
Abstract
Furin cleaves diverse types of protein precursors in the secretory pathway. The substrates for furin cleavage possess a specific 20-residue recognition sequence motif. In this report, based on the functional characterisation of the 20-residue sequence motif, we developed a furin cleavage site prediction tool, PiTou, using a hybrid method composed of a hidden Markov model and biological knowledge-based cumulative probability score functions. PiTou can accurately predict the presence and location of furin cleavage sites in protein sequences with high sensitivity (96.9%) and high specificity (97.3%). PiTou's prediction scores are biological meaningful and reflect binding strength and solvent accessibility of furin substrates. A prediction result is interpreted within cellular contexts: subcellular localisation, cellular function and interference by other dynamic protein modifications. Combining next-generation sequencing, PiTou can help with elucidating the molecular mechanism of furin cleavage-associated human diseases. PiTou has been made freely available at the associated website.
Similar content being viewed by others

Introduction
Furin cleaves inactive protein precursors in the secretory pathway and controls the activation of diverse types of functional proteins1,2. The known substrates that are activated by furin include both host proteins and pathogen proteins. The biological functional categories of furin substrates are diverse and include extracellular matrix proteins, signalling peptides, hormone, growth factors, serum proteins, transmembrane receptors, ion channels, bacterial toxins and viral fusion peptides3. Regulation of furin-mediated substrate cleavage plays a crucial role in embryogenesis, pathogen infection, neurologic disease and cancer3. In addition, the utility of furin cleavage-targeted selective anti-cancer drug delivery is also being explored4.
The execution of furin cleavage depends on the recognition of the furin cleavage site motif by the functional furin enzyme. The furin cleavage site motif was initially described as a four amino acid pattern: R-X-[K/R]-R↓1. However, this pattern does not explain all furin cleavage sites, e.g. the furin cleavage sites of the human albumin precursor VFRR↓DA5 and the human C-type natriuretic peptide precursor RLLR↓ DL6 cannot be described by the pattern R-X-[K/R]-R↓. On the other hand, a mutated form of Sindbis Virus PE2 protein RSKR↓LV contains the pattern R-X-[K/R]-R↓ but is not efficiently cleaved by furin7. In our previous work, the furin cleavage site was re-analysed and characterised as a 20 amino acid motif running from position P14 to position P6′, which can be divided into one core region (eight amino acids from P6–P2′) and two flanking solvent accessible regions (eight amino acids from P7–P14 and four amino acids from P3′–P6′)8. The core region (P6–P2′) fits into to the furin catalytic pocket and determines the binding strength. The flexible solvent accessible regions (P7–P14 and P3′–P6′) flank the core region. They provide the accessibility of the core region to the furin binding pocket and also facilitate conformational changes of the core region required by the dynamic furin cleavage process.
Our previous analysis indicated that the physical properties of this 20-residue motif are evolutionarily conserved across different organisms, including mammals, bacteria and viruses8,9. Furthermore, the biology underlying the relationship between the physical properties of furin cleavage sites, cellular function and viral infectivity has been analysed8. FurinDB, a database of 20-residue furin cleavage sites and associated drugs, was then constructed to provide a solid publicly available infrastructure for furin cleavage-related studies10. The functionally characterised 20-residue motif of the furin cleavage recognition site and FurinDB laid down an important theoretical foundation for the development of a reliable prediction tool for furin cleavage sites. In this report, we developed a furin cleavage site prediction tool: PiTou. PiTou can predict the presence and location of furin cleavage site on protein sequences. PiTou is designed based on the functional characterisation of the underlying biology of furin cleavage site motifs. The PiTou algorithm is implemented as a hybrid method that combines the advantages of both a machine learning-based hidden Markov model and a set of biological mechanism-based cumulative probability score functions. The performance of the prediction tool is high, with a sensitivity of 96.9% and specificity of 97.3%. PiTou's prediction scores are biological meaningful and they reflect binding strength and solvent accessibility of furin substrates. A prediction result also need to be interpreted within biological meaningful cellular contexts: subcellular localisation, cellular function and interference by other dynamic protein modifications. Combining next-generation sequencing, PiTou can help to discover the molecular mechanism underlying furin cleavage site-associated human diseases. PiTou has been made freely available at the associated website http://www.nuolan.net/reference.html.
