils were contacted with a drop of 10 mM Na2PdCl4 solution for (a) 1 min and (b) 3 min. The dark spots without Pd NPs (marked with circles) and with Pd NPs (marked with squares) in (a) are blisters where pitting corrosion has occurred. The insets of (a) and (b) show the magnified images of the Pd NPs on Al foil. (c) Al foil contacted with a drop of 10 mM CuF2, CuCl2, CuBr2, Cu(NO3)2 and CuSO4 aqueous solution for 10 min.
To confirm that pitting corrosion assists the use of Al as a reducing agent, Al foil was added into 20 mM aqueous solutions of CuF2, CuCl2, CuBr2, CuSO4 and Cu(NO3)2, of which F−, Cl−, Br− are known to enhance the pitting corrosion of Al foil with different rates4,5,6,7 and SO42− and NO3− do not5,6. Cu particles, confirmed by XRD (Supplementary Fig. S2), were produced in minutes in the solutions of CuF2, CuBr2 and CuCl2. In contrast, there were no notable changes in the solutions of CuSO4 and Cu(NO3)2 even after a much longer time, indicating that SO42− and NO3− do not cause pitting corrosion of Al, which is consistent with the literature reports5,6. The SEM images in Figure 3c show that only the Al foil after being in contact with drops of CuF2, CuBr2 and CuCl2 solutions (10 mM) for 10 min had Cu particles. The Al foils after being in contact with the CuSO4 and Cu(NO3)2 solutions do not have corrosion pits or Cu particles. Another observation is that the number of Cu particles increases as the size of the anions decreases, i.e., the Al foil with CuF2 has the most Cu particles and that with CuBr2 has the least. The reason is that a smaller anion can more easily transport through the oxide film during pitting corrosion5,6,7.
Mechanisms: atomic hydrogen (H) as the actual reducing agent
Water molecules can transport through the oxide film and react with the metallic Al beneath to produce H2 gas, in which H is generated as an intermediate20. Accordingly, H2 gas, H and metallic Al are the possible reducing agents in this reaction system. A controlled experiment with the passage of H2 gas overnight reveals that H2 cannot reduce an aqueous solution of Na2PdCl4. In contrast, H is more active than H2 and it can reduce metal oxides such as CuO at room temperature21. As mentioned above, Pd particles can form on the Al foil in the early stage of pitting corrosion, when the oxide film is not broken. Therefore, the electro-reduction of [PdClx(OH)y]2− in the solution does not obtain the electrons released from the Al electro-oxidation reaction beneath the non-conductive oxide film, but from the electro-oxidation reaction of the H on the top of the oxide film. The H is produced as an intermediate of the pitting corrosion of Al below the oxide film20. It cannot reduce aluminum oxide21 because of the very low standard reduction potential of , but it can diffuse through to the top of the oxide film22. The hydrogen electro-oxidation reaction continues on the top of the oxide film and the released electrons flow onto the surface of the formed Pd particles for further reduction of [PdClx(OH)y]2−, leading to the growth of the Pd particles. As mentioned above, the Pd particles will become detached when the oxide film beneath is broken and a new oxide film forms atop the metallic Al instantly (3 × 10−9 s at 25°C)12. The whole process is shown by the schematic drawing in Figure 4a. It was observed that the Pd particles develop into dendrites (left insert in Figure 3b), suggesting that the growth rate is limited by the diffusion of the solute species from the bulk solution to the surface of the particles23. It also suggests an abundant supply of the reductant, which is supported with the fast hydrogen electrooxidation reaction and the high mobility of electrons on and within the Pd particles.

Atomic hydrogen is the actual reducing agent.
(a) Schematic illustrating the reaction mechanism and (b) results of the reactions of stated metal salts with Al foil in H2O or EG at room temperature. Metal particles were produced with the compounds indicated green color and no metal particles were produced with the compounds indicated in red color.
