
Abstract
Eukaryotic elongation factor 1A (eEF1A) is the only protein modified by ethanolamine phosphoglycerol (EPG). In mammals and plants, EPG is attached to conserved glutamate residues located in eEF1A domains II and III, whereas in the unicellular eukaryote, Trypanosoma brucei, a single EPG moiety is attached to domain III. A biosynthetic precursor of EPG and structural requirements for EPG attachment to T. brucei eEF1A have been reported, but the role of this unique protein modification in cellular growth and eEF1A function has remained elusive. Here we report, for the first time in a eukaryotic cell, a model system to study potential roles of EPG. By down-regulation of EF1A expression and subsequent complementation of eEF1A function using conditionally expressed exogenous eEF1A (mutant) proteins, we show that eEF1A lacking EPG complements trypanosomes deficient in endogenous eEF1A, demonstrating that EPG attachment is not essential for normal growth of T. brucei in culture.
Similar content being viewed by others


Introduction
Eukaryotic elongation factor 1A (eEF1A) is an essential protein involved in the elongation step of protein translation, where it delivers aminoacyl-tRNAs to the A-site of the ribosome (reviewed by1). Besides its pivotal role during protein synthesis, eEF1A is involved in many other cellular processes such as signal transduction, nuclear export of proteins and mitochondrial tRNA import2,3,4; in addition, it interacts with components of the cytoskeleton5,6,7,8. This multi-functionality may not be surprising since eEF1A is one of the most abundant cellular proteins, comprising 1–3% of total cytosolic protein9, and, as a consequence, is present in large excess to its ligands in peptide synthesis (molar ratios for eEF1A:eEF1B and eEF1A:ribosomes of 10:1 and 25:1, respectively)10.
Covalent protein modifications are ubiquitous in eukaryotic cells, affecting protein folding, maturation, structure and sub-cellular localization; moreover, they are involved in the regulation of biological activities, including interactions with other molecules11,12,13. For eEF1A, several covalent modifications such as phosphorylation14,15, lysine methylation16,17 and C-terminal methyl-esterification18, have been reported to affect its biological activity during polypeptide synthesis; however, their precise roles are poorly understood (reviewed by19). In addition, mammalian and plant eEF1A is modified by two ethanolamine phosphoglycerol (EPG) moieties attached to conserved glutamic acid residues in domains II and III20,21,22. Similarly, EPG-modified eEF1A has also been described in the protozoan parasite, Trypanosoma brucei. T. brucei eEF1A (TbEF1A) is modified with only a single EPG moiety at Glu362 in domain III however, despite the fact that the second potential EPG modification site, Glu289 in domain II, is conserved between trypanosomes, mammals and plants23. Interestingly, Saccharomyces cerevisiae represents the only eukaryote so far where eEF1A seems not to be modified with EPG24. This lack of EPG attachment to eEF1A in S. cerevisiae, which represents one of the most commonly used and best studied eukaryotic model organisms, makes the search for enzymes involved in EPG attachment and the study of the function of EPG attachment more difficult and challenging.
A study using T. brucei as a model organism to investigate EPG biosynthesis and attachment to eEF1A showed that the ethanolamine moiety in EPG derives from the phospholipid phosphatidylethanolamine23. In addition, recently we showed that replacement of Glu362 in TbEF1A completely inhibited the addition of EPG, even if glutamate was replaced by aspartate, indicating that the enzyme mediating attachment of EPG, or its precursor molecule, is highly specific for Glu at this position25. Remarkably, although EPG attachment to eEF1A was first reported more than 20 years ago, the physiological role of this unique protein modification has remained elusive.
In the present study, we describe for the first time a model system to investigate potential roles of EPG in a eukaryotic cell. We chose T. brucei as model organism to knock down the expression of endogenous TbEF1A using inducible RNA interference (RNAi), which in T. brucei represents a powerful tool to down-regulate protein expression26 and results in complete growth arrest of TbEF1A-depleted parasites. By complementation experiments using conditionally expressed exogenous eEF1A proteins, we then assessed the potential of mutated eEF1A to rescue the lethal phenotype. Using this approach, we demonstrate that eEF1A mutant proteins lacking the EPG attachment sites restored growth of T. brucei procyclic culture forms depleted of endogenous TbEF1A.
Results
Since the role of EPG in eEF1A function is unknown, we decided to establish a model system that would allow the study of EPG function in cell growth. Development of the system using T. brucei procyclic forms in culture involved three steps: i) generation of an inducible RNAi cell line to deplete endogenous TbEF1A, ii) introduction of an inducible ectopic copy of wild type eEF1A into the RNAi-competent cell line, as proof-of-principle to demonstrate functional complementation of endogenous TbEF1A by an ectopic copy and iii) introduction of an inducible ectopic copy of mutated eEF1A lacking the EPG attachment site into the RNAi-competent cell line to study EPG function for parasite growth in culture.
