
Abstract
Angiotensin-I converting enzyme (ACE), a two-domain dipeptidylcarboxypeptidase, is a key regulator of blood pressure as a result of its critical role in the renin-angiotensin-aldosterone and kallikrein-kinin systems. Hence it is an important drug target in the treatment of cardiovascular diseases. ACE is primarily known for its ability to cleave angiotensin I (Ang I) to the vasoactive octapeptide angiotensin II (Ang II), but is also able to cleave a number of other substrates including the vasodilator bradykinin and N-acetyl-Ser-Asp-Lys-Pro (Ac-SDKP), a physiological modulator of hematopoiesis. For the first time we provide a detailed biochemical and structural basis for the domain selectivity of the natural peptide inhibitors of ACE, bradykinin potentiating peptide b and Ang II. Moreover, Ang II showed selective competitive inhibition of the carboxy-terminal domain of human somatic ACE providing evidence for a regulatory role in the human renin-angiotensin system (RAS).
Similar content being viewed by others

Introduction
Much attention has focused on the role of the systemic renin-angiotensin system (RAS) in mammalian blood homeostasis and cardiovascular function, but there is also tremendous and increasing interest in understanding the physiological and pathological roles of the local, or tissue, RAS1,2. The systemic RAS relies on a proteolytic cascade involving circulating renin/prorenin and angiotensin-I converting enzyme (ACE, EC 3.4.15.1), a carboxydipeptidase present on the external surface of endothelial cells of the lung, to generate the hypertensive and mitogenic octapeptide, angiotensin II (Ang II) from angiotensinogen (AGT). AGT is a serum α2-globulin that is released constitutively into the blood from the liver. Renin, an aspartyl protease secreted into the circulation from the kidney, catalyses the release of the decapeptide angiotensin I (Ang I) from the N-terminus of AGT. The last step in the production of Ang II is the cleavage of the dipeptide, His-Leu, from the C-terminus of Ang I by endothelial ACE3. Although ACE is a promiscuous peptidase, it does not cleave Ang II. This resistance to further hydrolysis by endothelial ACE is an important contribution to the success of Ang II as a systemic signalling peptide, ensuring that it is the major product of the RAS after passage through the pulmonary vascular bed4.
In contrast, tissue RAS is characterised by the presence of AGT, angiotensin processing enzymes and angiotensin receptors in a single tissue to form a fully functional RAS. These local systems for generating tissue Ang II have been identified in many mammalian tissues, including the heart, kidney, brain, bone marrow, pancreas and adipose tissues5. Local RAS in organs involved in regulating renal and cardiovascular physiology appear to augment circulating Ang II signalling and therefore might influence the progress of hypertension and cardiovascular disease1,6. Recently, evidence has been provided for an intracellular autocrine RAS that generates Ang II to control cell growth and for a functional RAS in human mitochondria where it has proposed roles in modulating respiration and nitric oxide (NO) production7. However, unlike the systemic RAS where renin secretion by the kidney is an important regulator of AGT processing, it is not at all clear how the production of Ang II by a tissue RAS is controlled.
The major form of vertebrate ACE (somatic ACE, sACE, a heavily glycosylated protein) comprises two very similar protein domains (N- and C-domains), each with a catalytic centre involving a zinc ion (for comprehensive reviews see8,9). The mammalian ACE gene arose from a gene duplication event during the course of vertebrate evolution10. Studies using domain-selective inhibitors and Ace knock-out mice have shown that the C-domain of mammalian sACE is mainly responsible for Ang II formation11,12. sACE is also known as kininase II from its contribution with carboxypeptidase N (kininase I), to the metabolic inactivation of the vasodilatory peptide bradykinin (BK) in the blood. This reaction is catalysed equally efficiently by both domains of sACE3. Another physiologically important substrate for sACE is Ac-SDKP, a peptide regulator of hematopoietic stem cell and fibroblast proliferation. The degradation of Ac-SDKP is exclusively carried out by the N-domain13. The importance of sACE in the metabolism of both circulating Ang II and BK is the physiological basis for the effectiveness of ACE inhibitors in reducing blood pressure14. However, current ACE inhibitors are not domain-specific and this results in elevated levels of BK which is widely accepted as mainly responsible for triggering side effects such as cough and angioedema15,16. Thus, C-domain selective ACE inhibitors are likely to reduce blood pressure by lowering Ang II, but have improved side effect profiles. The recently determined high-resolution crystal structures of ACE proteins has identified some of the differences in active site architecture of the N- and C- domains that appear to be responsible for some of the divergent catalytic properties of the two enzyme activities and revealed molecular interactions of domain selective synthetic ACE inhibitors17,18,19.
The first ACE inhibitors showing anti-hypertensive properties were members of a large family of natural bradykinin-potentiating peptides (BPPs) isolated from snake venoms20. Interestingly, some of these display selective potency towards the C-domain of sACE21. They characteristically possess an N-terminal pyroglutamate residue and a C-terminal Pro-Pro dipeptide and are strong competitive inhibitors of Ang I cleavage4,22,23. Current ACE inhibitors in clinical use today all possess an ionisable group that coordinates with the active site zinc and therefore must differ from the natural peptidic inhibitors in their mechanism of inhibition. Although several crystal structures of ACE proteins with bound inhibitors have been determined in recent years, there have been no structural data of the enzyme in complex with natural peptides.