Results
Performance of PiTou on the prediction of furin cleavage sites, its designing features and comparison with the other prediction method
Cross-validation is proven to be an effective method of evaluating the predictive performance of a prediction tool11. Leave-one-out cross-validation (LOOCV) was used to evaluate the sensitivity of PiTou on 131 known furin cleavage sites. The sensitivity from the cross-validation reached 96.9% ( false negative rate 3.1%, 127 out of total 131 sites) (Supplemental Information S1). The specificity was estimated using 4265 arginine sites that are not cleaved by furin in the experiments. A specificity of 97.3% (
false negative rate 3.1%, 127 out of total 131 sites) (Supplemental Information S1). The specificity was estimated using 4265 arginine sites that are not cleaved by furin in the experiments. A specificity of 97.3% ( false positive rate 2.7%, 4151 out of total 4265 sites) (Supplemental Information S2) was reached. The detailed results of cross-validation and specificity estimation are available as supplementary materials on the web site.
false positive rate 2.7%, 4151 out of total 4265 sites) (Supplemental Information S2) was reached. The detailed results of cross-validation and specificity estimation are available as supplementary materials on the web site.
The performance of PiTou was compared with the published furin cleavage prediction tool ProP method12. The sensitivity and specificity values of ProP were taken from the original paper. Compared with ProP, PiTou showed superior sensitivity and specificity ( Table 1 ). Unfortunately, a direct comparison of sensitivity and specificity of PiTou method and ProP method on a same independent test dataset is currently not possible due to two limitations: (1) the cross validation implementations of tools are not publicly available; (2) the number of known furin cleavage sites is small and no independent testing dataset of furin cleavage sites is available and can be used for comparing these two tools. Therefore, the sensitivity and specificity values taken from ProP publication and those of PiTou may not be easily compared. However, the designing features of PiTou is evidently distinct from that of ProP. The high sensitivity and high specificity of the PiTou furin cleavage site prediction tool benefitted from its designing features. The most important feature is that PiTou is entirely biological knowledge-based, but the prediction score is substantiated by a machine learning-based hidden Markov model and cumulative probability score functions (Methods). This designing feature evolved from understanding the 20-residue furin cleavage site motif responsible for recognition by furin8. The constraints imposed by the physical properties of the 20-residue furin cleavage motif and the 3D binding model of substrates to the furin catalytic domain were translated into the score functions of PiTou ( Figure 1 ). Biological and structural information on this 20-residue motif enhance our understanding of molecular biology of furin cleavage and thus improve the PiTou's prediction accuracy. On the contrary, the ProP method's designing feature is very different. The ProP method is a pure machine learning based method that entirely relies on automatic training process of neural networks12. The neural networks of ProP method consider the biology of furin cleavage and binding of substrates to the furin binding pocket as a black box ( Table 1 ).

The design of PiTou algorithm: the PiTou score function  is biological knowledge-based and it comprises of a machine learning-based hidden Markov model and a set of biological mechanism-based cumulative probability score functions.
is biological knowledge-based and it comprises of a machine learning-based hidden Markov model and a set of biological mechanism-based cumulative probability score functions. is the sum of two parts: the core region binding score
is the sum of two parts: the core region binding score  (calculated from eight amino acids at P6–P2′) and the flanking region solvent accessible score
(calculated from eight amino acids at P6–P2′) and the flanking region solvent accessible score  (calculated from eight amino acids at P7–P14 and four amino acids at P3′–P6′).