Certainly, Al can reduce metal compounds with positive standard reduction potentials and anions that can cause significant pitting corrosion of Al, shown in Figure 4b. It should be noted that some reactions were conducted in ethylene glycol (EG) to avoid the hydrolysis reaction in water. Some metal compounds with negative reduction potential, such as FeCl3 (E°Fe3+/Fe = −0.037 V), PbCl2 (E°Pb2+/Pb = −0.126 V), SnCl2 (E°Sn2+/Sn = −0.138 V), CoCl2 (E°Co2+/Co = −0.28 V), NiCl2 (E°Ni2+/Ni = −0.257 V) and FeCl2 (E°Fe2+/Fe = −0.447 V), were also tried. It was found that metallic particles were produced in the solutions of PbCl2 and SnCl2 but none in the solutions of FeCl3, CoCl2, NiCl2 and FeCl2. The FeCl3 solution is a unique case in that it is reduced to FeCl2 owing to E°Fe2+/Fe3+ = 0.77 V, which is more positive than E°Fe2+/Fe and therefore is the favored reaction. The lower limit of the standard reduction potential for metal compounds to be reduced with Al is located in the range of −0.138 to −0.257 V. This supports that H is the actual reducing agent because the reduction potential of H/H+ is about −0.17 and −0.24 V, respectively, at pH = 3 and 4 as calculated by the Nernst equation. All produced metallic particles were confirmed by XRD (Supplementary Fig. S3).
Applications: synthesis of metallic nanomaterials
In order to assess the potential application of using Al as a reducing agent for wet-chemical synthesis, we prepared two carbon supported metallic nanomaterials: 40 wt.% Pd on carbon support (Pd/C) as an electrocatalyst for formic acid electro-oxidation in fuel cells and intermetallic Cu2Sb/C as an anode material for lithium-ion batteries.
Both Al foil and powder were used for the preparation of Pd/C, but Al foil was chosen rather than powder for the following reasons: the reduction rate with Al foil is faster than with Al powder because the latter has a larger ratio of aluminum oxide to aluminum; it is easier to separate Al foil from the reaction products; and in the stirring reaction solution, Al foil has a lower moving speed than carbon black while Al powder has a speed similar to carbon black, implying a larger relative moving speed between Al foil and carbon black and thus a larger scrubbing force to detach the Pd NPs from the Al surface.
Since carbon black cannot be well dispersed in water, the preparation of Pd/C was carried out in EG. It was found that the reduction of 10 mM Na2PdCl4 by Al foil in EG was complete in about 1 h (Supplementary Fig. S4). The same mechanism occurs in EG solution as described above and it is supported by the change in pH and UV-Vis peaks with concentration (Supplementary Fig. S5). Accordingly, Pd/C was first synthesized in EG with Al foil at room temperature for 3 h. Unfortunately, the obtained Pd/C (denoted as Pd/C-EG), confirmed by XRD and EDX (Supplementary Fig. S6), has very large Pd particles as shown in Figure 5a.

TEM images of Pd/C catalysts, particle size analysis and SEM images of Al foil surfaces.
Pd/C prepared (a) in EG, (b) with PVP in water and (c) with FeCl2 in EG. (d) Particle size distribution histograms of Pd NPs on Pd/C-PVP and Pd/C-FeCl2. (e) SEM images of Al foil surfaces (left) before and (right) after the preparation of Pd/C-FeCl2.