In the present study, we have elucidated the crystal structure of human C-domain sACE in complex with two natural peptides, Ang II, the principal end-product of the RAS and with a snake venom inhibitor- bradykinin potentiating peptide (BPPb, a human C-domain specific peptide inhibitor)21. The structure of the complex with BPPb reveals for the first time the detailed molecular interactions in a zinc independent manner. We have also established that Ang II is a competitive inhibitor of human sACE, displaying strong selectivity towards the C-domain active site. In addition, the structure of the C-domain sACE-Ang II complex revealed the role of the penultimate Pro residue of Ang II in conferring resistance to hydrolysis. This inhibition of ACE by Ang II could provide a negative feedback mechanism contributing to homeostatic regulation of Ang II production in the tissue and cellular RAS.
Results
Crystal structures of human C-domain sACE - peptide complexes
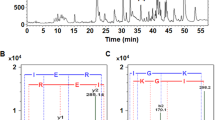
Human C-domain sACE was co-crystallised with Ang II and the bradykinin potentiating peptide (BPPb) at 2.0 and 2.6Å resolution, respectively (Fig. 1a,b; Table 1). The schematic model of the ACE active site with the various sub-site nomenclature described in this report is shown in Fig. 2a. The co-crystallisation of Ang I (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) with C-domain sACE resulted in conversion to Ang II (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe) which can be observed in the substrate-binding channel. In the C-domain sACE-Ang II peptide structure, electron density was observed for the main chain for a six-residue peptide (Fig 2b). Unambiguous difference density map for the side chains of sites P2 and P1 of the peptide (Val3, Tyr4) was interpreted as Ang II being present in two different (‘sliding’) conformations. In one conformation residues 1 to 6 were fitted (Asp-Arg-Val-Tyr-Ile-His) while residues 2-7 (Arg-Val-Tyr-Ile-His-Pro) were included for the second one with Val3 and Tyr4 shifting into the P3 and P2 positions, respectively (Fig 2f). This result was consistently observed in two independent experiments (i.e., two separate datasets were collected from two different crystals) and analysis of the electron density and B-factors indicated that 0.5 occupancy for each conformation is most likely. Based on molecular modelling we predict that the C-terminal (Pro-) Phe of Ang II could be accommodated in the binding pocket. It is likely that the side chain of Phe occupies the hydrophobic pocket surrounded by aromatic residues Phe512, Tyr146, Trp279 and Trp185 and the peptide main chain atoms extend into the solvent channel by displacing some of the bound solvent molecules towards a cluster of charged residues (Gln160, Glu162). For the BPPb peptide (pGlu-Gly-Leu-Pro-Pro-Arg-Pro-Lys-Ile-Pro-Pro) clear continuous electron density was observed for the entire molecule stretching across a major part of the active site cavity (Fig. 2c). The catalytic zinc ion at the active site is visible in the C-domain sACE-Ang II structure and provides an anchor point through direct coordination with the peptide backbone involving several bound water molecules (Fig. 2d,e; Table 2). Surprisingly, in the C-domain sACE-BPPb complex structure, the binding of BPPb caused the elimination of the zinc ion at the active site, involving significant conformational changes. In particular, His383 which normally provides a coordinating ligand to the zinc ion adopts a different rotomeric conformation making interactions with Gly414 and Asp415 (Fig. 1c). In addition, upon binding of the BPPb peptide (11-mer) in the active site, visible structural rearrangements of the two helices (α1 and α2) and the proximal loop region at the entrance to the cleft (to accommodate the large peptide) was observed (Fig. 1d). These significant structural changes were observed for the first time in the C-domain of sACE active site. In both structures, two chloride ions and an acetate ion (from the crystallisation medium) were observed near the active site and portions of the carbohydrate residues at N-glycosylation sites were evident (Fig. 1a).

(a) Substrate-bound human C-domain sACE crystal structure.C-domain ACE (cyan) in cartoon representation, with Ang II in red sticks, glycosylated carbohydrates in yellow sticks. The catalytic zinc ion is shown as a green sphere. (b) BPPb- bound C-domain ACE crystal structure. C-domain ACE (cyan) in cartoon representation, with BPPb in orange sticks. (c) Loss of the zinc ion coordination upon C-domain ACE binding to BPPb. Left panel, zinc coordination in the Ang II-bound C-domain ACE, with a classical tetrahedral motif, bound zinc ion as green sphere. Right panel, movement of the coordinating His383. (d) Conformational changes of C-domain ACE upon BPPb binding. Ang II and BPPb-bound C-domain ACE (cyan and blue respectively) in cartoon representation. Ang II and BPPb are shown as as red and orange sticks respectively. The zinc ion is only present in the Ang II-bound structure. Movement of the α1 and α2 helices is highlighted.