(calculated from eight amino acids at P7–P14 and four amino acids at P3′–P6′).  and
and  incorporate the physical properties of the 20-residue furin cleavage motif and the binding of substrates to the furin catalytic domain. The analysis of the 20-residue furin cleavage motif is described in previous publication8.
incorporate the physical properties of the 20-residue furin cleavage motif and the binding of substrates to the furin catalytic domain. The analysis of the 20-residue furin cleavage motif is described in previous publication8.
Integrating next generation sequence analysis and elucidating the molecular mechanisms of furin cleavage site-associated human diseases
Next-generation sequencing can rapidly sequence hundreds of genes and identify genomic mutations in the onset of human diseases or mutations which have emerged in the progression of a human disease, such as cancer13. With the accumulation of large amount of genomic data, one big challenge is to understand the cellular functional consequence of genetic mutations identified. A change in an amino acid in the 20-residue motif of a furin cleavage site can alter the pattern of the favoured physical properties of that specific position or region and thus affect the furin cleavage efficiency on the substrate. Malfunctions in furin cleavage efficiency will in turn cause human disease. Three examples of mutations resulting a loss or gain of furin cleavage sites are known, and almost all the known examples are associated with the molecular mechanism of a human disease: X-linked hypohydrotic ectodermal dysplasia, arrhythmogenic right ventricular dysplasia/cardiomyopathy disorder, prolonged thrombin time and a mild bleeding tendency ( Table 2 ).
In all three cases illustrated in table 2 ( Table 2 ), PiTou successfully predicted the loss of known functional furin cleavage sites or gain of not naturally occurring furin cleavage sites as the consequence of genomic mutations, thus in turn identified the molecular mechanism underlying some furin cleavage associated human diseases. In addition, all three examples showed that mutations at positions around furin cleavage sites can have dramatic consequences and cause diseases and disorders. Particularly, these examples provide three interesting insights:
-
1
Disease can be caused not only by the loss of a normal functional furin cleavage site, but also the gain of an aberrant furin cleavage site.
-
2
Both loss and gain of furin cleavage do not necessarily require mutations at the arginine at position P1 or P4.
-
3
A mutation directly present in the catalytic domain or the regulatory domain on a protein is not the only way that a genetic mutation can cause cellular functional consequence. A genetic mutation can also alter a short sequence motifs required by the interaction with other proteins and result in entirely different cellular phenotype. This concept was demonstrated by the genomic mutations resulted changes of the proteolytic cleavage of extracellular enzymes by furin.
Discussion
Given the high sensitivity (96.9%) and high specificity (97.3%), PiTou can be used to identify potential furin cleavage sites on various types of extracellular proteins, e.g. extracellular matrix proteins, signalling peptides, hormones, growth factors, serum proteins, transmembrane receptors, ion channels, bacterial toxins and viral proteins. The predicted cleavage sites will provide experimental groups with directions for the elucidation of the possible underlying cellular mechanisms of the observed cellular phenotype.
The final prediction score  of PiTou is an implementation of two main criteria (
Figure 1
):
of PiTou is an implementation of two main criteria (
Figure 1
):
-
A
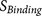 : the binding strength of the core region to the furin catalytic pocket
: the binding strength of the core region to the furin catalytic pocket -
B
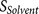 : the accessibility of the core region to the furin catalytic pocket, supported by the hydrophilicity and flexibility of the flanking regions.
: the accessibility of the core region to the furin catalytic pocket, supported by the hydrophilicity and flexibility of the flanking regions.
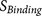 : the binding strength of the core region to the furin catalytic pocket
: the binding strength of the core region to the furin catalytic pocket 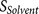 : the accessibility of the core region to the furin catalytic pocket, supported by the hydrophilicity and flexibility of the flanking regions.
