
Abstract
Glioblastoma multiforme (GBM) is a debilitating disease that is associated with poor prognosis, short median patient survival and a very limited response to therapies. GBM has a very complex pathogenesis that involves mutations and alterations of several key cellular pathways that are involved in cell proliferation, survival, migration and angiogenesis. Therefore, efforts that are directed toward better understanding of GBM pathogenesis are essential to the development of efficient therapies that provide hope and extent patient survival. In this review, we outline the alterations commonly associated with GBM pathogenesis and summarize therapeutic strategies that are aimed at targeting aberrant cellular pathways in GBM.
Similar content being viewed by others
Introduction
Glioblastoma multiforme (GBM, WHO grade 4) is the most frequently occurring malignant central nervous system tumor with a global incidence of 0.59–3.69 per 100 000.1 It is by far the most common and malignant of all glial tumors, and is associated with poor prognosis with a median patient survival of 12–15 months from diagnosis.2,3 Unfortunately, only around 3–5% of patients survive for a period of 3 years or more.4,5 Although GBM affect primarily the cerebral hemispheres of adult brains, they are much less common in children, where they affect specifically the brainstem region. GBMs are classified as either primary or secondary, roughly 90% of cases are primary and occur de novo in elderly patients. Secondary cases progress from lower grade astrocytomas and are more prevalent in younger patients. Primary and secondary GBMs have differing genetic profiles with IDH1 mutations being evident in secondary GBM and not primary.6 Common genetic alterations are associated with a loss of heterozygosity (LOH) of the chromosome arm 10q, that occur in 60–90% of GBM cases.7,8 Other alterations and deletions that affect the p53 gene could be as high as 85.3–87%.9,10 P53 alterations are more common in secondary GBMs than primary GBM tumors.6 Mutations in the epidermal growth factor receptor (EGFR) and in the platelet-derived growth factor receptor (PDGFR) are also associated with GBM pathogenesis and account for 40–57%(refs. 9–11) and 60%(ref. 12) subsequently. Other mutations target the gene of the mouse double minute homolog 2 (MDM2) (10–15%)13 and the phosphatase and tensin homolog (PTEN) gene (20–34%).14,15 Interestingly, genomic analyses performed by the Cancer Genome Atlas Research Network has revealed further alterations in key signaling pathways that contribute to the pathology of the disease. The RTK/Ras/PI3K signaling pathway was found to be altered in 86–89.6% and the pRB signaling pathway was found to be affected in 77–78.9% of GBM cases studied.9,10 It is important to add, that mutations encountered in GBM may not affect one single cellular pathway but may be the result of alterations in several of the pathways mentioned above. This adds further complexity to our understanding of GBM pathogenesis and results in additional complexity for the development of GBM therapies.
Regrettably, patients who are affected by GBM have a poor prognosis and existing therapies do not appear to be very efficient against GBM. The current gold standard for the treatment of GBM is palliative and includes surgery, adjuvant radiotherapy and temozolomide (TMZ) chemotherapy. Despite multimodal aggressive therapy, GBM is uniformly fatal with survival over 3 years being considered long-term.16 Due to the poor survival rate of GBM patients, it is imperative that novel avenues for therapy are explored in order to improve patient prognosis and eventually develop a cure to this fatal disease.
Receptor tyrosine kinase pathways
The tyrosine kinase receptors
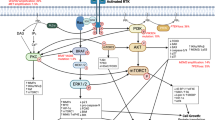
Receptor tyrosine kinases (RTKs) are a family of cell surface receptors, which act as receptors for growth factors, hormones, cytokines, neurotrophic factors and other extracellular signaling molecules. Upon activation by ligands, RTKs signal through two major downstream pathways Ras/MAPK/ERK and Ras/PI3K/AKT 17 (Figure 1). These pathways are involved in the regulation of cell proliferation, survival, differentiation and angiogenesis. In this review, we focus on six tyrosine kinase receptors; the epidermal growth factor receptor (EGFR), the vascular endothelial growth factor receptor (VEGFR), the platelet-derived growth factor receptor (PDGFR), the hepatocyte growth factor receptor (HGFR/c-MET), the fibroblast growth factor receptor (FGFR) and the insulin-like growth factor 1 receptor (IGF-1R).

Schematic representation of RTK activation and the resultant downstream signaling. Black arrows indicate activation whereas red arrows indicate inhibition.
Tyrosine kinase receptors share a similar structure that is composed of an extracellular ligand-binding domain, a hydrophobic transmembrane domain and an intracellular tyrosine kinase domain. They are activated by ligand binding which results in receptor dimerization and autophosphorylation of the tyrosine kinase domain. This event results in activation of two main downstream signaling pathways: Ras/MAPK/ERK and Ras/PI3K/AKT.18–24 Due to the ability of these receptors to activate downstream signaling pathways that are involved in proliferation, invasiveness, survival and angiogenesis, RTKs and their ligands are promising therapeutic targets for the treatment of GBM (Figure 1).
EGFR
EGFR belongs to a family of four tyrosine kinases that encompasses ErbB1 (EGFR, HER1), ErbB2 (Her-2, Neu), ErbB3 (Her-3) and ErbB4 (Her-4). Amplifications and mutations in EGFR (HER1) were detected in 45–57% of GBM cases studied9,10 indicating a causal role in the pathogenesis of GBM. EGFRs induce proliferation and have been implied in glioblastoma pathogenesis and resistance to treatment.25 Interestingly EGFR is not the only member of this family that is mutated in GBM. ErbB2/HER-2 mutation was also detected in 8–41% of GBM cases.9,26 A truncated mutant EGFR variant III (EGFRvIII) is frequently expressed in glioblastoma multiforme and is constitutively activated in a ligand independent manner, resulting in cell proliferation and survival. Despite the growth enhancing properties of the EGFRvIII, its expression has been linked to increased overall survival in patients.27,28 This could be explained by the fact that EGFRvIII is a neoantigen and this may result in the elicitation of an immune response.