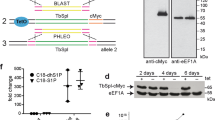
RNAi mediated knock-down of TbEF1A. Expression of TbEF1A was down-regulated in T. brucei 29-13 procyclic forms by targeting the intergenic region 1 located between the first and second tandemly-arranged TbEF1A gene on chromosome 10 of the T. brucei genome. After 3 days of induction of RNAi, growth of the parasite clone C5 stopped completely whereas uninduced cells proliferated normally (Fig. 1A). Northern blot analysis demonstrated that the addition of tetracycline resulted in disappearance of the corresponding mRNA (Fig 1B). Protein analysis by immunoblotting using anti-eEF1A antibody showed complete absence of eEF1A at days 2, 4 and 6 of induction (Fig. 1C, lower panel), whereas eEF1A was continuously present in control parasites incubated in the absence of tetracycline (Fig. 1C, upper panel). Similar results (growth curve, Northern blot, immunoblot) were obtained with a second clone (results not shown).

RNAi-mediated silencing of T. brucei eEF1A.
(A) To down-regulate TbEF1A expression, T. brucei procyclic forms were transfected with an RNAi plasmid targeting the intergenic region 1 of the tandemly repeated eEF1A genes. The selected clone (C5) was cultured in the absence (triangles) or presence (diamonds) of tetracycline for 7 days and diluted daily to a density 3×106 cells/ml. (B) Northern blots of total RNA extracted from parasites after three days of incubation in the absence (−) or presence (+) of tetracycline (tet) and hybridized with 32P-labeled probes (top); rRNA was used as loading control (bottom). (C) SDS-PAGE and immunoblot analysis of TbEF1A in parasites (clone C5) cultured in the absence (−) or presence (+) of tetracycline (tet) for 0, 2, 4 and 6 days and probed with α-EF1A antibody. Each lane contains lysate from 1×107 cells.
Complementation of TbEF1A-depleted T. brucei with ectopic wild type eEF1A. To study if ectopic eEF1A was able to complement TbEF1A-depleted T. brucei parasites and restore parasite growth in culture, a tetracycline-inducible ectopic copy of wild type eEF1A was introduced into the rRNA locus of the TbEF1A RNAi cell line C5. The addition of tetracycline to parasites in culture was expected to induce both the RNAi machinery directed against endogenous TbEF1A and the expression of the ectopic copy of eEF1A. The results show that growth of the selected clone (C5-E, with E referring to glutamic acid at position 362 of wild type TbEF1A) in the presence of tetracycline was similar to that of control cells incubated in the absence of the antibiotic (Fig. 2A). Northern blot analysis using a probe against eEF1A intergenic region 1 shows that the primary transcript was absent after induction with tetracycline for 3 days (Fig. 2B), demonstrating that RNAi against endogenous TbEF1A was effective (see also Fig. 1B). Similar results (growth curve, Northern blot) were obtained with a second clone (results not shown).

Complementation of TbEF1A-depleted T. brucei with wild-type eEF1A.
(A) T. brucei procyclic form clone C5-E1 transfected with an RNAi plasmid against TbEF1A (see Fig 1) and an inducible copy of wild-type eEF1A was grown in the absence (triangles) or presence (diamonds) of tetracycline for 7 days. Each day, the cultures were diluted to a cell density of 3×106 cells/ml. (B) Northern blots of total RNA extracted from parasites after three days of incubation in the absence (−) or presence (+) of tetracycline (tet) and hybridized with 32P-labeled probes (top); rRNA was used as loading control (bottom). (C) SDS-PAGE and immunoblot analysis of TbEF1A in C5-E parasites (upper panel) and HA-eEF1A in C5-haE parasites (lower panel) cultured in the presence of tetracycline for 0, 2, 4 and 6 days and probed using α-EF1A and α-HA antibody, respectively. Each lane contains lysate from 1×107 cells. (D) RT-PCR analysis of eEF1A transcript levels. cDNA generated from parasites cultured in the presence of tetracycline was amplified using primer pairs Tb2100_WT (Supplemental Table S1), hybridizing to ectopic TbEF1A (top panel), Tb2100_5′UTR and Tb2100_WT, hybridizing to endogenous TbEF1A (middle panel) and Tb3360, hybridizing to T. brucei mitochondrial elongation factor Tu (EFTu; as PCR control; bottom panel), respectively. Lanes containing cDNA, total RNA as negative control and genomic DNA as positive control, are indicated. The numbers above the panels refer to the culture time of the parasites in presence of tetracycline (in days).