: the accessibility of the core region to the furin catalytic pocket, supported by the hydrophilicity and flexibility of the flanking regions. The biological knowledge about the binding complex is reflected by  which contributes to both
which contributes to both  and
and  . As a consequence, by examining the output of the core region binding score
. As a consequence, by examining the output of the core region binding score  and the flanking region solvent accessible score
and the flanking region solvent accessible score  , PiTou not only predicts whether an arginine site is cleaved by furin, but also predicts the molecular mechanism by which furin cleavage can or cannottake place at a given arginine site, e.g. because the binding strength of core region is weak, resulting in low
, PiTou not only predicts whether an arginine site is cleaved by furin, but also predicts the molecular mechanism by which furin cleavage can or cannottake place at a given arginine site, e.g. because the binding strength of core region is weak, resulting in low  or because the accessibility of the core region is poor, resulting in low
or because the accessibility of the core region is poor, resulting in low  , etc. This design feature allows the possibility of using the PiTou score set
, etc. This design feature allows the possibility of using the PiTou score set  as an indicator to engineer the amino acids around a given arginine site with the 20-residue motif pattern8 to increase/decrease the binding strength score
as an indicator to engineer the amino acids around a given arginine site with the 20-residue motif pattern8 to increase/decrease the binding strength score  or the accessibility score
or the accessibility score  , thus increasing/decreasing desired furin cleavage efficiency, or to an extreme extent, to create/diminish a furin cleavage site that can possibly modify cell biology under disease conditions. This is conceptually different from pure machine learning-based ProP approaches that produce a prediction result, while our knowledge of the molecular mechanism of furin cleavage process is considered to be a black box, thus does not necessarily improved.
, thus increasing/decreasing desired furin cleavage efficiency, or to an extreme extent, to create/diminish a furin cleavage site that can possibly modify cell biology under disease conditions. This is conceptually different from pure machine learning-based ProP approaches that produce a prediction result, while our knowledge of the molecular mechanism of furin cleavage process is considered to be a black box, thus does not necessarily improved.
The positive prediction result of PiTou  indicates that an arginine site can be cleaved by furin; however, a prediction result needs to be interpreted within biological meaningful cellular contexts. There are three cellular contexts which still need to be considered:
indicates that an arginine site can be cleaved by furin; however, a prediction result needs to be interpreted within biological meaningful cellular contexts. There are three cellular contexts which still need to be considered:
-
1
The accessibility of furin cleavage sites in the context of subcellular localisation. Functional furin does not come into contact with cytosolic proteins because furin is an extracellular enzyme. Equally, for potential furin cleavage recognition sites present on transmembrane proteins, the cytosolic part of a transmembrane protein should not have the opportunity to come into contact with functional furin, whereas only the extracellular part of transmembrane proteins can be cleaved by furin in the secretory pathway. Subcellular localisation of a protein can be used to eliminate of false positive predictions.
-
2
Cellular functions of substrates. The most interesting examples are viral fusion peptides. The P3′–P6′ region of viral fusion peptides cannot be too hydrophilic or too hydrophobic because the virus appears to need a subtle balance between sufficient hydrophobicity required by the viral fusion process and sufficient hydrophilicity required by furin cleavage efficiency8. This biology of viral fusion with furin cleavage present an contradicted logic and unique challenge for the life cycle of virus. The motif analysis suggested that viruses have cunningly solved this dilemma by tuning the composition of small hydrophobic amino acids (glycine, alanine and proline) in the P3′–P6′ region8. As a consequence of the presence of hydrophobic amino acids, the average hydrophobicity scale of the P3′–P6′ region of these viral substrates is much higher than the average of mammalian and bacterial substrates (Figure 2, 0.005 versus −1.235, student t-test pvalue = 1.3E-004, calculated using the physical property EISD840101 consensus normalized hydrophobicity scale for amino acids)8,14. This hydrophobic stretch in the P3′–P6′ region of furin cleavage sites on viral spike proteins can sometimes result in a lower predicted score of PiTou. Therefore, a more careful consideration should be given when a query sequence has a viral origin.
