
Abstract
Dishevelled, a phosphoprotein scaffold, is a central component in all the Wnt-sensitive signaling pathways. In the present study, we report that Dishevelled is post-translationally modified, both in vitro and in vivo, via arginine methylation. We also show protein arginine methyl transferases 1 and 7 as the key enzymes catalyzing Dishevelled methylation. Interestingly, Wnt3a stimulation of F9 teratocarcinoma cells results in reduced Dishevelled methylation. Similarly, the methylation-deficient mutant of Dishevelled, R271K, displayed spontaneous membrane localization and robust activation of Wnt signaling; suggesting that differential methylation of Dishevelled plays an important role in Wnt signaling. Thus arginine methylation is shown to be an important switch in regulation of Dishevelled function and Wnt signaling.
Similar content being viewed by others


Introduction
Wnt signaling plays a critical role during embryonic development and aberrant activation of Wnt pathway leads to tumorigenesis1,2,3,4,5. Wnt ligands bind to their cognate receptors, Frizzleds (Fzs) and initiate multiple intracellular signaling cascades that include β-catenin-dependent and β-catenin-independent pathways. Canonically (β-catenin-dependent), Wnt stimulation leads to post-transcriptional and post-translational mechanism/s-mediated stabilization of β-catenin, which is otherwise subjected to proteasome-mediated degradation6,7,8,9. Stabilized β-catenin translocates into the nucleus where it functions as a co-activator with lymphoid enhancer factor (LEF)/T-cell factor (TCF) transcription factors10,11. Wnts also initiate non-canonical pathways (β-catenin-independent), which either leads to activation of small G-proteins and c-Jun N-terminal kinases (JNK) or cyclic guanosine monophosphate (cGMP)/PDE/calcium pathways12,13. Interestingly, all of these Wnt-sensitive pathways, which are highly conserved from fly to the humans14,15, operate simultaneously in the same cells16,17,18. However, the mechanism/s by which the Wnt signal segregates into distinct downstream signaling pathways is not well defined.
Dishevelled (Dvl) is a central component in all the Wnt-sensitive pathways19,20. Structurally, Dvls display three conserved domains: a DIX domain at the N-terminus; a PDZ domain in the mid region; and a DEP domain located between the PDZ domain and the C-terminus of Dvl19,20. While, the DIX domain enables possible dimerization of Dvl with other members of the Dvl family as well as with Axin, the PDZ and DEP domains provide docking sites for a large number of proteins that include protein kinases, phosphatases, adaptor proteins and most importantly Frizzleds with a C-terminal PDZ ligand like structure20,21. Together, the Dvl-interactome represents a “molecular toolbox” of signaling molecules, which is critical for Wnt-sensitive pathway activation19. Despite their large molecular weight [2 Mega Da22], Dvl-based supermolecular complexes are also dynamic. In cultured cells, Dvls were observed to form large cytoplasmic aggregates23, which quickly relocate to the cell membrane upon Wnt stimulation24. How the Wnt signal provokes the assembly of Dvl-based supermolecular complexes and their membrane localization remains unclear.
Post-translational modifications regulate protein function/s by altering protein-protein interactions25. We recently identify arginine methylation as a novel post-translational modification regulating Wnt signaling8,9. In arginine methylation, protein arginine methyl transferases (PRMTs) catalyze the addition of methyl group/s to the nitrogens of arginines within the protein/s26. We and others have also shown that methylated arginines either block or promote protein-protein interactions8,9,27,28.
In the present study, we report that Dvl3, one of the three Dvl isoforms encoded by the mammalian genome, is a novel PRMT substrate. Using in silico, biochemical and cell biological approaches we show that Dvl3 is post-translationally modified via arginine methylation. We also identify PRMT1 and PRMT7 as the key enzymes catalyzing Dvl3 methylation. Interestingly, activation of Wnt pathway resulted in reduced Dvl3 methylation. Consistent with the Wnt-stimulated reduction in Dvl methylation, methylation-deficient mutants of Dvl3 also displayed increased membrane localization and robust stimulation of Lef/Tcf-sensitive gene transcription, revealing a novel biological role for Dvl3 methylation in the regulation of Wnt signaling.