(a) Schematic model of the ACE active site.The relative positions of the enzyme subsites (Sn, S3, S2, S1, S1′, S2′), the residues of peptide inhibitors (Pn, P3, P2, P1, P1′, P2′) and the position of the catalytic zinc ion are shown. (b) Portions of the difference (Fo-Fc) electron density map for the bound Ang II peptide. Electron density map is contoured at 2.5σ level. The picture was created using a Fourier difference density map in which the peptide atoms were omitted. The two figures represent the interpreted structures of Ang II in two different (sliding) positions with 0.5 occupancy each for Ang II [1-6] and Ang II [2-7] respectively. (c) Portion of the difference (Fo-Fc) electron density map for the BPPb peptide (contoured at 2.5σ level). The picture was created from a Fourier difference density map in which the peptide atoms were omitted. (d) Detailed schematic of Ang II binding to C-domain ACE. Hydrogen bonds are highlighted (dashed lines) with Ang II in red and panel 1 with Ang II [1–6] and 2 with Ang II [2–7]. (e) Schematic diagram of BPPb binding to C-domain ACE. Hydrogen bonds are highlighted (dashed lines) with BPPb in orange. (f) Detailed binding of Ang II with the C-domain ACE. ACE in cartoon representation (cyan) with residues involved in binding highlighted (sticks). Ang II in red sticks, zinc ion as green sphere and water molecules in light blue are shown. Panel 1 with Ang II [1–6] and 2 with Ang II [2–7]. (g) Detailed binding of BPPb to C-domain ACE. ACE in cartoon representation (cyan) with residues involved in binding highlighted (sticks). BPPb in orange sticks, zinc ion as green sphere and water molecules in light blue. (h) Surface representation of Ang II binding (in red sticks) at the active site with electrostatic potential (red, blue for negative and positive potential respectively) computed with the APBS tool in PyMOL. The catalytic zinc ion is shown as a green sphere. (i) Surface representation of BPPb binding (in orange sticks) at the active site drawn as described in (h).
Description of peptide interactions
In the case of C-domain sACE-Ang II peptide complex, the observable part of the Ang II peptide docked with P2, P1, P1′ and P2′of the substrate occupying the S2, S1, S1′ and S2′ sub-sites (the S1 and S1′ subsites flank the catalytic site in the general model of peptidase substrate specificity24), respectively (Table 2, Figs. 2d,f,h). In either of the two configurations adopted by Ang II in the active site cleft, the substrate could not be cleaved, suggesting that Ang II is a true competitive inhibitor of Ang I conversion. The main chain of the C-terminal P2′ residue is clearly visible and anchors the peptide at the S2′ sub-site through hydrogen bonds with Gln281, Tyr520 and Lys511, while the side chain is expected to be stabilised through hydrophobic interaction with the surrounding aromatic residues (in particular Tyr523). The main chain of the P1′ residue from the Ang II peptide makes hydrogen bond interactions with surrounding residues His353, Ala354 and His513. At the P1 position, the residue of Ang II is involved in a tetrahedral coordination with the zinc ion and is stabilised by potential hydrogen bonds with His383, His387 and Tyr523, where the peptide binding channel at the catalytic site is constricted (Fig. 2h). The adjacent P2 residue is stabilised by strong interaction of its main chain with the main chain of Ala356 (through two hydrogen bonds). Clear electron density was visible up to P4 of Ang II, with P3 linked through a water molecule to Arg522, while P4 is positioned in a hydrophobic pocket (S4) in proximity to Trp357.
The structure of C-domain sACE-BPPb complex revealed interactions between the pyrrolidine ring of the two Pro residues in the penultimate and C-terminal positions (P1′ and P2′, respectively) and the amino acid side-chains that form the S1′and S2′ enzyme sub-sites, respectively (Table 2, Figs. 2e,g,i). A cluster of aromatic residues form this binding pocket (Phe457, Tyr523, Val380) and Tyr523 ‘stacks’ against the C-terminal Pro, thus enhancing the interaction with the protein atoms. The two C-terminal Pro residues (P1′ and P2′) are strongly anchored in the prime binding sites (S1′ and S2′) by hydrogen bonds with multiple residues. The P2′ terminal Pro in particular has strong interactions with Tyr520, Lys511 and Gln281. The interaction of the P1′ residue with His353 is dependent on a water molecule due to the movement of the loop region away from the catalytic pocket. Movement of His383 causes loss of coordination of the zinc ion with no steric hindrance by accommodating the bound peptide. The His383 position (normally coordinating with the zinc ion) observed in the native structure is partly occupied by an acetate ion in the present structure of C-domain sACE-BPPb complex (Fig. 1c). The Ile9 residue of the BPPb peptide makes a water-mediated coordination with Glu411. This residue is further stabilised by the S1 hydrophobic patch (composed of His353, Ala354, Phe512). The main chain of the adjacent Lys residue of the peptide (P2) interacts with Ala356 through two hydrogen bonds. All of the N-terminal residues of BPPb were visible. Pro7 is stabilised by hydrophobic interaction with a bulky Trp357 residue, while the Arg6 side chain makes interactions through two hydrogen bonds with Ser516 and Ser517 respectively. The main chain of BPPb is stabilised at Pro5 by hydrogen bond with the hydroxyl group of Tyr360 and residues Pro5 to Leu3 reside in a hydrophobic pocket (Trp59, Ile88). Finally the Gly and pyroGlu at the N-terminus of the BPPb peptide are anchored by hydrogen bonds with Asp121 and Lys118, respectively. The structure clearly demonstrates how longer peptide substrates can be accommodated in the C-domain of sACE.