-
3
The interference of furin cleavage by other dynamic modifications on an amino acid. This is a novel and interesting issue. An algorithm takes one letter symbol to represent an amino acid, e.g. A for alanine, R for arginine, S for serine, etc. However, the physical property of the same amino acid can be different under different conditions and the very same type of amino acid may lead to completely different cellular consequences. The statistical analysis of the physical properties of a substrate in the P1′ position indicated that the total volume of a substrate fitting into position P1′ could not exceed the total volume available in the narrow furin binding pocket at position P1′ and therefore position P1′ has preference for small hydrophilic residues such as serine8. Three small residues, i.e. serine, threonine and asparagine, have the potential to be glycosylated. The volume of serine, threonine and asparagine will increase after N-linked glycosylation on asparagine and O-linked glycosylation on serine or threonine. The glycosylated amino acids no longer possess the preferred physical properties required at the P1′ position. The same type of amino with the symbol N (asparagine) present at position P1′ can result in either efficient furin cleavage or defective furin cleavage, which can entirely depend on whether the side chain volume of N (asparagine) has been modified or not7. Therefore, the presence of small residues such as asparagine, serine or threonine at substrate position P1′ deserves particular attention in the analysis of furin cleavage-mediated viral infection. A sequence motif may be cleaved by furin in one cellular context (no glycosylation at position P1′), but the very same sequence motif may not be cleaved by furin in a different cellular context (glycosylation at position P1′).
The three discussions above (subcellular localisation of the region of substrates with transmembrane regions, hydrophobic tendencies in the P3′–P6′ region of viral fusion peptides and glycosylation interference with furin cleavage) showed that the cellular context and biological background are important for the interpretation of prediction results from a bioinformatics tool. Furthermore, they emphasise the important concept of studying the underlying molecular mechanism accompanying the design of a computational prediction tool rather than purely relying on analysing the sequence motif pattern with statistical models or machine learning-based methods. This concept not only limits to the development of furin cleavage site prediction tool, but may also apply to the development of other types of prediction tools such as clinical utility-related molecular diagnostic signatures15.

The hydrophobicity scale of the P3′–P6′ region of viral substrates (filled black box) is much higher than that of mammalian and bacterial substrates (white box), student t-test pvalue = 1.3E-004.The hydrophobicity is calculated using the physical property EISD840101 consensus normalized hydrophobicity scale for amino acids14.
PiTou has been demonstrated helpful for elucidating cellular functional consequence of genomic mutations and understanding the molecular mechanisms of furin cleavage site-associated human diseases. The number of genomic mutations resulting a loss or gain of a furin cleavage site associated with human disease may be underestimated. Because the furin cleavage site motif is comprised of about 20 residues (P14-P6′), not four residues (P4-P1) as previously thought, in theory, any mutation, deletion or insertion within the residues in these 20 positions can change the physical properties; this raises the possibility of losing or gaining a furin cleavage site or at least affecting furin cleavage efficiency. The PiTou furin cleavage prediction tool can serve as an efficient computational tool to screen and evaluate the possibility of an aberrant gain or loss of furin cleavage in the mutated protein sequence in patients with various disorders or diseases and thus help with elucidating the molecular mechanism of human diseases. Next-generation sequencing can identify thousands of genomic mutations in the progression of human diseases. PiTou can predict the functional consequence of these genomic mutations on furin cleavage efficiency. By combining next-generation sequencing and the PiTou furin cleavage site prediction tool, our fundamental understanding of the pathogenesis of human diseases has been enhanced.
The PiTou package is publicly available for download at www.nuolan.net/reference.html. We believe PiTou will provide a valuable publicly available computational tool to scientists in the field of molecular biology and molecular medicine.