Results
Dvl3 is a PRMT substrate
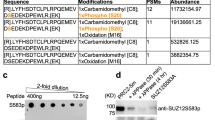
Proteins that harbor arginine-glycine rich regions (RG) are predominant targets for PRMTs. Using in silico searches, we have identified three ‘RG’ motifs within the primary protein sequence of Dvl3 (Fig. 1A). The identified RG-motifs are located in the PDZ domain (R271), in the linker 2 domain (R342) and in the C-terminus (R614) of Dvl3 (Fig. 1A). Alignment of Dvl3 sequences revealed that R271 and R614 residues are highly conserved across species, including mouse, human, xenopus, zebrafish and drosophila (Fig. 1B). While, R342 is restricted to human, mouse and xenopus Dvl3 sequences (Fig. 1B), indicating that these conserved “RG” motifs might have a functional role.

Dvl3 is a PRMT substrate.
(A) Primary amino acid sequences of Dvl3 were scanned for the presence of ‘RG’ rich regions and are represented in the figure. (B) Primary amino acid sequences of Dvl3 from mouse, human, Xenopus, zebrafish and Drosophila were aligned using ClustalW. Marked arginine residues are evolutionarily conserved.
Dvl3 is methylated by PRMTs in vitro and in vivo
As a first step to explore if Dvl3 is indeed a PRMT substrate, we tested if purified PRMTs could catalyze Dvl3 methylation. For these experiments, epitope tagged-PRMTs (HA-PRMT1, 2, 5,7,and 8) were expressed and purified from F9 teratocarcinoma cells (F9 cells) using anti-HA affinity matrix, while, Dvl3 (Myc-hDvl3) was expressed and purified from human embryonic kidney 293 (HEK293) cells. The methylation reaction includes the purified PRMTs, purified hDvl3 and 3H-S-adenosyl L-Methionine (SAMe) as the methyl donor (Fig. 2). Of all the PRMTs tested, only PRMT1, 7 and 8, but not PRMT2 and 5, could readily methylate Dvl3 (Fig. 2A). Interestingly, PRMT7 catalyzed stronger methylation of Dvl3 in comparison to PRMT1 or PRMT8 (Fig. 2A). Although there are similarities between PRMTs 1, 7 and 8 based on phylogenetic evolution29, a more common method of analyzing PRMT function is based on the end products produced26. PRMTs are classified into several families that include, type I enzymes (PRMT1, PRMT3, PRMT4/CARM1, PRMT6, PRMT8) and type II enzymes (PRMT5, PRMT7, PRMT10), which catalyze the formation of asymmetric or symmetric dimethyl arginines, respectively, on the terminal guanadino (ω) nitrogens26. It is very interesting and unique to observe both type I and type II PRMT-mediated methylation of Dvl3. However, the functional consequence/s of such differential methylation by type I and type II enzymes on Dvl3's function and Wnt signaling, remains to be discerned.

PRMTs catalyze Dvl3 methylation.
(A) PRMTs purified from F9 cells were used in an in vitro methylation reaction along with purified myc-hDvl3 (5 µl) and 1 µCi [3H]-SAMe as described in the methods. PRMTs purified from F9 cells treated with Wnt3a (20 ng/ml) were used in an in vitro methylation assay along with Dvl3 purified either from untreated (B) or HEK293 cells treated with Adenosine dialdehyde (C) as described in the methods. (D) F9 cells were labeled with [3H]-L-methyl methionine in the absence or presence of Wnt3a (20 ng/ml) as described in the methods. The methylation status of Dvl3 was revealed by anti-Dvl3 pull-downs, SDS-PAGE and fluorography. After fluorography, the blots were probed with anti-Dvl3 antibodies (lower panel). The amounts of Dvl3 methylated (fluorograph) and the total amount of Dvl3 immunoprecipitated (myc immunoblots) were quantified using Quantity one (BioRad) software and the normalized values (methylated Dvl3/total Dvl3) were represented in the figure as densitometry readings.