Immunoblot analysis of proteins extracted from eEF1A-complemented parasites C5-E indicates that expression of eEF1A was severely decreased after 2 days of incubation in the presence of tetracycline (Fig. 2C, upper panel). However, in contrast to the situation in the RNAi cell line lacking expression of ectopic eEF1A (see Fig. 1C, lower panel), strong bands of eEF1A were visible after 4 days and 6 days of tetracycline induction (Fig. 2C, upper panel), suggesting that they represent protein encoded by the ectopic copy of eEF1A. Because the anti-eEF1A antibody does not discriminate between endogenous and ectopically expressed eEF1A, we studied the time course of expression of an ectopic HA-tagged form of eEF1A that can subsequently be analyzed by immunoblotting using anti-HA antibody. The results showed that HA-tagged eEF1A in the cell line C5-haE can be detected as a faint band after 2 days of incubation in the presence of tetracycline and as strong bands after 4 and 6 days of induction (Fig. 2C, lower panel). These data show that re-appearance of eEF1A in clone C5-E after 4 and 6 days of incubation in the presence of tetracycline coincides with the time point of expression of HA-eEF1A in clone C5-haE, suggesting that the eEF1A bands in clone C5-E likely represent ectopically expressed wild type eEF1A. Unfortunately, clone C5-haE expressing HA-tagged eEF1A could not be used for further experiments since HA-eEF1A was unable to complement endogenous eEF1A and restore parasite growth (results not shown), indicating that the N-terminal HA-tag interfered with eEF1A function.
To further analyze which forms of eEF1A were transcribed in the C5-E parasites, we performed RT-PCR using primer pairs allowing discrimination between RNA derived from genomic TbEF1A and ectopic eEF1A. Primary transcripts from endogenous TbEF1A contain gene-specific UTR sequences flanking the tandemly arranged TbEF1A ORFs whereas these sequences are not present in the primary transcripts from the ectopic copy. The results using a primer pair hybridizing to the 5′UTR and TbEF1A ORF (Supplemental Table S1) show that mRNA originating from endogenous TbEF1A was present in clone C5-E at day 0 but absent after 2 days of incubation in the presence of tetracycline (Fig. 2D, middle panel). In contrast, the use of a primer pair hybridizing to the TbEF1A ORF showed the presence of the corresponding mRNA during the entire induction period (Fig. 2D, top panel), demonstrating that ectopic eEF1A was transcribed whereas transcription of endogenous TbEF1A was ablated.
Complementation of TbEF1A-depleted T. brucei with ectopic eEF1A lacking the EPG modification site. The successful complementation of the TbEF1A RNAi cell line with wild type eEF1A demonstrates that the system can be used to study the role of EPG attachment to eEF1A for growth of T. brucei procyclic forms in culture by using eEF1A mutant proteins lacking the EPG attachment site. Based on our previous findings25, we transfected TbEF1A RNAi cells with ectopic copies of eEF1A in which the glutamate residues at position 362 were replaced by aspartate (E362D) or glutamine (E362Q). The results show that growth of parasite clones transfected with ectopic eEF1A/E362D (clone C5-D) or eEF1A/E362Q (clone C5-Q) was identical to that of uninduced cells (Fig. 3A and 3B, upper panels). Northern blot analyses using a probe against TbEF1A intergenic region 1 showed that, following induction by tetracycline for 3 days, the endogenous eEF1A transcripts in the clones were effectively down-regulated (Fig. 3A and 3B, lower panels), demonstrating that RNAi against endogenous TbEF1A was effective. Similar results (growth curve, Northern blot) were obtained with one additional clone from each of the two transfections (results not shown). In addition, as expected immunoblot analyses demonstrated the presence of eEF1A in protein extracts from clones C5-D and C5-Q throughout the entire induction period (Fig. 3C). The decreased intensity of the bands after 2 days of induction with tetracycline reflects down-regulation of endogenous TbEF1A while expression of ectopic (mutated) eEF1A is induced (compare with Fig. 2C). The band at 37 kDa represents a degradation product of eEF1A occasionally seen in preparations23,25,27. Finally, the presence of transcripts originating from ectopic eEF1A together with the disappearance of mRNA derived from endogenous TbEF1A confirmed proper function of the constructs (Fig. 4A and 4B).

Complementation of TbEF1A-depleted T. brucei with eEF1A lacking the EPG attachment site.