Methods
Dataset of known furin cleavage sites
A dataset of 131 known furin cleavage sites was retrieved from FurinDB, a database of 20-residue furin cleavage sites, substrates and associated drugs10. Each site included is supported by experimental biochemical evidence10. The taxa of these substrates cover three different origins: virus, bacterial and mammals. All substrates included are cleaved by mammalian furin. The cellular function of the substrates in the dataset covers a representative functional spectrum: extracellular matrix proteins, signalling peptides, hormones, growth factors, serum proteins, transmembrane receptors, ion channels, bacterial toxins and viral fusion peptides3. The remaining 4265 arginine sites presented in the protein sequences of furin substrates but not reported to be cleaved by furin in the experiments were also collected as negative sites.
The set of 20 residues in the furin cleavage site were formatted and aligned into a multiple sequence alignment ( ). One important consideration is the cell biology of furin cleavage. Furin is an extracellular enzyme and furin cleavage takes place after the secreted signal peptide of a protein sequence is cleaved off. For a host protein precursor, when the location of a furin cleavage site is very close to the N-terminal, the overlapping region between the known secreted signal peptides and the P1P14 position of the furin cleavage site motif is substituted with gap symbols in the multiple sequence alignment.
). One important consideration is the cell biology of furin cleavage. Furin is an extracellular enzyme and furin cleavage takes place after the secreted signal peptide of a protein sequence is cleaved off. For a host protein precursor, when the location of a furin cleavage site is very close to the N-terminal, the overlapping region between the known secreted signal peptides and the P1P14 position of the furin cleavage site motif is substituted with gap symbols in the multiple sequence alignment.
Constructing the biological knowledge-based score function
PiTou is a biological knowledge-based furin cleavage site prediction tool that employs both a machine learning-based hidden Markov model and a set of biological knowledge-based cumulative probability score functions. The PiTou score function  is the sum of two parts, similar as the scheme for the knowledge-based prediction of short functional motifs16: the core region binding score
is the sum of two parts, similar as the scheme for the knowledge-based prediction of short functional motifs16: the core region binding score  (calculated from eight amino acids at P6–P2′) and the flanking region solvent accessible score
(calculated from eight amino acids at P6–P2′) and the flanking region solvent accessible score  (calculated from eight amino acids at P7–P14 and four amino acids at P3′–P6′). The overview of the design of PiTou algorithm is illustrated in figure 1 (
Figure 1
).
(calculated from eight amino acids at P7–P14 and four amino acids at P3′–P6′). The overview of the design of PiTou algorithm is illustrated in figure 1 (
Figure 1
).



Hidden markov model (HMM) provide robust a probabilistic model that comprise of states with emission probabilities and transition probabilities. A HMM can sensitively measure the similarity between residues in a query protein sequence with homologous residues in a target set of protein sequence17. A profile hidden Markov model  is constructed using the multiple sequence alignment of furin cleavage sites.
is constructed using the multiple sequence alignment of furin cleavage sites.  evaluates the similarity of the core region (P6–P2′) of a query sequence to the amino acid type occurrence frequency in the core regions (P6–P2′) of known furin cleavage site sequences. The score
evaluates the similarity of the core region (P6–P2′) of a query sequence to the amino acid type occurrence frequency in the core regions (P6–P2′) of known furin cleavage site sequences. The score  is the standard log-odd probabilities from this hidden Markov model
is the standard log-odd probabilities from this hidden Markov model  18.
18.