VEGFR
VEGF is a potent angiogenic protein that is known to increase vascular permeability. Although VEGF has a role in normal tissues, malignant transformation has been shown to induce VEGF expression.29 Under hypoxic conditions, the hypoxia inducible transcription factors (HIF1α and HIF1β) translocate to the nucleus and activate the VEGF gene (Figure 1.). Activation of VEGF leads to increased angiogenesis to counteract hypoxia.30 Glioblastoma multiforme tumors are often hypoxic and have increased VEGF expression that contributes to the irregular vasculature associated with GBM. GBM tissues have been shown to have very high levels of VEGF expression that is associated with an up-regulation of the VGFR receptor VEGFR2.31,32
PDGFR
PDGF/PDGFR signaling is involved in the development of normal tissues and its dysregulation contributes to oncogenesis. GBMs regularly exhibit a PDGF autocrine loop that is absent in normal brain tissues. This observation pinpoints to the importance of PDGF in GBM pathology.33,34 Data analyses from TCGA research network revealed amplification of platelet-derived growth factor receptor alpha (PDGFRα) in 10–13% of the cases studied.9,10 PDGFRα is the second most frequently amplified RTK in GBM behind EGFR. Glioblastoma multiforme has been shown to express all PDGF ligands (PDGF-A, PDGF-B, PDGF-C and PDGF-D) and the two cell surface receptors: PDGFR-α and PDGFR-β.21
HGFR/c-MET
Scatter factor (SF)/hepatocyte growth factor (HGF) is the activating ligand for HGFR/c-MET that have been shown to be secreted by brain tumor cells. HGFR/c-MET expression and activation in tumor cells and vascular endothelial cells, results in cellular proliferation and invasion.35 The association of HGFR/c-MET with proliferation and survival indicates its suitability as a target for GBM therapy. HGFR/c-MET amplification was detected in 1.6–4% of human GBMs studied.9,10 Expression of HGFR/c-MET has been linked with poor prognosis for GBM patients.36,37
FGFR
Humans have 22 FGFs (fibroblast growth factors) and four different FGF receptors (FGFR1, 2, 3 and 4).38 FGFR amplification was identified in 3.2% of the cases studied by TCGA.10 FGF2 has been shown to stimulate growth of cultured GBM cell lines and inhibition of FGFR signaling by RNA interference or by antibody blockade reduced GBM cell proliferation.39 FGFR1 has also been shown to be expressed at higher levels in brain tumors and when compared to adjacent normal brain tissue, suggesting a role for this receptor in tumorigenesis.40,41 FGF5 has also been shown to be overexpressed in GBM and this expression was linked to increased proliferation.41
IGF-1R
GBM cell lines and tissues have been shown to express the IGF-1R.42,43 IGF-1R was seen to be overexpressed in GBM, and this overexpression was linked to shorter survival and reduced responsiveness to temozolomide, hinting at the role of IGF-1R signaling in GBM pathogenesis.44
Targeting the tyrosine kinase receptors
Small-molecule kinase inhibitors
Many molecules that target the kinase domains of RTKs have been tested in the context of GBM (Table 1). Erlotinib is an EGFR tyrosine kinase inhibitor that prevents the autophosphorylation of the tyrosine kinase intracellular domain of EGFR.45 It has been tested in several phase II studies for GBM and in conjunction with temozolomide for newly diagnosed GBM. The combination of the two drugs was well tolerated by patients and resulted in improved survival.45 However, treatment with Erlotinib alone was not effective in patients with recurrent GBM.46 Gefitinib (ZD1839/Iressa) is also an EGFR tyrosine kinase inhibitor that has been shown to radiosensitize U251 GBM cells in vitro.47 When tested at phase II trial, Gefitinib did not lead to an improvement in overall and progression free survivals for patients with newly diagnosed GBM.48
Multiple kinase inhibitors such as AEE788 and Vandetanib target both EGFR and VEGFR tyrosine kinases (Table 1). When tested in GBM patients, these drugs appeared to have little efficacy or increased toxicity. AEE788 was shown to have highly toxic side effects and very little efficacy for the treatment of recurrent GBM at phase I clinical trial,49 whereas Vandetanib had very little effect in vitro on GBM cell lines. However, when combined with histone deacetylase inhibitors (HDACIs), Vandetanib reduced GBM cell proliferation in vitro.50 The incorporation of Vandetanib to the standard therapy regimen (surgery+chemotherapy+radiotherapy) in phase II trial, also yielded little effect on overall survival and resulted in early termination of trial.51 Lapatinib is another multiple kinase inhibitor that binds both EGFR and HER2 tyrosine kinases and prevents their activation. In a phase I/II trial for recurrent GBM, it was shown to have little effect on patients.52 However, CUDC-101 a multi-targeted EGFR/HDAC (histone deacetylase) inhibitor has been shown to enhance the radiosensitivity of GBM cell lines in vitro.53
Vatalanib (PTK787), Sorafenib and Tivozanib are VEGFR tyrosine kinase inhibitors that have been found to have little efficacy on GBM patients when administered individually (Table 1). Vatalanib (PTK787) is well tolerated by patients but it does not appear to result in tumor regression.54 Likewise, the combination of Sorafenib with standard therapy also resulted in little effect on the treatment efficacy for GBM at the phase II stage.55 In a phase II study for patients with recurrent GBM, Tivozanib had apparent anti-angiogenic effects, but failed to affect tumor volume.56
Cediranib (AZD2171), a VEGFR-2 tyrosine kinase inhibitor, has been used as a monotherapy or in combination with Lomustine chemotherapy for recurrent GBM in phase III trial. Cediranib failed to improve progression free survival as a monotherapy and in conjunction with Lomustine.57 Similarly, Vandetanib a dual inhibitor of VEGFR-2 and EGFR that was tested in a phase II trial for recurrent GBM also had little efficacy in patients with GBM.58