Earlier we have shown that methylation of G3BP1 (a Dvl3-associated protein) by PRMT1 is Wnt3a sensitive8. We probed next if PRMT1-mediated Dvl3 methylation is also Wnt3a sensitive. PRMT1 purified from un-stimulated F9 cells failed to methylate Dvl3 (Fig. 2B). On the contrary, PRMT1 isolated from Wnt3a-stimulated F9 cells catalyzed a profound and progressive increase in Dvl3 methylation (Fig. 2B). To ascertain if a methyl mark by another PRMT is a pre-requisite for Dvl3 methylation by PRMT1, we performed in vitro methylation assays on Dvl3 purified from HEK293 cells treated with a methyl transferase inhibitor, adenosine dialdehyde (Adox). Interestingly, PRMT1 catalyzed a similar Wnt3a-stimulated methylation on the hypomethylated Dvl3 substrate (Fig. 2C), suggesting that a prior methyl mark on Dvl3 is not obligate for methylation by PRMT1 (Fig. 2C).
To determine if Dvl3 is also methylated in vivo, we made use of metabolic labeling of cells with 3H-L-methyl methionine as described in the methods. Dvl3 pull-downs of labeled F9 cell lysates followed by fluorography revealed Dvl3 methylation (Fig. 2D). While, pull-downs performed on similar lysates with control IgG (cIgG) served as a negative control (Fig. 2D). Interestingly, Dvl3 pull-downs performed on lysates of F9 cells, which are labeled and stimulated with Wnt3a (3 h), displayed a remarkable reduction in Dvl3 methylation (Fig. 2D). These data clearly establish that Dvl3 is indeed a PRMT substrate and that Wnt stimulation provokes reduced Dvl3 methylation.
Differential methylation of Dvl3 by PRMT1 and 7
The protein sequence of Dvl3 encodes three “RG” motifs (R271, R342, R614, Fig. 1). To test whether R271, R342 and R614 are direct targets of PRMTs, we made methylation-deficient mutants of Dvl3 by substituting arginines with likely charged lysines (R271K, R342K, R614K). The methylation-deficient mutants were successfully expressed and purified in HEK293 cells (Fig. 3). The abilities of PRMT1 and PRMT7 to catalyze methylation of the wild type and methylation-deficient mutants of Dvl3 were later measured using in vitro methylation assays (Fig. 3). As shown earlier (Fig. 2), PRMT7 catalyzed efficient methylation of wild-type Dvl3 (Fig. 3). PRMT7, in strong contrast, failed to methylate R271K, R342K and R614K mutants of Dvl3 (Fig. 3). While, PRMT1 isolated from Wnt3a-stimulated F9 cell lysates catalyzed methylation of both wild-type and R271K mutant but not R342K or R614K (Fig. 3). Taken together, these data suggest that differential methylation by PRMT1 (at R342 and R614) and by PRMT7 (at R271, R342 and R614) might play an important role in Dvl3 function/s. It is interesting to note an increased methylation of R271K mutant of Dvl3 by Wnt3a-stimulated PRMT1 (Fig. 3). Although only a speculation, structural changes associated with the absence of basal methylation on R271 of Dvl3 (presumably by PRMT7) might facilitate greater access to PRMT1, resulting in increased methylation.

Differential methylation of Dvl3 by PRMT1 and PRMT7.
PRMT1 and PRMT7 were purified from F9 cells and were utilized in an in vitro methylation assay using purified myc-hDvl3 (wild-type) or its mutants R271K, R342K, or R614K as substrates and3H-SAMe as a methyl donor.