 is a physical property score, each
is a physical property score, each  score is calculated from a known physical property feature or biological feature presented on the furin cleavage site 20-residue motif8. Physical property values are retrieved from an AAindex database that stores various physical and biochemical properties of amino acids19. The 20-residue furin cleavage recognition site motif was formularised into 12
score is calculated from a known physical property feature or biological feature presented on the furin cleavage site 20-residue motif8. Physical property values are retrieved from an AAindex database that stores various physical and biochemical properties of amino acids19. The 20-residue furin cleavage recognition site motif was formularised into 12  functions (
Table 3
). Each
functions (
Table 3
). Each  results in a negative score or a zero score. The absolute value of a negative score reflects the degree of deviation from the evolutionally conserved physical property pattern in the 20-residue furin cleavage site motif: a larger deviation results in a large negative value and a smaller deviation results in a small negative value. There are two types of
results in a negative score or a zero score. The absolute value of a negative score reflects the degree of deviation from the evolutionally conserved physical property pattern in the 20-residue furin cleavage site motif: a larger deviation results in a large negative value and a smaller deviation results in a small negative value. There are two types of  functions16,20:
functions16,20:
 functions that evaluate
functions that evaluate  binding strength of the core region (P6–P2′) and
binding strength of the core region (P6–P2′) and  solvent accessibility of two flanking regions (P7–P14 and P3′–P6′)
solvent accessibility of two flanking regions (P7–P14 and P3′–P6′)All scores  ,
,  ,
,  ,
,  ,
,  are log-odd probabilities. Every single arginine site presented in the query protein sequence is considered as a potential furin cleavage site and the 20-residue sequence motif encompassing this arginine was evaluated using the prediction score function
are log-odd probabilities. Every single arginine site presented in the query protein sequence is considered as a potential furin cleavage site and the 20-residue sequence motif encompassing this arginine was evaluated using the prediction score function  . An arginine site with a predicted score
. An arginine site with a predicted score  is interpreted as a predicted furin cleavage site.
is interpreted as a predicted furin cleavage site.
References
Nakayama, K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 327 (Pt3), 625–635 (1997).
Molloy, S. S., Anderson, E. D., Jean, F. & Thomas, G. Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9, 28–35 (1999).
Thomas, G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 (2002).
Hajdin, K., D'Alessandro, V., Niggli, F. K., Schafer, B. W. & Bernasconi, M. Furin targeted drug delivery for treatment of rhabdomyosarcoma in a mouse model. PLoS. One. 5, e10445 (2010).
Brennan, S. O. & Nakayama, K. Furin has the proalbumin substrate specificity and serpin inhibitory properties of an in situ hepatic convertase. FEBS Lett. 338, 147–151 (1994).
Wu, C., Wu, F., Pan, J., Morser, J. & Wu, Q. Furin-mediated processing of Pro-C-type natriuretic peptide. J. Biol. Chem. 278, 25847–25852 (2003).
Klimstra, W. B., Heidner, H. W. & Johnston, R. E. The furin protease cleavage recognition sequence of Sindbis virus PE2 can mediate virion attachment to cell surface heparan sulfate. J. Virol. 73, 6299–6306 (1999).
Sun, T. A 20 Residues Motif Delineates the Furin Cleavage Site and its Physical Properties May Influence Viral Fusion. Biochemistry Insights 2, 9–20 (2009).
Sun, T. & Wu, J. Comparative study of the binding pockets of mammalian proprotein convertases and its implications for the design of specific small molecule inhibitors. Int J Biol Sci 6, 89–95 (2010).
Sun, T., Huang, Q., Fang, Y. & Wu, J. FurinDB: A Database of 20-Residue Furin Cleavage Site Motifs, Substrates and Their Associated Drugs. Int. J. Mol. Sci. 12, 1060–1065 (2011).
Cawley, G. C. & Talbot, N. L. Fast exact leave-one-out cross-validation of sparse least-squares support vector machines. Neural Netw. 17, 1467–1475 (2004).
Duckert, P., Brunak, S. & Blom, N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel 17, 107–112 (2004).
Voelkerding, K. V., Dames, S. A. & Durtschi, J. D. Next-generation sequencing: from basic research to diagnostics. Clin. Chem. 55, 641–658 (2009).
Eisenberg, D. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 53, 595–623 (1984).
Sun, T. et al. Biological functions of the genes in the mammaprint breast cancer profile reflect the hallmarks of cancer. Biomark. Insights. 5, 129–138 (2010).
Neuberger, G. A Framework for the Knowledge-Based Prediction of Short FunctionalMotifs from Amino Acid Sequence. 2006. Vienna university. Ref Type: Thesis/Dissertation.