Comparison of the two structures shows a partial common mode of peptide binding to C-domain of sACE. Similarity at the S2′and S1′ subsites appears to be an essential element with strong hydrogen bonds and hydrophobic interactions contributing to the binding of Pro7/His6 of Ang II and Pro11 of BPPb. Further, the backbone interactions at S2 are identical in both structures with Ala340 stabilising the main chain of the peptide. This arrangement of the orientation of the peptide backbone makes the peptide bond (P1 - P1′ linkage) resistant to cleavage. The major difference in the binding of the two peptides is the visible changes occurring at the catalytic site and the loss of the zinc ion. The movement of the side chains in the C-domain sACE-BPPb complex involves a network of solvent molecules, also interacting with the peptide.
The mode of recognition in the C-domain sACE-Ang II complex structure involves the main chain of the peptide taking control at the catalytic site by replacing a water molecule involved in the proteolysis mechanism. Furthermore, Glu384 (potentially acting as the catalytic base that protonates the amine product as it leaves) does not seem to be involved in any peptide interaction. All together, the peptides presented here show strong interactions with sACE via their backbone atoms and for the first time provide experimental evidence that direct coordination with the zinc ion is not necessary for preventing proteolysis.
Ang II inhibits sACE activity of human serum and is a selective inhibitor of the C-domain
The crystal structure of C-domain sACE in complex with Ang II provided direct structural evidence for Ang II being a competitive inhibitor of C-domain sACE activity. The ability of Ang II to inhibit the peptidyl dipeptidase activity of sACE was tested using human serum as the source of soluble two-domain enzyme and HHL as the substrate. The addition of Ang II to the reaction mixture resulted in inhibition of HHL hydrolysis with an IC50 value of 82 μM (Fig. 3a). Linearising this plot a Hill slope of 1 was obtained consistent with simple linear competitive inhibition. HHL is a substrate for both the N- and C- domains and therefore it was uncertain whether both domains were inhibited to similar extents. We thus studied the inhibition of each domain separately using soluble recombinant human N- and C- domain enzymes that had been expressed as truncated proteins in mammalian cells and purified to homogeneity. Ang II inhibited HHL hydrolysis by the C-domain with an IC50of 73 μM (Fig. 3b) and was competitive in nature with a dissociation constant (Ki) of 4 μM (Fig. 3c, Table 3), but was a poor inhibitor of the N-domain activity with only around 20% inhibition at the highest Ang II concentration (1 mM) (Fig. 3d). These results suggest that the reduced activity of serum ACE towards HHL in presence of Ang II is predominantly due to the inhibition of the C-domain. The Kifor inhibition of the C-domain is similar to the apparent Km of 3.5 μM for the conversion of Ang I to Ang II obtained using the same enzyme preparation (Table 3). This similarity suggested that product inhibition might play a physiological role in regulating the conversion of Ang I to Ang II by two-domain sACE. To investigate this possibility further, we determined directly the effect of exogenous Ang II on the conversion of Ang I to Ang II by human serum sACE by comparing the decrease in Ang I in the absence and presence of Ang II. Molar ratios of Ang I/Ang II of 1:1 and 1:2 resulted in 20 and 38% inhibition of Ang I hydrolysis, respectively (Fig. 3d).

(a) Inhibition of human sACE by Ang II.Serum was used as the source of soluble sACE. ACE activity was assayed using HHL as substrate in presence of various concentrations of Ang II as described in the Methods. Data is expressed as a percentage of uninhibited activity and each data point is the mean of three replicates. A Hill slope of 1 was obtained using GraphPad© , which is consistent with simple linear competitive inhibition. (b) Inhibition of human N- and C-domain ACE by Ang II. ACE activity of recombinant N-domain and C-domain was determined using HHL as substrate in presence of various concentrations of Ang II as described in the Supplementary material. Data is expressed as a percentage of uninhibited activity and each data point is the mean of three replicates. A Hill slope of 1 for the inhibition of C-domain ACE was obtained using GraphPad© , which is consistent with simple linear competitive inhibition. (c) Lineweaver-Burk plot showing the competitive nature of the inhibition by Ang II of the hydrolysis of HHL by C-domain ACE. (d) Ang II inhibits Ang I conversion by human sACE. The impact of Ang II (2.5 and 5 µM) on the conversion of Ang I (2.5 µM) to Ang II by human sACE (serum) was determined by quantifying the decline in Ang I using HPLC. The results are expressed as % hydrolysis and are the means ± SEM (n = 4). HPLC analysis showed that Ang II was metabolically stable in serum under the same conditions as used for the inhibitor studies.