R271K/R614K mutants of Dvl3 localizes to plasma membrane
Dvls form functional polymers/oligomers22,23 and when expressed in cell cultures form large cytoplasmic punctae30. We probed next, if the methylation-deficient mutants of Dvl3 are able to form cytosolic punctae. For these experiments, myc-tagged wild-type and myc-tagged methylation-deficient mutants of Dvl3 were transfected into HEK293 cells followed by immunocytochemistry with anti-myc antibodies (Fig. 4A). As expected, the wild-type Dvl3 formed large cytosolic aggregates (Fig. 4A). Strikingly, the R271K and R614K mutants of Dvl3 not only failed to form cytosolic aggregates but also spontaneously localized to the cell membrane (Fig. 4A). While, R342K mutant, similar to that of wild-type, formed cytosolic punctae (Fig. 4A). To further test the role of R-K substitutions in differential localization of Dvl3 mutants, we purified the membrane and cytosolic fractions of HEK293 cells transfected with wild-type and methylation-deficient mutants of Dvl3 (Fig. 4B). Western blotting of the membrane and the cytosolic fractions with anti-myc antibodies revealed high levels of R271K and R614K mutants in the membrane fractions when compared to wild-type Dvl3 (Fig. 4B). R342K mutant levels in the membrane fractions were indifferent to that of wild-type Dvl3 (Fig. 4B). Consistent to our observations in HEK293 cells, expression of wild-type and methylation-deficient mutants of Dvl3 in normal bronchial epithelial cells (Beas-2B, B2B) also showed increased levels of R271K and R614K in the membrane fractions in comparison to wild-type and R342K mutant of Dvl3 (Fig. 4C).

R271K mutant of Dvl3 localizes to plasma membrane.
(A) HEK293 cells transfected with empty vector, myc-hDvl3 (wild-type) or its mutants R271K, R342K, R614K were fixed, permeabilized and immunostained with anti-myc antibodies and Cy3-labeled secondary antibodies. Scale bar 5 µm. HEK293 cells (B) or bronchial epithelial cells (Beas-2B, C) were transfected either with empty vector, myc-hDvl3 (wild-type) or its methylation-deficient mutants, membrane and cytosolic fractions separated and immunoblots performed with anti-myc and anti-sodium/potassium ATPase antibodies as described in the methods. For quantifying the amounts of wild-type or methylation-deficient mutants of Dvl3 in the membrane fractions of Beas2B cell lysates, Quantity one (BioRad) software was used. Dvl3 amounts were normalized to their corresponding sodium potassium ATPase controls and are represented in the figure as densitometry readings.
R271K mutant of Dvl3 stimulates Wnt signaling
Over-expression of Dvls trigger Wnt signaling independent of Wnts7,31,32. We probed next if arginine-lysine (R-K) substitutions of Dvl3 alter their Wnt signaling abilities as determined by Lef/Tcf-sensitive luciferase activities (Fig. 5). Expression of wild-type Dvl3, as expected, induced a robust increase in Lef/Tcf-sensitive luciferase activities (Fig. 5A). Strikingly, expression of R271K mutant of Dvl3 induced a more profound increase in luciferase activities in comparison to wild-type Dvl3-induced effects (Fig. 5A). Expression of R342K or R614K mutants of Dvl3, also induced increased Lef/Tcf-dependent luciferase activities, albeit less efficiently than R271K mutant (Fig. 5A). The effects of methylation-deficient mutants of Dvl3 on Lef/Tcf-sensitive luciferase activities are not restricted to HEK293 cells, expression of wild-type or mutant forms of Dvl3 also induced similar effects in human bronchial epithelial cells (B2B, Fig. 5B). Thus, arginine methylation appears to be an important regulator of not only Dvl3 transport to the cell membrane but also activation of Wnt signaling.