Trisodium citrate and polyvinylpyrrolidone (PVP) were considered as stabilizers to reduce the particle size. The reactions were carried out in water since carbon black with these stabilizers can be dispersed well in water. The aqueous Na2PdCl4 solution with PVP became dark within a few minutes after the addition of Al foil and the reaction was complete in about 15 min. In contrast, the solution with trisodium citrate did not show a noticeable change in 2 h. The difference could be that PVP can interact with the metallic Pd atoms through its carbonyl group24 while trisodium citrate complexes with the Pd ions25. In addition, citrate can inhibit the pitting corrosion of Al by a competitive adsorption with Cl− on the oxide film26. Figure 5b and d shows that the Pd/C (denoted as Pd/C-PVP) prepared with a 10:1 ratio of PVP repeat unit to Na2PdCl4 in water at room temperature for 1 h results in Pd NPs with a mean particle diameter of 4.9 nm (additional XRD and EDX data in Supplementary Fig. S7), indicating a significant size reduction compared to that of Pd/C-EG shown in Figure 5a. However, the dispersion of Pd NPs on the carbon is irregular; some carbon black is not loaded with NPs. FeCl2 was also used as a stabilizer to synthesize Pd/C (denoted as Pd/C-FeCl2) in EG at room temperature for 3 h, which was confirmed by XRD and EDX results (Supplementary Fig. S8). The Pd NPs are stabilized by the adsorption of Fe2+. Figures 5c and d show that the Pd/C-FeCl2 prepared with a 10:1 ratio of Na2PdCl4 to FeCl2 has Pd NPs uniformly distributed on the carbon black with a mean diameter of 6.5 nm. Although Pd/C-FeCl2 has larger Pd NPs than Pd/C-PVP, the carbon black is more uniformly loaded with Pd NPs. Electrochemical evaluation (Supplementary Fig. S9) shows that Pd/C-FeCl2 and Pd/C-PVP have high activity toward formic acid electro-oxidation, consistent with the reports in the litereature27,28. Pitting corrosion occurrence during the preparation of Pd/C is shown by the SEM images of Al foil surfaces in Figure 5e.
Carbon-supported Cu2Sb (denoted Cu2Sb/C) was prepared using Al foil as a reducing agent in EG under an N2 atmosphere and confirmed by XRD and SEM-EDX mapping in Figure 6. The molar ratio of Cu to Sb in the product is 2.05 as determined by EDX, indicating complete reduction of both CuCl2 and SbCl3. Similar to Pd/C, the carbon black works as a scrubber to remove the formed Cu2Sb from Al foil. The quantitative reduction of Cu and Sb could be attributed to their similar reduction reaction rates. The crystallite size of Cu2Sb is about 20 nm, as determined by the Scherrer equation. The small crystalline size, in the presence of a carbon matrix, has the potential to buffer the large volume expansion of the anode during charge/discharge of lithium-ion batteries3.

(a) XRD pattern and (b) SEM and EDX mapping images of Cu2Sb/C.
Discussion
Our experimental results show that Al as a reducing agent in wet-chemical synthesis is activated by the pitting corrosion of Al, in which the produced atomic hydrogen as an intermediate of the pitting corrosion in protonic solvents provides the actual reducing function. This mechanism is dependent on two conditions: the formation of oxide film in the protonic solvent is instant and the oxide film is non-conductive. The former condition ensures that Al does not have a chance to directly react with the metal ions for reduction; the latter condition means that the electrons released from the electro-oxidation of Al cannot travel through the oxide film to participate in the electro-reduction of the metal ions on the top surface. For example, some reactive metals, such as Zn and Fe, do not follow this mechanism because their oxide films are semiconducting.
This discovered mechanism sheds new light on a classical general chemistry laboratory demonstration of the supposed displacement reactions, i.e., why does Al react with aqueous CuCl2 solution to produce Cu metal but not with CuSO4 or Cu(NO3)217,18? Here we have presented two reasons why the reaction of Al and CuCl2 is not a simple displacement reaction as widely believed17,18. First, the stated reaction does not occur with CuSO4 and Cu(NO3)2 at observable levels indicating the reaction is more complex and in fact requires specific anions to cause significant pitting corrosion as shown in this study. Second, if Al displaces Cu2+, then the standard reduction potential of Al (E°Al3+/Al = −1.662 V) suggests that Al will displace any ion with a more positive reduction potential; however this is not the case. As we have shown in Figure 4b, only those with a standard reduction potential at or above E°Sn2+/Sn = −0.138 V react with Al, leading to the conclusion that H provides the actual reducing function. Therefore, our findings question the mechanisms given in the chemistry text17,18 that there is a redox reaction between Al and Cu2+ ions. Instead, a more complicated series of reactions occur, culminating with the reaction between H and Cu2+ ions.
This new strategy is applicable to synthesize a range of metallic nanoparticles from suitable precursors with anions causing significant pitting corrosion of Al in protonic solvents, e.g., F−, Cl− and Br− as shown in this study and SCN−, ClO3−, ClO4−, gluconate anions for future studies. Al activated by pitting corrosion has the reducing capability of atomic hydrogen, which can reduce a wide range of metals as demonstrated in this study. It thus can be used to synthesize supported and unsupported metallic nanomaterials for wide applications, such as in energy conversion and storage devices.