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
Durbin, R., Eddy, S., Krogh, A. & Mitchison, G. Biological sequence analysis: probabilistic models of proteins and nucleic acids (Cambridge University Press,1998).
Kawashima, S., Ogata, H. & Kanehisa, M. AAindex: Amino Acid Index Database. Nucleic Acids Res. 27, 368–369 (1999).
Sun, T. Sequence-analytic characterization and prediction of furin cleavage recognition site based on a simple substrate-catalytic domain binding structural model. 2007. Graz University of Technology. Ref Type: Thesis/Dissertation.
Vincent, M. C., Biancalana, V., Ginisty, D., Mandel, J. L. & Calvas, P. Mutational spectrum of the ED1 gene in X-linked hypohidrotic ectodermal dysplasia. Eur. J. Hum. Genet. 9, 355–363 (2001).
Chen, Y. et al. Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia. Proc. Natl. Acad. Sci. U. S. A 98, 7218-7223 (2001).
Awad, M. M. et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Hum. Genet. 79, 136–142 (2006).
Brennan, S. O., Hammonds, B. & George, P. M. Aberrant hepatic processing causes removal of activation peptide and primary polymerisation site from fibrinogen Canterbury (A alpha 20 Val --> Asp). J. Clin. Invest 96, 2854–2858 (1995).
Zimmerman, J. M., Eliezer, N. & Simha, R. The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 21, 170–201 (1968).
Kuhn, L. A., Swanson, C. A., Pique, M. E., Tainer, J. A. & Getzoff, E. D. Atomic and residue hydrophilicity in the context of folded protein structures. Proteins 23, 536–547 (1995).
Fauchere, J. L., Charton, M., Kier, L. B., Verloop, A. & Pliska, V. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int. J. Pept. Protein Res. 32, 269–278 (1988).
Bull, H. B. & Breese, K. Surface tension of amino acid solutions: a hydrophobicity scale of the amino acid residues. Arch. Biochem. Biophys. 161, 665–670 (1974).
Karplus, P. A. & Schulz, G. E. Prediction of Chain Flexibility in Proteins - A Tool for the Selection of Peptide Antigens. Naturwissenschaften 72, 212–213 (1985).
Acknowledgements
This work was stared as Sun Tian's PhD thesis at TUGraz and fund by GENAU Bioinformatics Integration Network PhD programme (2005–2007) and NSFC grant 11072080. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Wang Huajun and Sun Tian thank Mr.Deng Huixian for his inspiration.
Author information
Authors and Affiliations
Contributions
Sun Tian and Jianhua Wu designed and implemented C++ version of the algorithm, finalised and compared different machine learning methods. Sun Tian and Wang Huajun implemented algorithm, wrote the PiTou software package codes and tested it. Sun Tian provided additional analysis of biology related to PiTou prediction. Sun Tian draft the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Information table S1 table S2
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tian, S., Huajun, W. & Wu, J. Computational prediction of furin cleavage sites by a hybrid method and understanding mechanism underlying diseases. Sci Rep 2, 261 (2012). https://doi.org/10.1038/srep00261
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00261
This article is cited by
-
A live dengue virus vaccine carrying a chimeric envelope glycoprotein elicits dual DENV2-DENV4 serotype-specific immunity
Nature Communications (2023)
-
Comparison of pathogenicity of subtype H9 avian influenza wild-type viruses from a wide geographic origin expressing mono-, di-, or tri-basic hemagglutinin cleavage sites
Veterinary Research (2020)
-
Whole genome characterisation of quail deltacoronavirus detected in Poland
Virus Genes (2019)
-
Furin inhibitor protects against neuronal cell death induced by activated NMDA receptors
Scientific Reports (2018)
-
Oocyte-specific deletion of furin leads to female infertility by causing early secondary follicle arrest in mice
Cell Death & Disease (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