Methylation-deficient mutants of Dvl3 stimulate Lef/Tcf-sensitive gene transcription.
HEK293 cells (A) or bronchial epithelial cells (B) were transiently co-transfected with super8xTOPFLASH and either myc-hDvl3 (wild-type) or its methylation-deficient mutants. The lysates were later assayed for luciferase activities as described in the methods. The data represents mean values ± S.E.M. from three independent experiments. **, p< 0.01; versus control (wild-type).
Discussion
In the present study, we report a novel role for Dvl3 methylation in Wnt signaling. It is interesting to note that R271, located in the PDZ domain, is predominantly methylated by PRMT7. Since, the PDZ domain provides docking sites for critical regulatory proteins19,20, we speculate that methylation of R271 might favor binding of Dvl3-associated proteins (DAPs)'s, which prevent membrane movement of Dvl3. Wnt stimulation, on the contrary, disrupts PRMT-mediated methylation of Dvl3 (Fig. 2D), releasing Dvl3-DAP interaction, ultimately leading to trafficking of Dvl3 to the cell membrane. Our hypothesis stemmed from the solved solution structure of PDZ domain of mDv1121. R271 resides in the flexible linker region between β-sheets “B” and “C”. Owing to methylation, R271 becomes strongly hydrophobic, which might trigger Dvl3 movement into a non-polar environment either through protein aggregation or protein interaction/s. The failure of R271K mutant to form cytoplasmic punctae and also to spontaneously relocate to the cell membrane (Fig. 4) strongly supports our hypothesis. However, the molecular details of these altered interactions either with known interactors or novel binding partners, upon methylation at R271, R342 and R614 remains to be discerned.
It is also interesting to note that while, PRMT7 catalyze basal methylation of Dvl3, PRMT1, on the contrary catalyze Dvl3 methylation upon Wnt3a stimulation (Fig. 2). In addition, PRMT1 catalyzed a similar methylation of Dvl3 isolated from untreated HEK293 cells or HEK293 cells treated with a methyl transferase inhibitor (Fig. 2). These data indicate that prior methylation by PRMTs, for e.g., on R271 by PRMT7, might not be obligate for Dvl3 methylation by PRMT1, or PRMT1 might specifically target only un-methylated Dvl3 substrates or both. Furthermore, the increase in Dvl3 methylation by PRMT1 later in the Wnt3a time course (4–6 hours) might as well represent a switching “off” of Wnt signal. Thus, differential methylation of Dvl3 by PRMT1 and PRMT7 appears to be a “switch” in regulating Dvl3 function and Wnt signaling.
In summary, the present report focuses on a single point: Dvl3 is a novel PRMT substrate. The identification of Dvl3 as a PRMT substrate adds a new and an important dimension to Dvl biology. This report also represents the beginning towards expanding our understanding of Dvl biology in relation to fundamental processes critical during embryogenesis and cancer. In order to better appreciate the role of Dvl3 methylation in Wnt signaling, which are beyond the scope of this report, is to examine if Dvl3 is poorly methylated or demethylated upon Wnt stimulation and whether PRMT7 activity is blocked by Wnt signaling. Our findings also provide key insights into therapeutic strategies of Dvl3-mediated cancers. The expression levels of all the Dvl isoforms, particularly Dvl3, are high in lung cancers33,34,35. However, the methylation status of Dvl3 in lung cancers is unknown and can serve as a biomarker for early detection of the disease.