The multiple kinase inhibitor Sunitinib is an inhibitor of VEGF, PDGFR, FLT1, FLT1/KDR, FLT3 and the RET kinases.59 In a phase II study for recurrent glioblastoma multiforme, Sunitinib was found to be unsuitable as a monotherapy with all patients’ disease progressing despite treatment.60
PDGFRα, PDGFRβ, Bcr-Abl, c-FMS and c-Kit tyrosine kinases can be targeted using the kinase inhibitor Imatinib (Gleevec/ST1571). This molecule disrupts the ligand-receptor autocrine loops for PDGFR.61 Likewise, this drug appeared to have little beneficial activity for GBM patients in phase II study.62 On the other hand, Tyrphostin (AG-1296), also a PDGFR-α, PDGFR-β, c-Kit, FMS-like tyrosine kinase 3 and a BEK tyrosine kinase inhibitor, was shown to reduce GBM cell viability in vitro and to have anti-tumor activity in a murine xenograft model of GBM.63 Tandutinib which targets PDGFR-β, FMS-like tyrosine kinase 3 and c-Kit, was tested in phase II trial in patients with recurrent GBM however this trial was halted due to the drug’s lack of efficacy.64 Other multi-kinase inhibitors such as Lenvatinib (E7080) and Nintedanib that inhibit VEGFR, FGFR and PDGFR kinases were tested in phase II studies. Although only Lenvatinib appeared to have modest activity on recurrent GBM patients, therapy with this inhibitor was accompanied with high toxicity in GBM treated patients.65–67
XL-184 (BMS-907351/Cabozantinib) is an oral inhibitor of c-MET, VEGFR-2 and RET,68 and it also has an inhibitory effect on KIT, FLT3 and TEK.69 Initial results from a phase II trial using XL-184 are promising, but further research is required to fully test its efficacy for GBM.68 Other molecules such as Foretinib and SGX-523 inhibit HGFR/c-MET tyrosine kinase and have been shown to reduce tumor growth in vitro and in vivo when using a GBM murine xenograft model.70,71 PD173074 is another multiple tyrosine kinase inhibitor that inhibits FGFR and VEGFR tyrosine kinases. PD173074 showed GBM growth inhibitory effects in vitro39 and as a result this drug might be of benefit for GBM patients. PQ401, GSK1838705A, PPP (picropodophyllin/AXL1717) and NVP-AEW541 are IGF-1R tyrosine kinase inhibitors that have all shown promising results pre-clinically (Table 1). PQ401 has been shown to suppress GBM cell growth and migration in vitro.72 GSK1838705A induced apoptosis of GBM cells in vitro, and when these cells were implanted in nude mice GSK1838705A had similar anti-GBM activity.73 PPP (Picropodophyllin/AXL1717) was shown to inhibit the growth of GBM cell lines that led to in vivo regression of intracranial xenografts.74 NVP-AEW541 induces apoptosis in GBM cell lines in vitro when co-administered with Dasatinib (a Bcr-Abl tyrosine kinase inhibitor).75 BMS-536924 is an ATP competitive IGF-1R/IR (insulin receptor) inhibitor that has shown promising anti-tumor properties in vitro and when tested on Temozolomide (TMZ) resistant GBM cells.76
These small-molecule inhibitors have been widely studied in many cancers, with varying degrees of success, however the clinical trial data for GBM shows that very few of these molecules have a significant anti-tumor response, and thus other components of the RTK receptors are being considered as therapeutic targets.
Antibody therapies targeted at RTKs’ extracellular domain
Whilst many therapies target the kinase domain of RTKs, the extracellular domain is also a viable target when using antibody therapies. These molecules are being used as antagonists of the ligand-binding domains of RTKs with the aim of preventing ligand-binding and subsequent activation of the kinase domains. A monoclonal EGFR targeting antibody known as Cetuximab has been utilized as a therapy for GBM. This antibody targets the extracellular domain of EGFR, and acts as an antagonist that prevents the activation of RTKs and therefore, inhibits tumor malignancy.77 Cetuximab has been tested as a salvage therapy for patients who have failed to respond to surgery, radiation therapy and chemotherapy. Although this monotherapy proved to be well tolerated, its activity for recurrent glioblastoma multiforme was minimal at phase II clinical trial.78 Other antibodies such as Ornartuzumab have been used to target the extracellular domain of the HGFR/c-MET receptor and this has been shown to inhibit orthotopic U87 GBM xenograft tumor growth.79 MK-0646 (H7C10/F50035/Dalotuzumab) a humanized monoclonal IGF-1R antibody that acts as an antagonist, has also been shown to reduce cell proliferation and to induce apoptosis.80 Although these antibody therapies are still in their relative infancy compared to the small-molecule inhibitors of the RTK kinases, early research has been promising in the context of GBM. It is important to note that due to their large size antibodies do not freely cross the blood–brain barrier, thus there is a need to engineer antibodies to enable them to cross the blood–brain barrier and access GBM tumors. Only around 0.1–0.2% of circulating antibodies have been shown to penetrate the blood–brain barrier.81 Bispecific antibodies consist of two different single chain Fv fragments connected by a linker. Directed antibodies with optimized binding to the transferrin receptor have been used to cross the blood–brain barrier in both murine and primate models.82 These engineered antibodies are exciting new therapeutics that enable the crossing of the blood–brain barrier and direct targeting of tumor cells. Alternatively antibodies can also be delivered directly into the brain using Ommaya reservoirs or at the time of surgery to bypass the blood–brain barrier.