Molecular basis for the Ang II selectivity for the C-domain of sACE
Recent findings have shown that the major site of angiotensin I cleavage is at the C-domain active site in sACE11,12, while both domains cleave bradykinin with equal efficiency25. These findings have given rise to the pursuit of C-domain selective inhibitors which, it is hoped, will provide control of blood pressure, while leaving the N-domain active site to prevent bradykinin accumulation and the associated side effects9,26.
It has been shown previously that bradykinin potentiating peptide, BPPb, has about 260-fold C-domain selectivity21. The selectivity of the BPPs is thought to be mediated by the P2 residue. Thus, the presence of the P2 Lys in BPPb, as opposed to the Gln, Pro and Pro in the P2 position in BPPa, BPP2 and BPPc, respectively, drives the C-domain selectivity of this peptide21.
The most potent C-domain selective inhibitor to date is the phosphinic peptide analogue RXPA380, which is approximately 3000-fold more C-domain selective26. Based on the crystal structure of C-domain in complex with RXPA380 inhibitor and mutagenesis studies, the potency of this inhibitor has been attributed largely to hydrophobic interactions between the P2 and P2′ groups with Phe391 and Val380, respectively18,27. In the case of the former Phe 391 is replaced by the more polar Tyr369 in the N-domain, which may present a steric clash with the P2 Phe of RXPA380. Additionally, the C-domain S2 pocket residue Glu403 is replaced by Arg381 in the N-domain and may also cause steric interference with the P2 group, further contributing to the C-domain selectivity of this inhibitor.
In order to decipher the molecular features for the C-domain selectivity of BPPb and Ang II peptides for sACE, models of the interaction of BPPb and Ang II with the N-domain of human ACE was generated using the known crystal structure of the N- domain of sACE with the aid of C-domain sACE-BPPb and Ang II as templates. Structural alignment in the binding pocket (Table 4) indicated that all the equivalent C-domain residues identified in the binding of Ang II are conserved in the N-domain. However, with regard to the BPPb peptide, the additional interactions of the peptide, located upstream of the catalytic site (i.e., S4–S9, Tables 2,4) highlight residues that are unique to C-domain sACE. These C-domain specific interactions provide a molecular basis for the domain selectivity of the BPPb peptide. The P2 Lys residue of BPPb (reported to be important for C-domain selectivity) is not involved in any interaction with the C-domain specific S2 residues and protrudes into the solvent channel. Most of the interactions observed with the RXPA380 inhibitor appear to be present apart from strong bonds with C-domain sACE at the catalytic site through Glu384 and Glu411.
Discussion
We report the first molecular structures of natural peptides in complex with a human ACE protein. These structures suggest a novel mechanism for the inhibition of human ACE by Pro-rich peptides from snake venoms. Importantly, they provide evidence (supported by biochemical data) that Ang II competitively and selectively inhibits the C-domain active site of human sACE.
BPPb is one of several blood-pressure lowering peptides isolated from snake venom and has been shown to be a highly selective inhibitor of the C-domain21. BPPb has two sequential prolines at the C-terminus, a feature shared with several other BPPs from snake venom4,22,23. The cyclic nature of the proline side-chain restricts free rotation around the Cα-N bond and the lack of an N-hydrogen prevents the formation of H-bonds with carbonyls. These properties, unique amongst the natural amino acids, can have a profound effect on peptide structure and the way in which peptides interact with the substrate binding sites of peptidases. Products of promiscuous proteases often become, in turn, competing substrates until no more susceptible peptide bonds remain. In the case of Ang II, the penultimate C-terminal position confers resistance to further cleavage by ACE and ensures that Ang II is the principal product of angiotensinogen metabolism in the pulmonary circulation. Our data provides insight into how resistance is achieved. Position P2′ of Ang II (His6/Pro7) is an important residue that anchors the peptide in the substrate cleft through strong interactions with the S1′ subsite and positions the penultimate peptide bond to some distance from key side-chain groups involved in catalysis. The peptides are trapped in the catalytic channel through interaction with their main chain atoms and ACE protein has the flexibility to accommodate longer substrates. The observed shift in the position of the shorter Ang II peptide and the minimal influence on the interactions from the substrate side chains, are indicative of the ability for ACE to cleave a range of other peptides25. Furthermore, the sliding mechanism suggests the process in which Ang I could bind ACE during proteolysis and the flexible molecular recognition of the resulting product.
Previously, Ang II has been shown to inhibit a somatic ACE isolated from rabbit lung with a Ki of 130 µM28 . In the present study, we find that AII is a highly selective competitive inhibitor of the C-domain of human sACE. This is not only of interest from the viewpoint of RAS regulation, but also from an evolutionary perspective. There is strong evidence that the conversion of Ang I to Ang II is primarily the responsibility of the C-terminal domain of sACE and that the different substrate specificity of the N-domain is important for other physiological roles, such as metabolic inactivation of Ac-SDKP and BK11,12,13. The advantage of possessing two enzyme activities with divergent substrate specificity is clear, in that it allows the C-domain to evolve as an efficient Ang I processing enzyme with the potential for domain-specific control.