Methods
Purification of Dvl3
HEK293 cells were transiently transfected with either myc-hDvl3 or methylation-deficient mutants of hDvl3 (6 µg) in 100 mm culture dishes. For purifying hypomethylated form of Myc-hDvl3, Dvl3 transfected cells were treated with Adenosine dialdehyde (Adox, 10 µM) for 16 h. The cells were later lysed in 1 ml of lysis buffer (1x PBS, 1% Nonidet P-40, 0.5% Sodium deoxycholate, 0.1% SDS, 1 µg/ml leupeptin, 1 µg/ml aprotonin and 1 µg/ml phenlymethylsulphonyl fluoride). The lysates were then utilized to immunoprecipitate Dvl3 using anti-myc agarose (60 µl beads/1 ml of lysate, A7470, Sigma) at 4°C for 16 h. After 16 h, the immunoprecipitates were washed thrice in RIPA buffer [20 mM Tris (pH 8.0), 150 mM NaCl, 5 mM EDTA and 1% Triton-X-100] and the bound myc-Dvl3 was eluted with 2 volumes of 1 mg/ml c-myc peptide twice. The eluted protein was pooled, concentrated to ~ 30 µl using Amicon ultra centrifugal filters and subsequently used in in vitro methylation experiments.
In vitro methylation assays
In vitro methylation assays were performed as described earlier8. Briefly, F9 cells were transiently transfected with HA-PRMTs (6 µg) in 100 mm culture dishes. After 24 h, the cells were treated with Wnt3a for different durations followed by cell lysis. The lysates were then utilized to purify PRMT1 using anti-HA antibodies and protein A sepharose CL-4B (17-0780-01, GE Life Sciences). After 16 h, the immunoprecipitates were washed thrice in RIPA buffer and once in methylation buffer (50 mM Tris pH 8.5, 20 mM KCl, 10 mM MgCl2, 1 mM β-mercaptoethanol, 100 mM Sucrose). Finally, purified PRMT1 was incubated with 10 µl of methylation reaction buffer containing 5 µl of purified myc-hDvl3 or its mutants and 1 µCi of S-adenosyl-L-[methyl-3H] methionine (NEN radiochemicals, 250 µCi, 9.25 MBq), at 30°C for 1 h. After 1 h, the reactions were stopped by addition of SDS sample loading buffer, boiled and separated on a SDS-PAGE gel. The gel was then fixed (45% methanol, 10% acetic acid in water, 30 min), amplified (Autofluor, National Diagnostics, 2 h), dried and fluorography was performed.
In vitro methylation assays
In vivo methylation assay for Dvl3 was performed as described earlier8. Briefly, F9 cells were seeded in 100 mm culture dishes and grown to confluence (24 h). After 24 h, the cells were washed once with PBS and protein translation was inhibited by incubating with 100 µg/ml cycloheximide and 40 µg/ml chloramphenicol in DMEM medium with 10% FBS for 30 min at 37°C. After 30 min, the cells were washed once with methionine free DMEM. Cell labeling mixture consisting of methionine free DMEM supplemented with 100 µg/ml cycloheximide and 40 µg/ml chloramphenicol and 60 µCi of L-[methyl-3H] methionine was added to the cells in the absence or presence of Wnt3a (20 ng/ml) and incubated at 37°C for 3 h. After 3 h of metabolic labeling, the cells were lysed in a lysis buffer. The methylation status of Dvl3 was later revealed by anti-Dvl3 pull-downs, SDS-PAGE and fluorography. After fluorography the blots were probed with anti-Dvl3 antibodies. Densitometric measurements of the bands were performed by using ChemiDoc XRS gel imaging system and Quantity one software (BioRad). The amounts of Dvl3 methylated (fluorograph) and the total amount of Dvl3 immunoprecipitated (myc immunoblots) were quantified and the normalized values (methylated Dvl3/total Dvl3) were represented in the figure.
Luciferase assays
HEK293 cells or normal bronchial epithelial cells (Beas2B), were co-transfected with myc-Dvl3 or its mutants with super 8xTOPFLASH (M50) luciferase reporter (gift from Dr. Randall Moon, Univ. of Washington). After 24 h, the cells were directly lysed in the culture plates by addition of 1x cell culture lysis reagent (Promega, Madison, WI). Luciferase activities of the lysates were later measured as described earlier8.