(A, B) T. brucei procyclic forms carrying an RNAi plasmid against TbEF1A (see Fig. 1) were transfected with an inducible copy of eEF1A in which the EPG attachment site, Glu362, has been mutated to aspartate (E362D; clone C5-D; panel A) or glutamine (E362Q; C5-Q; panel B). Parasites were cultured in the absence (triangles) or presence (diamonds) of tetracycline for 7 days and diluted each day to a cell density of 3×106 cells/ml (top panels in A, B). The lower panels in A and B show Northern blots of total RNA extracted from parasites after three days of incubation in the absence (−) or presence (+) of tetracycline (tet) and hybridized with 32P-labeled probes (top); rRNA was used as loading control (bottom). (C) SDS-PAGE and immunoblot analysis of eEF1A in C5-D (upper panel) and C5-Q (lower panel) parasites cultured in the presence of tetracycline for 0, 2, 4 and 6 days and probed using α-EF1A antibody. Each lane contains lysate from 1×107 cells.

RT-PCR analysis of eEF1A transcript levels in mutant parasites expressing EPG-deficient eEF1A.
cDNA generated from eEF1A/E362D (A) and eEF1A/E362Q (B) parasites (see Fig 3) cultured in the presence of tetracycline was amplified using primer pairs Tb2100_WT (Supplemental Table S1), hybridizing to mutated ectopic eEF1A (top panels), Tb2100_5′UTR and Tb2100_WT, hybridizing to endogenous TbEF1A (middle panels) and Tb3360, hybridizing to T. brucei elongation factor Tu (EFTu; as PCR control; bottom panels), respectively. Lanes containing cDNA, total RNA as negative control and genomic DNA as positive control, are indicated. The numbers above the panels refer to the culture time of the parasites in presence of tetracycline (in days).
Taken together, the results of the growth curves, Northern blots, immunoblots and RT-PCR experiments demonstrate that the mutated forms of eEF1A, lacking the EPG attachment site, are able to complement endogenous TbEF1A and restore normal growth of TbEF1A-depleted T. brucei procyclic forms in culture.
Discussion
Normally, a given protein modification is present simultaneously on many different proteins within a cell. Thus, approaches to knock out enzymes or other essential components involved in the synthesis or attachment of a modification may not only affect the protein of interest but many other proteins as well. Remarkably, eEF1A is the only protein so far that is modified with EPG20,22,28. However, the biosynthetic route for the synthesis and attachment of EPG is still unknown, although the phospholipid, phosphatidylethanolamine, has been identified as the donor of the ethanolamine moiety of EPG23. Furthermore, the function of EPG is completely unknown. In the present report, we describe for the first time a model system to study possible roles of EPG in eEF1A function in a eukaryotic cell. As model organism we chose the protozoan parasite, T. brucei, since i) EPG in TbEF1A has been structurally characterized23, ii) TbEF1A, in contrast to eEF1A in other eukaryotes, contains a single EPG modification site only23, iii) point mutations of the EPG attachment site of TbEF1A, Glu362, have been shown to abolish EPG binding25 and iv) T. brucei is amenable to genetic manipulation, such as RNAi, inducible gene expression and knock-out or knock-in strategies29,30,31.
First we generated a tetracycline-inducible T. brucei cell line, in which expression of endogenous TbEF1A was ablated by RNAi directed against the intergenic region 1, located between the first and second TbEF1A gene on chromosome 10. Induction of RNAi by tetracycline resulted in complete disappearance of TbEF1A mRNA and protein, resulting in growth arrest of parasites in culture. These results are in line with a study in which expression of TbEF1A was down-regulated by RNAi directed against the ORF of TbEF1A4. In a second step, we successfully rescued the growth defect by introducing a tetracycline-inducible ectopic copy of wild type eEF1A into the RNAi cell line. After tetracycline-dependent ablation of endogenous TbEF1A and expression of ectopic eEF1A, growth of T. brucei procyclic forms was unchanged compared to uninduced control cells. The result demonstrates that ectopically expressed wild type eEF1A is able to complement endogenous TbEF1A function in parasites in culture. Finally, we studied if eEF1A, in which the EPG attachment site, Glu362, was replaced by Asp or Gln, was able to rescue eEF1A-depleted RNAi parasites. The results show that this is the case, i.e. both eEF1A/E362D and eEF1A/E362Q restored growth of T. brucei procyclic forms depleted of endogenous TbEF1A, indicating that attachment of EPG to TbEF1A is not necessary for normal growth of T. brucei under standard culture conditions.