As a heterogeneous reducing agent for synthesis of supported metallic nanomaterials, the first concern is how to remove the formed metallic particles from the Al surface while avoiding aggregation and successfully transferring the particles to the support. Previously, ultrasonication29 and a scrubbing brush30 have been used to remove the particles from Cu or Fe foils. As demonstrated in this study, the transfer of Pd NPs from the Al surface to the support surface is simply achieved by the interaction between the carbon support and the Al surface. The second concern is how to control the particle size and shape to tune the properties of the nanomaterials. To approach this, introducing a stabilizer is a very common method. We thus investigated the feasibility by comparing several stabilizers: PVP, trisodium citrate and FeCl2 in the preparation of Pd/C. The results show that highly dispersed, finely sized Pd/C can be obtained with stabilizers of PVP and FeCl2. There is no Pd particle produced with trisodium citrate mainly due to the inhibition of pitting corrosion of Al by the competitive adsorption of citrate with Cl− on the oxide film26. Similarly, a stabilizer preferable to adsorb on certain metal crystalline planes can be adopted to tune the particle shape only if it does not inhibit the pitting corrosion of Al. For example, cetyltrimethylammonium bromide is a widely used surfactant to synthesize cubic-like metal nanoparticles. It can provide bromide anions in an aqueous solution, which can activate the Al as a reducing agent. It should also be noted that the reduction efficiency using Al as a reducing agent is somewhat lowered due to the formation of molecular hydrogen (H2) by some of the atomic hydrogen produced from the reaction of Al with water or ethylene glycol; the molecular hydrogen cannot reduce Na2PdCl4 in an aqueous solution at room temperature.
Alloys and intermetallics show many unique properties that pure metals do not, allowing these materials a wider range of applications and the ability to customize properties by tailoring the composition. This strategy can also be extended to prepare bimetallic and multi-component metallic materials within the range of reducing capability. The intermetallic Cu2Sb, one unique material, has been synthesized previously3,31,32,33 with the potential application as a high capacity lithium ion battery anode material. These synthesis methods require long reaction times3,33, high temperatures31,33, specialized equipment32, or hazardous and toxic chemicals31. In our study, the intermetallic compound Cu2Sb/C has been synthesized by a simple, “green” method: adding Al foil as the reducing agent to an EG solution of the metal chlorides at room temperature without the use of specialized equipment.
Our findings could stimulate the use of other reactive metals with oxide films as reducing agents. Further optimization of the reaction conditions could help to realize further control of the size and shape of the metallic nanoparticles and enhance our ability to prepare a wider array of bimetallic and ternary compounds. Besides, as a reducing agent, Al activated by pitting corrosion can also be applied for wet-chemical synthesis of non-metallic chemicals.
Methods
Synthesis
Aluminum (Al) foil (24 μm thick, Fisher Sci.) was cut into pieces of approximately 1 cm × 3 cm prior to the use in the reactions. Al powder (325 mesh, Alfa Aesar), metal compounds, polyvinylpyrrolidone (PVP, MW = 40000, MP Biomedicals) and FeCl2·4H2O were used as received. The solvent was deionized water or ethylene glycol (EG) for different metal compounds.
A solution of the desired metal compounds (AgF, AgNO3, BiCl3/EG, CoCl2·6H2O, Cu(NO3)2·3H2O, CuBr2, CuCl2, CuF2, CuSO4·5H2O, FeCl2·4H2O, FeCl3·6H2O, H2PtCl6·6H2O, HAuCl4·xH2O, InCl3·xH2O, K2PtCl4, Na2PdCl4, NiCl2·6H2O, PbCl2, PdCl2, SbCl3/EG and SnCl2·2H2O/EG) was prepared in either deionized water (CuCl2 and all others) or EG (BiCl3, CuCl2, InCl3, PbCl2, SbCl3 and SnCl2). In most cases, a 20 mM solution was prepared. The required amount of Al foil was then added to the stirring solution to result in a molar ratio between Al and the metal compound of 10:1. The produced metallic powders were separated by centrifugation followed by washing in either deionized water or isopropanol.