Therapies directed at RTK ligands
Antibodies have also been used to ‘trap’ the ligands that activate RTK signaling pathways. Bevacizumab is a humanized murine monoclonal antibody that binds VEGF and prevents its binding to the receptor. This antibody was granted accelerated approval by the FDA (food and drug administration) in 2009 for the treatment of patients with progressive or recurrent GBM. Despite its approval, Bevacizumab has been shown to have little efficacy for newly diagnosed GBM. The addition of Bevacizumab to the current course of therapy conveys no benefit for overall patient survival.83 Aflibercept is another VEGF ‘trap’ that binds VEGF and prevents its interaction with the receptor. In phase II trial Aflibercept appeared to have little activity for recurrent GBM patients with only 7.7% of patients experiencing progression free survival after 6 months.84 Rilotumumab (AMG102) is an anti-HGF monoclonal antibody that binds HGF and prevents its binding to the HGFR/c-MET, and consequent activation of downstream targets. When combined with temozolomide in vitro, Rilotumumab has been proven to inhibit the growth of U87MG glioblastoma multiforme cells.85 In a phase II clinical study, this antibody showed little effect for the treatment of recurrent glioblastoma multiforme.86 Although targeting these ligands is an attractive avenue for GBM therapy, the efficacy of these therapies has been limited. This may be due to factors such as RTK receptors being mutated and constitutively active, such as mutations encountered in EGFRvIII. The blood–brain barrier may also present an issue for these antibody therapies, preventing their tumor penetration, inhibiting their anti-tumor effects.
RTK downstream signaling pathways
The PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway is activated by transmembrane tyrosine kinase growth factor receptors, transmembrane integrins and G-protein-coupled receptors (Figure 1). Upon activation of these receptors, functional PI3K translocates to the plasma membrane and leads to the production of phosphatidylinositol 3,4,5-triphosphate (PIP3) from phosphatidylinositol bisphosphate (PIP2).87,88 PIP3 activates serine/threonine kinase phosphoinositide-dependent kinase 1 (PDK1) and AKT (at threonine 308).87,88 Phosphatase and tensin homolog (PTEN) acts to counteract PI3K signaling by dephosphorylating PIP3 to PIP2.89 Activated Akt phosphorylates the FOXO subfamily, which inhibits the transcription of several pro-apoptotic proteins, it can also inhibit apoptosis by phosphorylating and inactivating pro-apoptotic proteins such as BAD and GSK3.88,90 Other functions include the phosphorylation and degradation of the inhibitor of κB (IκB), and which results in increased nuclear factor kappa B (NF-κβ) activity and transcriptional stimulation of pro-survival genes,91 it also modulates MDM2, which inhibits P53 (an activator of cell-cycle arrest).92
Akt directly and indirectly leads to activation of mTOR which is present in two distinct complexes: mTORC1 and mTORC2. mTORC1 is composed of mTOR, Raptor, mLST8 and PRAS40. mTORC1 activates S6K1 and subsequently S6, resulting in increased cell proliferation and growth. It also leads to the inhibition of eIF4E binding protein 1 (4E-BP1), which allows the formation of eukaryotic initiation factor 4F (eIF4F) and protein translation.93 mTORC2 is composed of mTOR, Rictor, Sin1 and mLST8 and its role is less understood.93 It has been found that mTORC2 activates PKC, promoting its kinase activity.94 It is also thought that mTORC2 may take part in cell survival and cytoskeletal organization.95 mTOR has been shown to regulate hypoxia-inducible factor 1α (HIF1α), leading to downstream activation of vascular endothelial growth factor (VEGF) secretion and increased angiogenesis.96
The Ras/MAP/ERK pathway
This signaling pathway is activated by cell surface receptors and regulates the activity of many cellular factors involved in angiogenesis, cell proliferation, migration and survival (Figure 1). The activation of Ras protein by the exchange of GDP with GTP, results in the activation of MAP kinases that also activate downstream ERK via phosphorylation.97 This pathway is often activated in certain tumors by mutations in cytokine receptors such as Flt-3, Kit, Fms or by overexpression of wild-type or mutated receptors.98 Activation of the Ras/MAP/ERK pathway also leads to activation of HIF-1α, which promotes tumorigenesis and activation of VEGF.99
RTK signaling pathways in GBM pathogenesis
A large percentage of mutations and deletions in the RTK signaling pathways are evident in numerous cancers including GBM. The RTK/Ras/PI(3)K pathway was found to be altered in 86–90% of GBM cases studied.9,10 Combined activation of the Ras and AKT pathways has been shown to induce glioblastoma tumor formation in mice.100 The AKT signaling pathway plays a pivotal role in the progression of grade III anaplastic astrocytoma to grade IV glioblastoma multiforme. AKT expressing tumors appear to grow at a faster rate than non-AKT expressing tumors.101 Furthermore, inhibition of the PI3K/AKT pathway has been shown to inhibit the growth of GBM cells,102 further highlighting the importance of this pathway in GBM pathogenesis.