Immunocytochemistry
HEK293 cells were transiently transfected with myc-hDvl3 or its mutants. After 24 h, the cells were fixed, permeabiized and stained with anti-myc antibodies followed by staining with Cy3-labeled anti-mouse secondary antibodies as described earlier7.
Membrane fractionation
HEK293 cells or Beas2B cells were transiently transfected with myc-hDvl3 or its mutants (6 µg/100 mm dishes). After 24 h, the cells were collected in a hypotonic lysis buffer and the membrane and cytosol fractions were separated as described earlier24. The amounts of Dvl3 and its mutants present in the membrane fractions were later evaluated by immunoblotting with anti-myc antibodies and anti-sodium potassium ATPase antibodies. Densitometric measurements of the bands were performed by using ChemiDoc XRS gel imaging system and Quantity one software (BioRad). Dvl3 amounts were normalized to their corresponding sodium potassium ATPase controls and are represented in the figure.
References
Polakis, P. Wnt signaling and cancer. Genes Dev 14, 1837–1851 (2000).
Logan, C. Y. & Nusse, R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20, 781–810 (2004).
Moon, R. T., Kohn, A. D., De Ferrari, G. V. & Kaykas, A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet 5, 691–701 (2004).
Malbon, C. C. Beta-catenin, cancer and G proteins: not just for frizzleds anymore. Sci STKE 2005, pe35 (2005).
Winn, R. A. et al. Restoration of Wnt-7a expression reverses non-small cell lung cancer cellular transformation through frizzled-9-mediated growth inhibition and promotion of cell differentiation. J Biol Chem 280, 19625–19634 (2005).
Angers, S. & Moon, R. T. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 10, 468–477 (2009).
Bikkavilli, R. K. & Malbon, C. C. Dishevelled-KSRP complex regulates Wnt signaling through post-transcriptional stabilization of beta-catenin mRNA. J Cell Sci 123, 1352–1362 (2010).
Bikkavilli, R. K. & Malbon, C. C. Arginine methylation of G3BP1 in response to Wnt3a regulates {beta}-catenin mRNA. J Cell Sci 124, 2310–2320 (2011).
Bikkavilli, R. K. & Malbon, C. C. Wnt3a-stimulated LRP6 phosphorylation is dependent upon arginine methylation of G3BP2. J Cell Sci 125, 2446–2456 (2012).
Molenaar, M. et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 86, 391–399 (1996).
Behrens, J. et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638–642 (1996).
Wang, H. Y. & Malbon, C. C. Wnt signaling, Ca2+ and cyclic GMP: visualizing Frizzled functions. Science 300, 1529–1530 (2003).
Bikkavilli, R. K. & Malbon, C. C. Mitogen-activated protein kinases and Wnt/beta-catenin signaling: Molecular conversations among signaling pathways. Commun Integr Biol 2, 46–49 (2009).
Mlodzik, M. Planar cell polarization: do the same mechanisms regulate Drosophila tissue polarity and vertebrate gastrulation? Trends Genet 18, 564–571 (2002).
Boutros, M., Paricio, N., Strutt, D. I. & Mlodzik, M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell 94, 109–118 (1998).
Habas, R., Dawid, I. B. & He, X. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev 17, 295–309 (2003).
Bikkavilli, R. K., Feigin, M. E. & Malbon, C. C. G alpha o mediates WNT-JNK signaling through dishevelled 1 and 3, RhoA family members and MEKK 1 and 4 in mammalian cells. J Cell Sci 121, 234–245 (2008).
Bikkavilli, R. K., Feigin, M. E. & Malbon, C. C. p38 mitogen-activated protein kinase regulates canonical Wnt-beta-catenin signaling by inactivation of GSK3beta. J Cell Sci 121 3598-3607 (2008).
Malbon, C. C. & Wang, H. Y. Dishevelled: a mobile scaffold catalyzing development. Curr Top Dev Biol 72, 153-166 (2006).