The typical synthesis for Pd/C (40 wt.%) in EG (denoted as Pd/C-EG) proceeds as follows. Carbon black (90 mg, Vulcan XC-72R, Cabot) was dispersed in 30 mL EG by vigorously stirring for at least 3 h. A EG solution of Na2PdCl4 (5.638 mL, 0.1 M) was added drop-wise into the carbon dispersion and stirred for 1 h. Al foil, at a 10:1 molar ratio, was then added and the suspension was kept stirring for 3 h at room temperature in air. To remove a majority of the unreacted Al, the suspension was kept sedentary for 15 min. The upper suspension was then centrifuged at 20,000 g for 15 min. The precipitated Pd/C-EG powders were then redispersed in a solution of 20 mL water and 5 mL ethanol, into which 1.0 M sulfuric acid was added to result in a final concentration of 0.02 M and stirred overnight to remove the residual Al and Al oxides. The Al-free Pd/C-EG powder was then centrifuged and washed in a water/ethanol solution by stirring for 1 h a total of three times. Lastly, the washed sample was dried under vacuum at room temperature.
To prepare Pd/C employing FeCl2 as a stabilizer (denoted as Pd/C-FeCl2), FeCl2·4H2O was added to an EG solution of Na2PdCl4 (0.1 M) at a 10:1 molar ratio of Na2PdCl4 to FeCl2 and the solution was stirred for 1 h before being added to the carbon black dispersion. As for the Pd/C prepared with PVP as a stabilizer (denoted as Pd/C-PVP), carbon black was dispersed in water with PVP at a 10:1 molar ratio of PVP repeat unit to Na2PdCl4 by stirring for 3 h and then aqueous Na2PdCl4 solution (0.1 M) was added and stirred for 1 h.
The intermetallic Cu2Sb supported on carbon black (acetylene black, 100% compressed, Alfa Aaser) was prepared in a similar manner to Pd/C-EG. Briefly, 100 mg carbon was dispersed in EG to result in a 20 wt.% composite with Cu2Sb. After dissolving 432.2 mg CuCl2 and 366.7 mg SbCl3, Al foil was added at a 25:1 molar ratio of Al to Cu + Sb and the reaction took place under an N2-atmosphere for 4 h. After straining the unreacted Al, the mixture was filtered and washed with isopropanol before drying in a vacuum oven overnight.
Physical characterization
The pH of the solutions was measured by a Corning 313 pH/Temperature meter. The concentrations of Na2PdCl4 solutions were characterized by the Pd d–d spin-forbidden transition peaks between 420 – 450 nm in UV-Vis spectra, recorded with a Cary 5000 UV-Vis-NIR spectrophotometer (Varian) using ultramicro UV-cuvettes (Fisher Sci.). To determine the reaction conversion with time, Al foil was added to a 10 mM Na2PdCl4 solution with a 10:1 molar ratio of Al to Na2PdCl4 for different times. The reaction solution was centrifuged at 10,000 g for 5 min and the supernatant was characterized by UV-Vis.
X-ray diffraction (XRD) patterns of the samples were recorded with a Philips APD 3520 diffractometer with Cu Kα radiation (λ = 0.15418 nm) and analyzed with the JADE 9.0 software package (Rigaku). The scanning electron microscopy (SEM) images for Al foils and Cu2Sb/C were taken, respectively, with JEOL-JSM5610 and Hitachi S5500. The compositions of the samples were determined using INCA software (Oxford Instruments) to quantify the energy-dispersive X-ray spectroscopy (EDS) spectra obtained by an EDS attachment (Oxford instruments) on the JEOL SEM. Transmission electron microscope (TEM) images were acquired with a JEOL 2010F operated at 200 keV to characterize the morphology of the samples. The particle size distributions were obtained by analyzing the TEM images using ImageJ software (NIH).
References
Lin, G. et al. Universal preparation of novel metal and semiconductor nanoparticle-glass composites with excellent nonlinear optical properties. J. Phys. Chem. C 115, 24598–24604 (2011).