Inhibitors of the PI3K/AKT/mTOR signaling pathway are also affected in GBM. As an example, PTEN is mutated or deleted in approximately 36–44% of GBM cases.9,10,103 Loss of PTEN function has also been linked to immune evasion seen in GBM tumors, with mutations of PTEN being linked to increased expression of the immune suppressive checkpoint PD-L1.104 Another example is the tumor suppressor Neurofibromin 1 (NF1) that inhibits Ras.105 NF1 has a region that is highly homologous to the catalytic domain of Ras GTPase-activating protein (p120GAP), and consequently, it stimulates Ras GTPase, which leads to Ras bound GTP hydrolysis into GDP, and the inactivation of Ras activity.106 NF-1 is involved in the development of GBM as evidenced by the correlation between neurofibromatosis type-1 (a disease characterized by NF-1 mutation) and GBM occurrence.107
Targeting RTK signaling pathways in GBM
PI3K
Although therapies targeting PI3K in GBM have shown promising results in vitro and in vivo using xenograft models, their clinical efficacy remain to be tested and/or proven. PX-866 (Sonolisib) is an irreversible PI3K inhibiting drug that has been shown to inhibit angiogenesis and invasion of GBM cells in vitro. Although the drug did not induce apoptosis of GBM cells, it did cause cell cycle arrest.108 This drug was tested in a phase II trial for recurrent glioblastoma and was well tolerated but 73% of patients treated had disease progression.109 Other inhibitors such as XL765 (SAR245409) and GDC-0084, dual PI3K/mTOR inhibitors, have anti-GBM effects in vitro and in vivo but their efficacy in clinical trials must be tested and presented.110,111
mTOR
Several mTOR inhibitors have been trialed for GBM with differing results. As an example, Temsirolimus (CCI-779), Sirolimus (Rapamycin) and Everolimus (RAD001) are mTOR inhibitors that were shown to have little efficacy on GBM treatment. Temsirolimus failed to show efficacy for recurrent GBM in Phase II clinical trial.112 Sirolimus also had little efficacy for treatment of recurrent GBM patients even when combined with the EGFR tyrosine kinase inhibitor Erlotinib.113 Similarly, Everolimus did not convey a significant survival benefit when combined with temozolomide and radiotherapy in a phase II trial for newly diagnosed GBM patients.114 On the other hand, AZD2014 (Vistusertib), CC-223 (TORKi) and Palomid 529, which are dual mTORC1/mTORC2 inhibitors, have shown therapeutic promise. AZD2014 (Vistusertib) radiosensitized glioblastoma stem-like cells in vitro and in vivo.115 As a result of these promising preclinical results, participants are being recruited for a phase I/II clinical trial and from previously treated GBM patients (clinical trial ID: NCT02619864). CC-223 (TORKi) was found to exhibit anti-tumor effects in a murine xenograft model of GBM (utilizing U87MG cells)116 and Palomid 529 hindered GBM tumor growth in an orthotopic murine tumor model.117
RAS targeting by aminobisphosphonates: nanotech-based strategies
Ras is another valid therapeutic target for the treatment of GBM. Aminobisphosphonates are promising anti-cancer therapeutics, these drugs are thought to disrupt cancer proliferation, invasion, survival and pro-angiogenic activity by inhibiting the synthesis of farnesyl and geranyl lipidic residues, which in turn prevents protein isoprenylation. Ras is a farnesylated protein that it is inhibited by aminobisphosphonates, this inhibition prevents Ras GTPase activity and prevents downstream signaling.118 Zoledronic acid (ZOL) is an aminobisphosphonate that has anti-cancer effects, however it is mainly used to treat bone metastases as it accumulates in the bone, as a result novel methods are required to deliver this drug extra-skeletally.119 Nanotechnology can be utilized to help prevent bone accumulation of ZOL and ensure blood–brain barrier penetration of the drug. Salzano G et al. developed self-assembling nanoparticles that target transferrin receptors via incorporation of transferrin known as Tf-PLCaPZ. Tf-PLCaPZ encapsulates zolderonic acid and delivers it across the blood–brain barrier. Tf-PLCaPZ showed significant in vitro LN229 cell growth inhibition, Tf-PLCaPZ also showed anti-tumor activity in vivo in a U373MG xenograft model.120,121 These promising preclinical results make ZOL an exciting potential therapy for GBM.
The RAF serine/threonine kinase
Raf is a component of the Ras/Raf/MEK/ERK signaling pathway that can be targeted for GBM treatment (Figure 1). Sorafenib, a Raf kinase inhibitor, has been tested in combination with Erlotinib (an EGFR tyrosine kinase inhibitor) and in a phase II trial for patients with recurrent GBM (Table 2). This combinational therapy did not appear to have the desired beneficial effects, as it failed to reach the goal of a 30% improved survival time. It was postulated that this may be due to pharmacokinetic interaction between the drugs which reduces their efficacy.122
Protein kinase C (PKC)
Protein kinase C (PKC) family members regulate several cellular responses including gene expression, protein secretion, cell proliferation, and the inflammatory response. Tamoxifen is an inhibitor of PKC that has been tested as a therapeutic compound for GBM. In a phase I study, Tamoxifen was well tolerated but when combined with radiotherapy it did not appear to radiosensitize GBM tumors, as was observed in vitro.123 In a phase II trial combining high-dose Tamoxifen and radiotherapy, it was found that this molecule did not increase survival of patients.124 Worryingly, it was reported that high-dose tamoxifen treatment was linked with multifocal glioblastoma recurrence which mainly occurred in patients who responded to the Tamoxifen treatment.125 Enzastaurin is an inhibitor of the PKCβ and PI3K/AKT pathways that has been tested in phase I and phase II clinical trials. When compared with the alkylating chemotherapeutic drug Lomustine in a phase III trial for recurrent GBM, Enzastaurin did not display better efficacy.126
The tumor suppressor p53
The p53 pathway is altered in a large variety of cancers, with GBM being no exception. 87% of cases studied by the Cancer Genome Atlas Research Network had alteration of the p53 signaling pathway, with p53 being mutated or deleted in 28–35% of cases.9,10 The p53 protein pathway is involved in the activation of genes that are implicated in cell cycle arrest and apoptosis (Figure 2).127 Stress signals, such as DNA damage, hypoxia, heat shock and cold shock elicit a p53 response. These stress signals also result in the activation of mouse double minute 2 homolog (MDM2), a protein that degrades p53.128 The p53 protein activates p21 that inhibits Cdk4/Cyclin D and Cdk2/Cyclin E complexes and prevents their cell cycle progression.129 Upon p53 activation the transcription of Cyclin B is also reduced, preventing cell cycle progression.130 Due to the importance of p53 in GBM pathogenesis, a gene therapy approach has been used to restore p53 expression. SGT-53 is a nanocomplex that delivers wild-type p53 to tumor cells. It was shown to sensitize Temozolomide resistant tumor cells to treatment in vitro and in vivo.131 Introduction of wild-type p53 into Temozolomide resistant GBM cells resulted in a reduction of MGMT protein expression and this may explain the improved responsiveness to TMZ observed.131 In a phase I trial, intratumoral delivery of wild-type p53 gene using an adenovirus (Ad-p53) caused apoptosis of transfected tumor cells, indicating a beneficial anti-tumor effect.132

A schematic representation of the role of p53 and pRB signaling in response to stress signals. Black arrows indicate activation whereas red arrows indicate inhibition.