The non-essentiality of EPG attachment to TbEF1A for parasite growth in culture raises the question why this unique protein modification has evolved and has been maintained in eukaryotes during evolution. The synthesis and attachment of EPG likely involves multiple gene products and metabolites and thus, is a costly event for a cell to maintain. Similar considerations also apply for two other unique protein modifications, diphthamide attachment to eukaryotic elongation factor 2 (eEF2) and hypusine linkage to eukaryotic initiation factor 5A (eIF5A) (reviewed by32). Interestingly, although these modifications – like EPG – have been reported for the first time more than 30 years ago33,34, their roles in protein function have also remained elusive. Similar to the situation for EPG attachment to eEF1A in T. brucei, modification of eEF2 with diphthamide is not strictly required for growth of yeast and mammalian cells35,36,37,38, despite the fact that this modification is conserved from archaea to humans. It has been proposed that essential functions of diphthamide may only become evident under certain conditions, e.g. in the context of a multi-cellular organism or during stress39,40,41. For instance, S. cerevisiae strains lacking diphthamide attachment to eEF2 were found to have increased frequency in ribosomal (-)1 frame shifting42. Furthermore, Chinese hamster ovary cells lacking genes involved in diphthamide biosynthesis were three-fold more sensitive towards ricin than wild type cells, suggesting that diphthamide may protect ribosomes from ribosome-inactivating proteins43. Accordingly, it is possible that the roles for EPG in eEF1A function will only become apparent under stress conditions, or in the natural environment of the parasite. In contrast to EPG and diphthamide, hypusine has been reported to be essential for cell viability in S. cerevisiae44,45,46, Leishmania donovani47 and multi-cellular organisms48,49. Nevertheless, its role in eIF5A function, which recently has been shown to promote translation elongation rather than translation initiation (reviewed by50) is not known.
Methods
Unless otherwise specified, all reagents were of analytical grade and were from Merck (Darmstadt, Germany), Sigma-Aldrich (Buchs, Switzerland) or ICN Biomedicals (Tägerig, Switzerland). DNA polymerase was obtained from Invitrogen (Basel, Switzerland). Restriction enzymes were purchased from Roche Diagnostics (Rotkreuz, Switzerland), Fermentas (St. Leon-Rot, Germany), or New England Biolabs (Ipswich, MA). [α-32P]dCTP and Bio-Max MS films were from GE Healthcare, Kodak MBX films and TransScreen-HE intensifying screen from Kodak (Lausanne, Switzerland).
Constructs
RNAi directed against TbEF1A was done using the pALC14 vector, a pLew-100-based stem loop construct containing a puromycin resistance gene51. The two fragments inserted in opposite direction into pALC14 represent the intergenic region 1 between the tandemly arranged TbEF1A genes, Tb927.10.2090 and Tb927.10.2100 (TriTrypDB, Tb92710v5, location 546408-547232). For tetracycline-inducible expression of the ectopic copy of TbEF1A, the open reading frame (ORF) of Tb927.10.2100 was first subcloned into pBluescript. Subsequently, the glutamate residue at position 362 was mutated to aspartate or glutamine, as previously described25. Hemagglutinin (HA)-tagged TbEF1A was cut out from pEGhaEF(1-449)25 using restriction enzymes BamHI and HindIII. Wild type, HA-tagged and mutated TbEF1A ORFs were then cloned between BamHI and HindIII restriction sites of pALC14 carrying a blasticidin gene. All constructs were sequenced before transfection. The primers used to amplify the TbEF1A intergenic region 1 and TbEF1A ORF, as well as those used for site-directed mutagenesis, are listed in Supplemental Table S1. All constructs were linearized with NotI prior to transfection.
Cell culture
T. brucei procyclic form strain 29–13 was grown at 27°C in SDM79 supplemented with 15% fetal bovine serum and 25 μg/ml hygromycin and 15 μg/ml G-418 to maintain constitutive expression of the T7 RNA polymerase and the tetracycline repressor29. Parasites were grown to a maximum density of 1.3×107 cells/ml.
Transgenic cell lines
RNAi cell lines against TbEF1A intergenic region 1 were selected with 2 μg/ml puromycin. One of the positive clones, named C5, was selected for transfections with the plasmids allowing tetracycline-inducible ectopic expression of TbEF1A proteins. The resulting transfectants were selected in the presence of 10 μg/ml blasticidin. Formation of double-stranded RNA and conditional expression of TbEF1A proteins were induced by the addition of 1 μg/ml tetracycline. Stable transfection, selection of transfectants and production of clonal cell lines were done as described previously25.