Yoon, S. & Manthiram, A. Sb-MOx-C (M = Al, Ti, or Mo) Nanocomposite anodes for lithium-ion batteries. Chem. Mater. 21, 3898–3904 (2009).
Applestone, D., Yoon, S. & Manthiram, A. Cu2Sb-Al2O3-C nanocomposite alloy anodes with exceptional cycle life for lithium ion batteries. J. Mater. Chem. 22, 3242–3248 (2012).
Szklarska-Smialowska, Z. Pitting Corrosion of Metals (National Association of Corrosion Engineers, Houston, 1986).
Rehim, S. S. A., Hassan, H. H. & Amin, M. A. Corrosion and corrosion inhibition of Al and some alloys in sulphate solutions containing halide ions investigated by an impedance technique. Appl. Surf. Sci. 187, 279–290 (2002).
Lenderink, H. J. W., Linden, M. V. D. & De Wit, J. H. W. Corrosion of aluminum in acidic and neutral solutions. Electrochim. Acta 38, 1989–1992 (1993).
Mazhar, A. A., Badawy, W. A. & Abou-Romia, M. M. Impedance studies of corrosion resistance of aluminum in chloride media. Surface and Coatings Technology 29, 335–345 (1986).
Amin, M. A., Abd El-Rehim, S. S., El-Sherbini, E. E. F., Mahmoud, S. R. & Abbas, M. N. Pitting corrosion studies on Al and Al–Zn alloys in SCN− solutions. Electrochim. Acta 54, 4288–4296 (2009).
Amin, M. A. Metastable and stable pitting events on Al induced by chlorate and perchlorate anions–Polarization, XPS and SEM studies. Electrochim. Acta 54, 1857–1863 (2009).
Amin, M. A., Abd El Rehim, S. S. & El-Lithy, A. S. Pitting and pitting control of Al in gluconic acid solutions–Polarization, chronoamperometry and morphological studies. Corros. Sci. 52, 3099–3108 (2010).
Amin, M. A., Abd El Rehim, S. S. & El-Lithy, A. S. Corrosion, passivation and breakdown of passivity of Al and Al–Cu alloys in gluconic acid solutions. Electrochim. Acta 55, 5996–6003 (2010).
McCafferty, E. Sequence of steps in the pitting of aluminum by chloride ions. Corros. Sci. 45, 1421–1438 (2003).
Muñoz, A. G. & Bessone, J. B. Pitting of aluminum in non-aqueous chloride media. Corros. Sci. 41, 1447–1463 (1999).
McCafferty, E. The electrode kinetics of pit initiation on aluminum. Corros. Sci. 37, 481–492 (1995).
Tomcsányi, L., Varga, K., Bartik, I., Horányi, H. & Maleczki, E. Electrochemical study of the pitting corrosion of aluminum and its alloys–II. Study of the interaction of chloride ions with a passive film on aluminum and initiation of pitting corrosion. Electrochim. Acta 34, 855–859 (1989).
Atanasoska, L. D., Dražić, D. M., Despić, A. R. & Zalar, A. Chloride ion penetration into oxide films on aluminum: Auger and XPS studies. J. Electroanal. Chem. Interfacial Electrochem. 182, 179–186 (1985).
Hutchings, K. Classic Chemistry Experiments (Royal Society of Chemistry, London, 2000).
Summerlin, L. R., Borgford, C. L. & Ealy, J. B. Chemical Demonstrations: A Sourcebook for Teachers (American Chemical Society, Washington, DC, 1988).
Espinosa-Alonso, L., de Jong, K. P. & Weckhuysen, B. M. A UV-Vis micro-spectroscopic study to rationalize the influence of Cl− (aq) on the formation of different Pd macro-distributions on γ-Al2O3 catalyst bodies. Phys. Chem. Chem. Phys. 12, 97–107 (2010).
Sun, Y.-l., Tian, Y. & Li, S.-F. Theoretical study on reaction mechanism of aluminum-water system. Chin. J. Chem. Phys. 21, 245 (2008).
Bergh, A. A. Atomic hydrogen as a reducing agent. Bell Syst. Tech. J. 44, 261–71 (1965).