Gao, C. & Chen, Y. G. Dishevelled: The hub of Wnt signaling. Cell Signal 22, 717–727 (2010).
Wong, H. C. et al. Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell 12, 1251–1260 (2003).
Yokoyama, N., Golebiewska, U., Wang, H. Y. & Malbon, C. C. Wnt-dependent assembly of supermolecular Dishevelled-3-based complexes. J Cell Sci 123, 3693–3702 (2010).
Schwarz-Romond, T. et al. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol 14, 484–492 (2007).
Yokoyama, N., Yin, D. & Malbon, C. C. Abundance, complexation and trafficking of Wnt/beta-catenin signaling elements in response to Wnt3a. J Mol Signal 2, 11 (2007).
Bedford, M. T. & Richard, S. Arginine methylation an emerging regulator of protein function. Mol Cell 18, 263–272 (2005).
Bedford, M. T. Arginine methylation at a glance. J Cell Sci 120, 4243–4246 (2007).
Bedford, M. T. & Leder, P. The FF domain: a novel motif that often accompanies WW domains. Trends Biochem Sci 24, 264–265 (1999).
Bedford, M. T. et al. Arginine methylation inhibits the binding of proline-rich ligands to Src homology 3, but not WW, domains. J Biol Chem 275, 16030–16036 (2000).
Krause, C. D. et al. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther 113, 50–87 (2007).
Schwarz-Romond, T., Merrifield, C., Nichols, B. J. & Bienz, M. The Wnt signalling effector Dishevelled forms dynamic protein assemblies rather than stable associations with cytoplasmic vesicles. J Cell Sci 118, 5269–5277 (2005).
Axelrod, J. D., Miller, J. R., Shulman, J. M., Moon, R. T. & Perrimon, N. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev 12, 2610–2622 (1998).
Winn, R. A. et al. Antitumorigenic effect of Wnt 7a and Fzd 9 in non-small cell lung cancer cells is mediated through ERK-5-dependent activation of peroxisome proliferator-activated receptor gamma. The Journal of biological chemistry 281, 26943–26950 (2006).
Wei, Q. et al. Dishevelled family proteins are expressed in non-small cell lung cancer and function differentially on tumor progression. Lung Cancer 62, 181–192 (2008).
Zhao, Y. et al. Dishevelled-1 and dishevelled-3 affect cell invasion mainly through canonical and noncanonical Wnt pathway, respectively and associate with poor prognosis in nonsmall cell lung cancer. Mol Carcinog 49, 760–770 (2010).
Uematsu, K. et al. Activation of the Wnt pathway in non small cell lung cancer: evidence of dishevelled overexpression. Oncogene 22, 7218–7221 (2003).
Acknowledgements
This work was supported by a Merit Award from the U.S. Department of Veterans Affairs, NIH grants R01CA1385282522717 and 5R21CA153268-02 to RW and USPHS Grant DK30111 from the NIDDK, National Institutes of Health to CCM.
Author information
Authors and Affiliations
Contributions
RKB conceived, designed and performed most of the experimental work and data analysis. SA, MVS, MS and NK assisted with the experiments. CCM and RW coordinated the project and assisted in planning the experiments and data analysis. RKB, CCM and RW wrote and approved the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Bikkavilli, R., Avasarala, S., Vanscoyk, M. et al. Dishevelled3 is a novel arginine methyl transferase substrate. Sci Rep 2, 805 (2012). https://doi.org/10.1038/srep00805
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00805
This article is cited by
-
Gene regulatory network from cranial neural crest cells to osteoblast differentiation and calvarial bone development
Cellular and Molecular Life Sciences (2022)
-
Phosphorylation-induced changes in the PDZ domain of Dishevelled 3
Scientific Reports (2021)
-
Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response
Nature Communications (2020)
-
Non-histone protein methylation as a regulator of cellular signalling and function
Nature Reviews Molecular Cell Biology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