Northern blot analysis
Total RNA for Northern blotting was isolated using a SV Total RNA Isolation System (Promega, Madison, WI) and 10 μg of the total RNA was separated on formaldehyde-agarose gels (1% agarose, 2% formaldehyde in 20 mM 3-(N-morpholino)propanesulfonic acid). To control for equal loading, ribosomal RNA was visualized on the same gel by ethidium bromide staining. Subsequently, the RNA was transferred to positively charged nylon membrane, Hybond-N+ (GE Healthcare, Glattbrugg, Switzerland). 32P-Labeled probe against polycistronic TbEF1A mRNA was made by random priming using the PCR product of the TbEF1A intergenic region 1 (Prime-a-Gene Labeling System, Promega). Hybridization was performed overnight at 60°C in 0.5 M Na2HPO4, pH 7.2, containing 7% (w/v) SDS, 1% (w/v) bovine serum albumin and 0.9 mM EDTA. The membrane was analyzed by autoradiography using Bio-Max MS film and a TransScreen-HE intensifying screen.
Immunoblot analysis
Proteins were extracted using Nonidet P-40 as described before25. Extracted proteins were separated by glycine-SDS-polyacrylamide gel electrophoresis (SDS-PAGE) under reducing conditions using 12% polyacrylamide gels52. Semi-dry blotting was performed as previously described25. A mouse monoclonal antibody against eEF1A (Upstate, Lake Placid, NY) and a secondary rabbit anti-mouse IgG antibody conjugated to horseradish peroxidase (Dako, Baar, Switzerland) were used at dilutions of 1:5000. Labeled proteins were detected by enhanced chemiluminescence (Pierce, Lausanne, Switzerland).
Reverse transcription PCR analysis
To assess TbEF1A transcript levels, reverse transcription-PCR (RT-PCR) was performed. Total RNA was collected from parasites at the time when the induction using tetracycline was started (day 0) and 48 hrs (day 2), 96 hrs (day 4) and 144 hrs (day 6) after induction. The synthesis of complementary DNA (cDNA) from single-stranded RNA was done according to the manufacturer's guidelines using SuperScript® II Reverse Transcriptase and Oligo(dT)20 Primer (Invitrogen) using 1μg of extracted total RNA. Subsequent amplification of cDNA was done using 20 cycles of denaturation (95°C for 30 s), annealing (50°C for 30 s) and extension (72°C for 60 s). The cDNA originating from the transcripts of both endogenous and ectopic TbEF1A was detected using primers Tb2100_WT forward and Tb2100_WT reverse (Supplemental Table S1), which match the nucleotide sequences located at the beginning and end, respectively, of the TbEF1A ORF. To detect the cDNA originating from endogenous TbEF1A only, primer Tb2100_5′UTR (Supplemental Table S1) was used in combination with the above mentioned reverse primer. Total RNA extract was used as a negative control. To control for equal loading, RT-PCR was done in parallel with a primer pair (Tb3360 forward and reverse; Supplemental Table S1) binding to the ORF of T. brucei elongation factor Tu (TriTrypDB, Tb927.10.13360). As a positive control for the PCR reaction and for the size of the amplified sequence, genomic DNA from the T. brucei strain 29-13 was used. Reaction products were semi-quantified by electrophoresis using 0.8% agarose gel.
References
Merrick, W. C. Eukaryotic protein synthesis: still a mystery. J Biol Chem 285, 21197–21201 (2010).
Panasyuk, G., Nemazanyy, I., Filonenko, V., Negrutskii, B. & El'skaya, A. V. A2 isoform of mammalian translation factor eEF1A displays increased tyrosine phosphorylation and ability to interact with different signalling molecules. Int J Biochem Cell Biol 40, 63–71 (2008).
Khacho, M. et al. eEF1A is a novel component of the mammalian nuclear protein export machinery. Mol Biol Cell 19, 5296–5308 (2008).
Bouzaidi-Tiali, N., Aeby, E., Charriere, F., Pusnik, M. & Schneider, A. Elongation factor 1a mediates the specificity of mitochondrial tRNA import in .T. brucei. EMBO J 26, 4302–4312 (2007).
Liu, G. et al. Interactions of elongation factor 1alpha with F-actin and beta-actin mRNA: implications for anchoring mRNA in cell protrusions. Mol Biol Cell 13, 579–592 (2002).
Murray, J. W., Edmonds, B. T., Liu, G. & Condeelis, J. Bundling of actin filaments by elongation factor 1 alpha inhibits polymerization at filament ends. J Cell Biol 135, 1309–1321 (1996).
Gross, S. R. & Kinzy, T. G. Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat Struct Mol Biol 12, 772–778 (2005).
Gross, S. R. & Kinzy, T. G. Improper organization of the actin cytoskeleton affects protein synthesis at initiation. Mol Cell Biol 27, 1974–1989 (2007).
Merrick, W. C. & Hershey, J. W. B. The pathway and mechanism of eukaryotic protein synthesis in .Translational Control, edited by J. W. B. Hershey, M. B. Mathews, & N. Sonnenberg (Cold Spring Harbor Laboratory Press, New York, 1996), pp. 31–69.