A key homeostatic mechanism for blood pressure control by the systemic RAS involves regulation of renin release from the juxtaglomerular cells of the kidney together with metabolism by ACE2 and aminopeptidases (Fig. 4). The ability of Ang II to be an effective competitor with Ang I for the active site of the C-domain of sACE provides the potential for an additional regulatory mechanism that is independent of renin release. This is unlikely to be relevant in the endothelial production of Ang II as the circulating substrate concentration is very low. However, Ang II inhibition might be physiologically relevant in tissues known to operate a local cellular and intracellular RAS, such as the heart and kidney, where Ang II levels are much greater29. Homeostatic regulation has the potential for ensuring sufficient substrate for alternative Ang I-metabolising enzymes (Fig. 4). For example, Ang I is efficiently cleaved by neprilysin, an ectopeptidase with broad tissue distribution and enrichment in the kidney, lung and regions of the brain, at the Pro7-Phe8 peptide bond to generate angiotensin (1–7), a vasodilatory and anti-fibrotic peptide that operates antagonistically to Ang II by activating the Mas receptor30.

Schematic illustrating the roles of aminopeptidase (AP), carboxypeptidases (ACE and ACE2) and neprilysin (NEP) in the metabolism of angiotensin peptides.
Ang 1–7 (Asp-Arg-Val-Tyr-Ile-His-Pro), which can be formed either by the carboxypeptidase activity of ACE2 on Ang II (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe) or by endopeptidase cleavage of Ang I (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) by neprilysin, antagonises the negative actions of Ang II on cardiovascular and renal physiology. The opposing effects of Ang II and Ang 1–7 are carried out primarily via the AT1 and Mas receptors (R), respectively. Ang III (Arg-Val-Tyr-Ile-His-Pro-Phe) is the principal active peptide in the brain RAS and its formation is regulated by aminopeptidase A41. Homeostatic regulation of Ang I/Ang II levels in the kidney or heart through product inhibition of C-domain ACE might not only be important for regulating direct actions of the peptide, but also for the formation of Ang 1–7 and Ang 2–10 from Ang I by NEP and aminopeptidase A, respectively . The C-domain of sACE is primarily responsible for the formation of Ang II, but the relative contributions of the two domains of sACE to the hydrolysis of Ang 2–10 and Ang 1–9 are not known.
In summary, we have elucidated the molecular interactions that underpin the inhibitory properties of two natural peptides towards ACE. Of special note is the observation that Ang II is a potent competitive and selective inhibitor of the C-domain of human sACE and that it binds in two different (‘sliding’) conformations. In addition, we have shown that BPPb, a C-domain specific ACE inhibitor binds at the active site involving several sub-sites in a zinc-independent manner, thus providing a molecular description of a new mode of binding compared with all the ACE-inhibitor complex structures described so far. These features could be utilized for structure-based design of more efficacious second generation C-domain specific sACE inhibitors. The relative affinities of the C-domain active site for Ang I and Ang II are very similar, suggesting that the rate of converting Ang I to Ang II, which is mainly carried out by the C-domain of sACE, might as part of a local RAS be regulated by the relative concentrations of these two peptides.
Methods
Chemicals
All chemicals including BPPb and Ang I peptides were purchased from Sigma-Aldrich.
Enzymes
N- and C-domain human ACE proteins were generated by expression in cultured mammalian CHO cells (human ACE) and purified to homogeneity as described previously19,31. Human serum was obtained from NHS Blood and Transplant, Seacroft, Leeds, U.K.
Assay of ACE activity
This was determined in 0.1 M HEPES buffer, pH 7.5, that also included 10 μM ZnSO4 0.3 M NaCl for human N- and C-domains using either hippuryl-histidyl-leucine (HHL, 5 mM, final concentration) or Ang I (25 µM, final concentration) as the substrate. HPLC with UV-detection (214 nm) was employed to quantify the release of hippuric acid from the hydrolysis of HHL and the conversion of Ang I to Ang II, as described previously32. When Ang II was tested as an inhibitor of Ang I conversion to Ang II, ACE activity was determined by comparing the decline in Ang I in the absence and presence of a known initial concentration of Ang II. The contribution of non-ACE activity (6.4%) of human serum to the metabolism of Ang I was determined by using 10 µM captopril to inhibit both domains of sACE. Recombinant enzymes were diluted in the reaction buffer containing 0.1 mg/ml of bovine serum albumin to give final concentrations of 12.5 or 25 ng/µl for C-domain human ACE and 940 ng/µl for N-domain human ACE. Serum was diluted 1 in 20 with 0.1 M HEPES buffer, pH 7. Ang ll enzyme assays were performed at 25°C and were terminated by the addition of trifluoroacetic acid (final concentration, 2% v/v). Non-linear regression analysis of kinetic data was performed using GraphPad© software.
X-ray crystallography
Native C-domain sACE was pre-incubated with either BPPb or Ang I peptide (protein:peptide molar ratio of 1:14) on ice for 4 hours before crystallisation. Two independent crystallisation experiments were performed with different batches of Ang I. Co-crystals were obtained with 2 µl of the C-domain sACE-peptide sample (10 mg/ml in 50 mM HEPES pH 7.5, 0.1 mM PMSF) mixed with equal volume of reservoir solution (50 mM sodium acetate pH 4.7, 10 μM zinc sulphate and 15% PEG 4000) and suspended above the well as a hanging drop. Diffraction quality crystals of C-domain sACE-peptide complex appeared approximately after a week.