The tumor suppressor pRb
The pRB pathway suppresses cell cycle entry and progression via its interaction with the transcription factor E2F, leading to down regulation of genes involved in cell cycle progression.133–135 The pRB pathway was altered in 78–79% of GBM cases studied with RB gene deletion or mutation in 7.6–11% of cases.9,10 As a result therapies have been developed to reactivate pRb. PD0332991 (Palbocilib) is an inhibitor of Cdk4/6, that prevents the downstream inhibition of pRb (Figure 2). PD0332991 (Palbocilib) has been shown to inhibit the growth of intracranial GBM xenograft tumors.136
O6-methylguanine-DNA methyltransferase (MGMT)
MGMT is an enzyme that conveys a resistance to temozolomide chemotherapy (the standard chemotherapy of choice for GBM. MGMT acts alone to remove the methyl lesions caused by temozolomide.137 A single MGMT molecule removes the O6-methylation on guanine in a single step and transfers the methyl group from the oxygen in the DNA to a cysteine residue in the active site of MGMT.137 The binding of the methyl group irreversibly inactivates MGMT. Once the methyl group is bound MGMT is ubiquitinated and degraded by the proteasome.138 The repair of O6-methylation on guanine is biphasic with an initial fast repair phase followed by a slower phase caused by the depletion and subsequent synthesis of MGMT.138 O6-benzylguanine blocks the active site of MGMT, inactivating it and allowing methyl adducts to accumulate. In a preclinical model using xenotransplanted nude mice, the combination of O6-benzylguanine with temozolomide or carmustine (BCNU), amplified the anti-tumor effects of these chemotherapeutic agents.139 These promising preclinical results were unfortunately not replicated at the clinical trial phase. O6-benzylguanine did not re-sensitize temozolomide resistant patients to temozolomide chemotherapy as expected and as a result the drug was not investigated past the phase II trial stage.140,141
TGF-β signaling
TGF-β is a cytokine that exerts its effects on many cell types and is involved in the regulation of cell growth, immunity, cell death and cell adhesion.142 TGF-β binds to TGF-β receptor II (TGF-ΒRII) resulting in the formation of a heterodimer with the TGF-β receptor I (TGF-ΒRI) and leading to the phosphorylation of TGF-ΒRI.143 This event results in TGF-ΒRI the phosphorylation and activation of SMAD proteins. Once activated, the SMADs form complexes that in turn regulate the expression of target genes,143 it is important to note that TGF-β also signals via non-SMAD pathways.144 TGF-β signaling has been shown to facilitate Ras/Raf/MEK/ERK signaling via the increased GTP loading of Ras. TGF-β has also been shown to activate the PI3K/AKT/mTOR pathway.145 In healthy conditions TGF-β acts as a tumor suppressor, inhibiting proliferation, as a result mutations in the TGF-β signaling pathway, lead to an insensitivity to this cell growth prevention.143 Aberrant TGF-β signaling results in inflammation, invasion, metastasis, angiogenesis and immune escape. In GBM the TGF-β pathway is dysregulated and contributes to pathogenesis and progression.143 GBM cells have been shown to secrete TGF-β2 that also suppresses the anti-GBM immune response.146
AP12009 (Trabedersen) is a TGF-β2-specific antisense oligonucleotide, that when delivered using convection enhanced delivery (CED) resulted in a longer median overall survival (in phase I/II trial).147 SB-431542, LY2109761 and LY364947 (HTS466284) are inhibitors of the TGF-βR1 tyrosine kinase that have been tested in the GBM setting. SB-431542 has been shown to inhibit GBM cell growth, and motility in vitro.148 LY2109761 delivery in conjunction with radiotherapy improves GBM tumor responsiveness to radiotherapy in an orthotopic murine model.149 LY2109761 also has been shown to delay tumor growth in murine xenografts when used as a monotherapy and when combined with TMZ chemotherapy.149 LY364947 (HTS466284) has also been shown to increase the sensitivity of GBM cells to radiotherapy.150
Conclusions
Glioblastoma multiforme is an elusive disease with a dismal prognosis, and alternative therapies are required to improve the prognosis for patients. Genomic analyses of GBM uncovered several dysregulations of key cellular signaling pathways that constitute attractive targets for therapy. Targeting individual components of these pathways using small-molecule inhibitors and antibodies has provided varying levels of success in the treatment of GBM. Therefore, it may be more advantageous to target multiple elements of various signaling pathways, to eradicate GBM. It is also important to note that tumor cells are heterogeneous, and a targeting strategy that is aimed at multiple pathways would constitute a more efficient therapy. Many therapies also fail to have beneficial effects due to the blood–brain barrier and the presence of active efflux pumps that prevent drug entry into the brain. One such example of receptor tyrosine kinase inhibitors that have low brain penetration rates are Erlotinib and Gefitinib which have cerebrospinal fluid penetration rates as low as 2.8–4.4% and 1.1–1.3% respectively.151 The drug transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) have been shown to reduce brain penetration of Erlotinib explaining the relatively poor results seen in the GBM setting.152 Recent advances in nanoparticle delivery of drugs have enabled the delivery of drugs previously incapable of crossing the blood–brain barrier, reach the brain parenchyma and thus, enable effective targeting of intracranial tumors.153,154 The combination of focused ultrasound with microbubbles has also been shown to allow drugs to penetrate the blood–brain barrier.155–157 This technique may allow RTK inhibitors to cross the blood–brain barrier more efficiently and therefore enhance their effects. Immunotherapy may also be used as an alternative therapy with targeted immune cells crossing the blood–brain barrier. Numerous promising immunotherapies using peptide-targeted vaccines are entering clinical trials and preliminary results are proving to be beneficial for patients.158,159 Dendritic cell vaccines also showed encouraging results at the clinical trial stage.160 With these novel therapies comes hope for the future treatment of GBM.