Kaur, K. J. & Ruben, L. Protein translation elongation factor-1 alpha from Trypanosoma brucei binds calmodulin. .J Biol Chem 269, 23045–23050 (1994).
Walsh, C. T., Garneau-Tsodikova, S. & Gatto, G. J., Jr Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 44, 7342–7372 (2005).
Seet, B. T., Dikic, I., Zhou, M. M. & Pawson, T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 7, 473–483 (2006).
Ahrné, E., Müller, M. & Lisacek, F. Unrestricted identification of modified proteins using MS/MS. Proteomics 10, 671–686 (2010).
Chang, Y. W. & Traugh, J. A. Insulin stimulation of phosphorylation of elongation factor 1 (eEF-1) enhances elongation activity. Eur J Biochem 251, 201–207 (1998).
Peters, H. I., Chang, Y. W. & Traugh, J. A. Phosphorylation of elongation factor 1 (EF-1) by protein kinase C stimulates GDP/GTP-exchange activity. Eur J Biochem 234, 550–556 (1995).
Dever, T. E., Costello, C. E., Owens, C. L., Rosenberry, T. L. & Merrick, W. C. Location of seven post-translational modifications in rabbit elongation factor 1 alpha including dimethyllysine, trimethyllysine and glycerylphosphorylethanolamine. J Biol Chem 264, 20518–20525 (1989).
Fonzi, W. A., Katayama, C., Leathers, T. & Sypherd, P. S. Regulation of protein synthesis factor EF-1 alpha in Mucor racemosus. Mol Cell Biol 5, 1100–1103 (1985).
Zobel-Thropp, P., Yang, M. C., Machado, L. & Clarke, S. A novel post-translational modification of yeast elongation factor 1A. Methylesterification at the C terminus. J Biol Chem 275, 37150–37158 (2000).
Ejiri, S. Moonlighting functions of polypeptide elongation factor 1: from actin bundling to zinc finger protein R1-associated nuclear localization. Biosci Biotechnol Biochem 66, 1–21 (2002).
Whiteheart, S. W., Shenbagamurthi, P., Chen, L., Cotter, R. J. & Hart, G. W. Murine elongation factor 1alpha is posttranslationally modified by novel amide-linked etanolamine-phosphogycerol moieties. J Biol Chem 264, 14334–14341 (1989).
Ransom, W. D., Lao, P.-C., Gage, D. A. & Boss, W. F. Phosphoglycerylethanolamine posttranslational modification of plant eukaryotic elongation factor 1alpha. Plant Physiol 117, 949–960 (1998).
Rosenberry, T. L. et al. Biosynthetic incorporation of [3H]ethanolamine into protein synthesis elongation factor 1alpha reveals a new post-translational protein modification. J Biol Chem 264, 7096–7099 (1989).
Signorell, A., Jelk, J., Rauch, M. & Bütikofer, P. Phosphatidylethanolamine is the precursor of the ethanolamine phosphoglycerol moiety bound to eukaryotic elongation factor 1A. .J. Biol. Chem. 283, 20320–20329 (2008).
Cavallius, J., Zoll, W., Chakraburtty, K. & Merrick, W. C. Characterization of yeast EF-1 alpha: non-conservation of post-translational modifications. Biochim Biophys Acta 1163, 75–80 (1993).
Greganova, E., Heller, M. & Bütikofer, P. A structural domain mediates attachment of ethanolamine phosphoglycerol to eukaryotic elongation factor 1A in Trypanosoma brucei. .PLoS One 5, e9486 (2010).
Bellofatto, V. & Palenchar, J. B. RNA interference as a genetic tool in trypanosomes. Methods Mol Biol 442, 83–94 (2008).
Regmi, S.,, . Rothberg, K. G.,, . . Hubbard, J. G. &, . . Ruben, L., . The RACK1 signal anchor protein from Trypanosoma brucei associates with eukaryotic elongation factor 1A: a role for translational control in cytokinesis. .Mol Microbiol 70, 724–745 (2008).
Tisdale, E. J. & Tartakoff, A. M. Extensive labeling with [3H]ethanolamine of a hydrophilic protein of animal cells. J Biol Chem 263, 8244–8252 (1988).
Wirtz, E., Leal, S., Ochatt, C. & Cross, G. A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99, 89–101 (1999).
Ullu, E., Tschudi, C. & Chakraborty, T. RNA interference in protozoan parasites. Cell Microbiol 6, 509–519 (2004).
Wirtz, E. & Clayton, C. Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science 268, 1179–1183 (1995).