X-ray diffraction data for C-domain sACE-peptide complexes were collected on PX station IO4 at Diamond Light Source (Oxon, UK). 25% PEG 4000 was added to the drop as a cryoprotectant to keep the crystal at constant temperature under the liquid nitrogen jet during data collection. For each complex one hundred images were collected by using a PILATUS-2M detector (Dectris, Swizerland). Raw data images were processed and scaled with MOSFLM33 and SCALA using the CCP4 suite34. Initial phases for structure solution were obtained using the molecular replacement routines of PHASER program35. The atomic coordinates of native C-domain sACE [PDB code 1O8A17] were used as a search model for structure determination. The resultant models were refined using REFMAC536. Five per cent of reflections were separated as Rfree set and used for cross validation37. Manual adjustments of model were carried out using COOT38. Water molecules were added at positions where Fo–Fc electron density peaks exceeded 3σ and potential hydrogen bonds could be made. Based on visible electron density interpretation, BPPb/Ang II peptide was incorporated in the structure and further refinement was carried out. Validation was conducted with the aid of program MOLPROBITY39. There were no residues in the disallowed region of the Ramachandran plot. Crystallographic data statistics are summarized in Table 1. All figures were drawn with PyMOL (DeLano Scientific LLC, San Carlos, CA, USA) and rendered with POV-ray. Hydrogen bonds were verified with the program HBPLUS40.
References
Bader, M. Tissue renin-angiotensin-aldosterone systems: Targets for pharmacological therapy. Annu. Rev. Pharmacol. Toxicol. 50, 439–465 (2010).
Kumar, R., Singh, V. P. & Baker, K. M. The intracellular renin-angiotensin system: a new paradigm. Trends Endocrinol. Metab. 18, 208–214 (2007).
Corvol, P., Eyries, M. & Soubrier, F. in Handbook of Proteolytic Enzymes edited by A. J. Barrett, N. D. Rawlings and J. F. Woessner (Elsevier Academic Press, Amsterdam, 2004), Vol. 1, pp. 332–346.
Soffer, R. L. Angiotensin-converting enzyme and the regulation of vasoactive peptides. Annu. Rev. Biochem. 45, 73–94. (1976).
Paul, M., Poyan Mehr, A. & Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 86, 747–803 (2006).
Navar, L. G., Kobori, H., Prieto, M. C. & Gonzalez-Villalobos, R. A. Intratubular renin-angiotensin system in hypertension. Hypertension 57, 355–362 (2011).
Abadir, P. M. et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc. Natl. Acad. Sci. U S A 108, 14849–14854 (2011).
Acharya, K. R., Sturrock, E. D., Riordan, J. F. & Ehlers, M. R. ACE revisited: a new target for structure-based drug design. Nat. Rev. Drug Discov. 2, 891–902 (2003).
Watermeyer, J. M., Kroger, W. L., Sturrock, E. D. & Ehlers, M. R. W. Angiotensin-converting enzyme - New insights into structure, biological significance and prospects for domain-selective inhibitors. Current Enzyme Inhibition 5, 134–147 (2009).
Hubert, C., Houot, A. M., Corvol, P. & Soubrier, F. Structure of the angiotensin I-converting enzyme gene - Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem. 266, 15377–15383 (1991).
Fuchs, S. et al. Angiotensin-converting enzyme C-terminal catalytic domain is the main site of angiotensin I cleavage in vivo. Hypertension 51, 267–274 (2008).
van Esch, J. H. et al. Selective angiotensin-converting enzyme C-domain inhibition is sufficient to prevent angiotensin I-induced vasoconstriction. Hypertension 45, 120–125 (2005).
Rousseau, A. et al. The hemoregulatory peptide N-acetyl-Ser-Asp-Lys-Pro is a natural and specific substrate of the N-terminal active site of human angiotensin-converting enzyme. J. Biol. Chem. 270, 3656–3661 (1995).
Todd, P. A. & Heel, R. C., Enalapril . A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in hypertension and congestive heart failure. Drugs 31, 198–248 (1986).
Israili, Z. H. & Hall, W. D. Cough and angioneurotic edema associated with angiotensin-converting enzyme inhibitor therapy. A review of the literature and pathophysiology. Ann Intern Med 117, 234–242 (1992).
Burnakis, T. G. & Mioduch, H. J. Combined therapy with captopril and potassium supplementation. A potential for hyperkalemia. Arch Intern Med 144, 2371–2372 (1984).
Natesh, R., Schwager, S. L., Sturrock, E. D. & Acharya, K. R. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 421, 551–554 (2003).
Corradi, H. R. et al. The structure of testis angiotensin-converting enzyme in complex with the C-domain-specific inhibitor RXPA380. Biochemistry 46, 5473–5478 (2007).