References
Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan JS . Epidemiology of gliomas. In: Raizer J, Parsa A (eds). Current Understanding and Treatment of Gliomas 1st edn. Springer International Publishing: Switzerland, 2015, pp 1–14.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352: 987–996.
Wen PY, Kesari S . Malignant gliomas in adults. N Engl J Med 2008; 359: 492–507.
Ohgaki H . Epidemiology of brain tumors. Methods Mol Biol 2009; 472: 323–342.
Martinez R, Schackert G, Yaya-Tur R, Rojas-Marcos I, Herman JG, Esteller M . Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme. J Neurooncol 2007; 83: 91–93.
Ohgaki H, Kleihues P . The definition of primary and secondary glioblastoma. Clin Cancer Res 2013; 19: 764–772.
Waugh MG . Chromosomal instability and phosphoinositide pathway gene signatures in glioblastoma multiforme. Mol Neurobiol 2016; 53: 621–630.
von Deimling A, Louis DN, von Ammon K, Petersen I, Hoell T, Chung RY et al. Association of epidermal growth factor receptor gene amplification with loss of chromosome 10 in human glioblastoma multiforme. J Neurosurg 1992; 77: 295–301.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455: 1061–1068.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–477.
Desai R, Suryadevara CM, Batich KA, Farber SH, Sanchez-Perez L, Sampson JH . Emerging immunotherapies for glioblastoma. Expert Opin Emerg Drugs 2016; 21: 133–145.
Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev 2010; 24: 2205–2218.
Shapiro WR, Green SB, Burger PC, Mahaley Jr MS, Selker RG, VanGilder JC et al. Randomized trial of three chemotherapy regimens and two radiotherapy regimens and two radiotherapy regimens in postoperative treatment of malignant glioma. Brain Tumor Cooperative Group Trial 8001. J Neurosurg 1989; 71: 1–9.
Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN et al. PTEN mutations in gliomas and glioneuronal tumors. Oncogene 1998; 16: 2259–2264.
Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 2001; 93: 1246–1256.
Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M et al. Long-term survival with glioblastoma multiforme. Brain 2007; 130 (Pt 10): 2596–2606.
Regad T . Targeting RTK signaling pathways in cancer. Cancers (Basel) 2015; 7: 1758–1784.
Scaltriti M, Baselga J . The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 2006; 12: 5268–5272.
Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell 2003; 12: 541–552.
Roskoski R Jr. VEGF receptor protein-tyrosine kinases: structure and regulation. Biochem Biophys Res Commun 2008; 375: 287–291.
Nazarenko I, Hede SM, He X, Hedren A, Thompson J, Lindstrom MS et al. PDGF and PDGF receptors in glioma. Ups J Med Sci 2012; 117: 99–112.
Andrae J, Gallini R, Betsholtz C . Role of platelet-derived growth factors in physiology and medicine. Genes Dev 2008; 22: 1276–1312.
Ma PC, Maulik G, Christensen J, Salgia R . c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev 2003; 22: 309–325.
Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B . The IGF-I receptor in cell growth, transformation and apoptosis. Biochim Biophys Acta 1997; 1332: F105–F126.
Huang PH, Xu AM, White FM . Oncogenic EGFR signaling networks in glioma. Sci Signal 2009; 2: re6.
Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J Natl Cancer Inst 2015; 108.
Montano N, Cenci T, Martini M, D'Alessandris QG, Pelacchi F, Ricci-Vitiani L et al. Expression of EGFRvIII in glioblastoma: prognostic significance revisited. Neoplasia 2011; 13: 1113–1121.
Heimberger AB, Suki D, Yang D, Shi W, Aldape K . The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med 2005; 3: 38.
McMahon G . VEGF receptor signaling in tumor angiogenesis. Oncologist 2000; 5 (Suppl 1): 3–10.
Shibuya M . Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct 2001; 26: 25–35.
Steiner HH, Karcher S, Mueller MM, Nalbantis E, Kunze S, Herold-Mende C . Autocrine pathways of the vascular endothelial growth factor (VEGF) in glioblastoma multiforme: clinical relevance of radiation-induced increase of VEGF levels. J Neurooncol 2004; 66: 129–138.
Joensuu H, Puputti M, Sihto H, Tynninen O, Nupponen NN . Amplification of genes encoding KIT, PDGFRalpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol 2005; 207: 224–231.
Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA . Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res 2002; 62: 3729–3735.
Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 1992; 52: 3213–3219.
Abounader R, Laterra J . Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 2005; 7: 436–451.
Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Koh JS et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009; 115: 140–148.
Petterson SA, Dahlrot RH, Hermansen SK, K A Munthe S, Gundesen MT, Wohlleben H et al. High levels of c-Met is associated with poor prognosis in glioblastoma. J Neurooncol 2015; 122: 517–527.
Itoh N, Ornitz DM . Evolution of the Fgf and Fgfr gene families. Trends Genet 2004; 20: 563–569.
Loilome W, Joshi AD, ap Rhys CM, Piccirillo S, Vescovi AL, Gallia GL et al. Glioblastoma cell growth is suppressed by disruption of Fibroblast Growth Factor pathway signaling. J Neurooncol 2009; 94: 359–366.
Morrison RS, Yamaguchi F, Bruner JM, Tang M, McKeehan W, Berger MS . Fibroblast growth factor receptor gene expression and immunoreactivity are elevated in human glioblastoma multiforme. Cancer Res 1994; 54: 2794–2799.