Greganova, E., Altmann, M. & Bütikofer, P. Unique modifications of translation elongation factors. FEBS J 278, 2613–2624 (2011).
Van Ness, B. G., Howard, J. B. & Bodley, J. W. ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J Biol Chem 255, 10710–10716 (1980).
Shiba, T., Mizote, H., Kaneko, T., Nakajima, T. & Kakimoto, Y. Hypusine, a new amino acid occurring in bovine brain. Isolation and structural determination. Biochim Biophys Acta 244, 523–531 (1971).
Kimata, Y. & Kohno, K. Elongation factor 2 mutants deficient in diphthamide formation show temperature-sensitive cell growth. J Biol Chem 269, 13497–13501 (1994).
Phan, L. D., Perentesis, J. P. & Bodley, J. W. Saccharomyces cerevisiae elongation factor 2. Mutagenesis of the histidine precursor of diphthamide yields a functional protein that is resistant to diphtheria toxin. .J Biol Chem 268, 8665–8668 (1993).
Liu, S., Milne, G. T., Kuremsky, J. G., Fink, G. R. & Leppla, S. H. Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol Cell Biol 24, 9487–9497 (2004).
Ivankovic, M., Rubelj, I., Matulic, M., Reich, E. & Brdar, B. Site-specific mutagenesis of the histidine precursor of diphthamide in the human elongation factor-2 gene confers resistance to diphtheria toxin. Mutat Res 609, 34–42 (2006).
Chen, C. M. & Behringer, R. R. Ovca1 regulates cell proliferation, embryonic development and tumorigenesis. Genes Dev 18, 320–332 (2004).
Liu, S. et al. Dph3, a small protein required for diphthamide biosynthesis, is essential in mouse development. Mol Cell Biol 26, 3835–3841 (2006).
Kruse, M. et al. Inhibition of CD83 cell surface expression during dendritic cell maturation by interference with nuclear export of CD83 mRNA. J Exp Med 191, 1581–1590 (2000).
Ortiz, P. A., Ulloque, R., Kihara, G. K., Zheng, H. & Kinzy, T. G. Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J Biol Chem 281, 32639–32648 (2006).
Gupta, P. K., Liu, S., Batavia, M. P. & Leppla, S. H. The diphthamide modification on elongation factor-2 renders mammalian cells resistant to ricin. Cell Microbiol 10, 1687–1694 (2008).
Schnier, J., Schwelberger, H. G., Smit-McBride, Z., Kang, H. A. & Hershey, J. W. Translation initiation factor 5A and its hypusine modification are essential for cell viability in the yeast Saccharomyces cerevisiae. .Mol Cell Biol 11, 3105–3114 (1991).
Wohl, T., Klier, H., Ammer, H., Lottspeich, F. & Magdolen, V. The HYP2 gene of Saccharomyces cerevisiae is essential for aerobic growth: characterization of different isoforms of the hypusine-containing protein Hyp2p and analysis of gene disruption mutants. .Mol Gen Genet 241, 305–311 (1993).
Sasaki, K., Abid, M. R. & Miyazaki, M. Deoxyhypusine synthase gene is essential for cell viability in the yeast Saccharomyces cerevisiae. FEBS Lett 384, 151–154 (1996).
Chawla, B. et al. Identification and characterization of a novel deoxyhypusine synthase in Leishmania donovani. J Biol Chem 285, 453–463 (2010).
Sugimoto, A. High-throughput RNAi in Caenorhabditis elegans: genome-wide screens and functional genomics. Differentiation 72, 81–91 (2004).
Nishimura, K., Lee, S. B., Park, J. H. & Park, M. H. Essential role of eIF5A-1 and deoxyhypusine synthase in mouse embryonic development. Amino Acids in press (2011).
Kaiser, A. Translational control of eIF5A in various diseases. Amino Acids in press (2011).
Bochud-Allemann, N. & Schneider, A. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. .J Biol Chem 277, 32849–32854 (2002).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 (1970).
Acknowledgements
The work was supported by Swiss National Science Foundation grant 31003A-130815 to P.B. We thank I. Roditi and D. Purple for comments on the manuscript and valuable input and P. Mäser for support.
Author information
Authors and Affiliations
Contributions
EG and PB designed the experiments and wrote the manuscript. EG performed the experiments.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Greganova, E., Bütikofer, P. Ethanolamine phosphoglycerol attachment to eEF1A is not essential for normal growth of Trypanosoma brucei. Sci Rep 2, 254 (2012). https://doi.org/10.1038/srep00254
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00254
This article is cited by
-
Oncogenic activation of EEF1A2 expression: a journey from a putative to an established oncogene
Cellular & Molecular Biology Letters (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