Anthony, C. S. et al. The N domain of human angiotensin-I converting enzyme: the role of N-glycosylation and the crystal structure in complex with an N-domain specific phosphinic inhibitor RXP407. J. Biol. Chem. 285, 35685–35693 (2010).
Erdos, E. G. The ACE and I: how ACE inhibitors came to be. FASEB J 20, 1034–1038 (2006).
Cotton, J. et al. Selective inhibition of the C-domain of angiotensin I converting enzyme by bradykinin potentiating peptides. Biochemistry 41, 6065–6071 (2002).
Cheung, H. S. & Cushman, D. W. Inhibition of homogeneous angiotensin-converting enzyme of rabbit lung by synthetic venom peptides of Bothrops jararaca. Biochim Biophys Acta 293, 451–463 (1973).
Cushman, D. W. et al. Inhibition of angiotensin-coverting enzyme by analogs of peptides from Bothrops jararaca venom. Experientia 29, 1032–1035 (1973).
Rawlings, N. D., Morton, F. R., Kok, C. Y., Kong, J. & Barrett, A. J. (2008) MEROPS: the peptidase database.Nucleic Acids Res. 36 Database issue, D320–325.
Jaspard, E., Wei, L. & Alhenc-Gelas, F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J. Biol. Chem. 268, 9496–9503 (1993).
Georgiadis, D. et al. Structural determinants of RXPA380, a potent and highly selective inhibitor of the angiotensin-converting enzyme C-domain. Biochemistry 43, 8048–8054 (2004).
Kroger, W. L. et al. Investigating the domain specificity of phosphinic inhibitors RXPA380 and RXP407 in angiotensin-converting enzyme. Biochemistry 48, 8405–8412 (2009).
Tsai, B-S. & Peach, M. J. Angiotensin homologs and analogs as inhibitors of rabbit pulmonary angiotensin-converting enzyme (1977). J Biol Chem 252, 4674–4681 (1977).
Campbell, D. J. et al. Effect of reduced angiotensin-converting enzyme gene expression and angiotensin-converting enzyme inhibition on angiotensin and bradykinin peptide levels in mice.Hypertension. 43, 854–859 (2004).
Rice, G. I. et al. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 383, 45–51 (2004).
Gordon, K. et al. Deglycosylation, processing and crystallization of human testis angiotensin-converting enzyme. Biochem. J. 371, 437–442 (2003).
Lamango, N. S. & Isaac, R. E. Identification and properties of a peptidyl dipeptidase in the housefly, Musca domestica, that resembles mammalian angiotensin- converting enzyme. Biochem. J. 299, 651–657 (1994).
Leslie, A. G. W. & Powell, H. R. Processing diffraction data with Mosflm. Evolving Methods for Macromolecular Crystallography 245, 41–51 (2007).
CCP4, The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50, 760–763 (1994).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Murshudov, G. N., Vagin, A. A. & Dodson, E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997).
Brünger, A. T. Free R-value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 (1992).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35 (Web Server issue) W375–383 (2007).
McDonald, I. K. & Thornton, J. M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793 (1994).
Marc, Y. & Llorens-Cortes, C. The role of the brain renin-angiotensin system in hypertension: Implications for new treatment. Prog. Neurobiol. 95, 89–103 (2011).
Acknowledgements
This work was supported by the Medical Research Council (UK) through a project grant (number G1001685) and a Wellcome Trust (UK) equipment grant (number 088464) to K.R.A. We thank the scientists at PX station IO4, Diamond Light Source, Didcot, Oxon (UK) for their support during X-ray diffraction data collection.
Author information
Authors and Affiliations
Contributions
G.M. carried out all the crystallography experiments. S.L.U.S. prepared human ACE proteins. K.R.A. and G.M. analysed all the structural data. R.E.I. performed all the kinetic experiments and analysed the data. R.E.I., K.R.A. and E.D.S. wrote the manuscript. K.R.A. and R.E.I. conceived of the study, supervised the work. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Protein Data Bank accession codes. The atomic coordinates and structure factors for C-domain sACE-Ang II (code 4APH) and C-domain sACE-BPPb (code 4APJ) complexes have been deposited in the Protein Data Bank (PDB).
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Masuyer, G., Schwager, S., Sturrock, E. et al. Molecular recognition and regulation of human angiotensin-I converting enzyme (ACE) activity by natural inhibitory peptides. Sci Rep 2, 717 (2012). https://doi.org/10.1038/srep00717
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00717
This article is cited by
-
Analysis of protonation equilibria of some alanyl dipeptides in water and aqueous ethanol mixtures
Amino Acids (2023)
-
ACE inhibitory effect of the protein hydrolysates prepared from commercially available nori product by pepsin–trypsin digestion
European Food Research and Technology (2022)
-
Structural characterization and in-silico analysis of Momordica charantia 7S globulin for stability and ACE inhibition
Scientific Reports (2020)
-
Health Promoting Effects of Food-Derived Bioactive Peptides: A Review
International Journal of Peptide Research and Therapeutics (2020)
-
Broken Rice as a Potential Functional Ingredient with Inhibitory Activity of Renin and Angiotensin-Converting Enzyme(ACE)
Plant Foods for Human Nutrition (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