Yamada-Takamura, Y., Koch, F., Maier, H. & Bolt, H. Hydrogen permeation barrier performance characterization of vapor deposited amorphous aluminum oxide films using coloration of tungsten oxide. Surface and Coatings Technology 153, 114–118 (2002).
Shikazono, N. Precipitation mechanisms of barite in sulfate-sulfide deposits in back-arc basins. Geochim. Cosmochim. Acta 58, 2203–2213 (1994).
Nemamcha, A., Moumeni, H. & Rehspringer, J. L. PVP Protective mechanism of palladium nanoparticles obtained by sonochemical process. Phys. Procedia 2, 713–717 (2009).
Lo, S. H. Y., Wang, Y.-Y. & Wan, C.-C. Synthesis of PVP stabilized Cu/Pd nanoparticles with citrate complexing agent and its application as an activator for electroless copper deposition. J. Colloid Interface Sci. 310, 190–195 (2007).
Rudd, W. J. & Scully, J. C. The function of the repassivation process in the inhibition of pitting corrosion on aluminum. Corros. Sci. 20, 611–631 (1980).
Wang, R., Liao, S. & Ji, S. High performance Pd-based catalysts for oxidation of formic acid. J. Power Sources 180, 205–208 (2008).
Wang, Z. B., Yuan, G. H., Zhou, K., Chu, Y. Y. & Chen, M. Effect of pH value and temperatures on performances of Pd/C catalysts prepared by modified polyol process for formic acid electrooxidation. Fuel Cells 11, 309–315 (2011).
Wu, C., Mosher, B. P. & Zeng, T. Rapid synthesis of gold and platinum nanoparticles using metal displacement reduction with sonomechanical assistance. Chem. Mater. 18, 2925–2928 (2006).
Wu, C. & Zeng, T. Size-tunable synthesis of metallic nanoparticles in a continuous and steady-flow reactor. Chem. Mater. 19, 123–125 (2006).
Kieslich, G., Birkel, C. S., Stewart, A., Kolb, U. & Tremel, W. Solution synthesis of nanoparticular binary transition metal antimonides. Inorg. Chem. 50, 6938–6943 (2011).
Mosby, J. M. & Prieto, A. L. Direct electrodeposition of Cu2Sb for lithium-ion battery anodes. J. Am. Chem. Soc. 130, 10656–10661 (2008).
Sarakonsri, T., Johnson, C. S., Hackney, S. A. & Thackeray, M. M. Solution route synthesis of InSb, Cu6Sn5 and Cu2Sb electrodes for lithium batteries. J. Power Sources 153, 319–327 (2006).
Acknowledgements
This work was supported by the Office of Naval Research MURI grant No. N00014-07-1-0758 and Welch Foundation grant F-1254. The authors would like to thank Dr. Xinsheng Zhao for the discussion on formic acid oxidation catalyzed by Pd/C and Kristen Bateman for her assistance with some of the sample preparation.
Author information
Authors and Affiliations
Contributions
W.L. recognized the phenomenon and both W.L. and T.C. carried out the experiments while A.M. supervised the research work. All the three authors designed the experiments and contributed to the writing of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, W., Cochell, T. & Manthiram, A. Activation of Aluminum as an Effective Reducing Agent by Pitting Corrosion for Wet-chemical Synthesis. Sci Rep 3, 1229 (2013). https://doi.org/10.1038/srep01229
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01229
This article is cited by
-
Current collectors corrosion behaviours and rechargeability of TiO2 in Aqueous Electrolyte Aluminium-ion batteries
Journal of Applied Electrochemistry (2023)
-
A general fruit acid chelation route for eco-friendly and ambient 3D printing of metals
Nature Communications (2022)
-
One step preparation of copper–silver self-catalyzed hybrid conductive ink with reduced sintering temperature for flexible electronics
Journal of Materials Science: Materials in Electronics (2019)
-
Synthesis of hydroxylated fatty amide from underutilized seed oil of Khaya senegalensis: a potential green inhibitor of corrosion in aluminum
Journal of Analytical Science and Technology (2018)
-
Biogenic corrosion inhibitor on mild steel protection in concentrated HCl medium
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