Allerstorfer S, Sonvilla G, Fischer H, Spiegl-Kreinecker S, Gauglhofer C, Setinek U et al. FGF5 as an oncogenic factor in human glioblastoma multiforme: autocrine and paracrine activities. Oncogene 2008; 27: 4180–4190.
Sandberg-Nordqvist AC, Stahlbom PA, Reinecke M, Collins VP, von Holst H, Sara V . Characterization of insulin-like growth factor 1 in human primary brain tumors. Cancer Res 1993; 53: 2475–2478.
Schlenska-Lange A, Knupfer H, Lange TJ, Kiess W, Knupfer M . Cell proliferation and migration in glioblastoma multiforme cell lines are influenced by insulin-like growth factor I in vitro. Anticancer Res 2008; 28 (2A): 1055–1060.
Maris C, D'Haene N, Trepant AL, Le Mercier M, Sauvage S, Allard J et al. IGF-IR: a new prognostic biomarker for human glioblastoma. Br J Cancer 2015; 113: 729–737.
Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol 2009; 27: 579–584.
Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol 2010; 12: 95–103.
Stea B, Falsey R, Kislin K, Patel J, Glanzberg H, Carey S et al. Time and dose-dependent radiosensitization of the glioblastoma multiforme U251 cells by the EGF receptor tyrosine kinase inhibitor ZD1839 ('Iressa'). Cancer Lett 2003; 202: 43–51.
Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int J Radiat Oncol Biol Phys 2011; 80: 347–353.
Reardon DA, Conrad CA, Cloughesy T, Prados MD, Friedman HS, Aldape KD et al. Phase I study of AEE788, a novel multitarget inhibitor of ErbB- and VEGF-receptor-family tyrosine kinases, in recurrent glioblastoma patients. Cancer Chemother Pharmacol 2012; 69: 1507–1518.
Jane EP, Premkumar DR, Addo-Yobo SO, Pollack IF . Abrogation of mitogen-activated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J Pharmacol Exp Ther 2009; 331: 327–337.
Lee EQ, Kaley TJ, Duda DG, Schiff D, Lassman AB, Wong ET et al. A Multicenter, Phase II, randomized, noncomparative clinical trial of radiation and temozolomide with or without vandetanib in newly diagnosed glioblastoma patients. Clin Cancer Res 2015; 21: 3610–3618.
Thiessen B, Stewart C, Tsao M, Kamel-Reid S, Schaiquevich P, Mason W et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother Pharmacol 2010; 65: 353–361.
Schlaff CD, Arscott WT, Gordon I, Camphausen KA, Tandle A . Human EGFR-2, EGFR and HDAC triple-inhibitor CUDC-101 enhances radiosensitiviy of GBM cells. BRJ 2015; 2: 105–119.
Gerstner ER, Eichler AF, Plotkin SR, Drappatz J, Doyle CL, Xu L et al. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. J Neurooncol 2011; 103: 325–332.
Hainsworth JD, Ervin T, Friedman E, Priego V, Murphy PB, Clark BL et al. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer 2010; 116: 3663–3669.
Kalpathy-Cramer J, Chandra V, Da X, Ou Y, Emblem KE, Muzikansky A et al. Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J Neurooncol 2016; 131: 603–610.
Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 2013; 31: 3212–3218.
Kreisl TN, McNeill KA, Sul J, Iwamoto FM, Shih J, Fine HA . A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro Oncol 2012; 14: 1519–1526.
Balana C, Gil MJ, Perez P, Reynes G, Gallego O, Ribalta T et al. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: results of a phase II study. Target Oncol 2014; 9: 321–329.
Neyns B, Sadones J, Chaskis C, Dujardin M, Everaert H, Lv S et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol 2011; 103: 491–501.
Kilic T, Alberta JA, Zdunek PR, Acar M, Iannarelli P, O'Reilly T et al. Intracranial inhibition of platelet-derived growth factor-mediated glioblastoma cell growth by an orally active kinase inhibitor of the 2-phenylaminopyrimidine class. Cancer Res 2000; 60: 5143–5150.
Wen PY, Yung WK, Lamborn KR, Dahia PL, Wang Y, Peng B et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin Cancer Res 2006; 12: 4899–4907.
Li H, Zheng J, Guan R, Zhu Z, Yuan X . Tyrphostin AG 1296 induces glioblastoma cell apoptosis in vitro and in vivo. Oncol Lett 2015; 10: 3429–3433.
Batchelor TT, Gerstner ER, Ye X, Desideri S, Duda DG, Peereboom D et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-beta tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro Oncol 2016; 19: 567–575.
Norden AD, Schiff D, Ahluwalia MS, Lesser GJ, Nayak L, Lee EQ et al. Phase II trial of triple tyrosine kinase receptor inhibitor nintedanib in recurrent high-grade gliomas. J Neurooncol 2015; 121: 297–302.
Muhic A, Poulsen HS, Sorensen M, Grunnet K, Lassen U . Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. J Neurooncol 2013; 111: 205–212.
Reardon DA, Pan E, Fan J, Mink J, Barboriak DP, Vredenburgh JJ et al. 417PD A phase II trial of the multitargeted kinase inhibitor lenvatinib (E7080) in patients (pts) with recurrent glioblastoma (GBM) and disease progression following prior bevacizumab treatment. Ann Oncol 2012; 23 (Suppl 9): ix144–ix151.
Wen PY, Prados M, Schiff D, Reardon DA, Cloughesy T, Mikkelsen T et al. Phase II study of XL184(BMS 907351), an inhibitor of MET, VEGFR2, and RET, in patients (pts) with progressive glioblastoma (GB). J Clin Oncol 2010; 28 (15_suppl): 2006.
Zhang Y, Guessous F, Kofman A, Schiff D, Abounader R . XL-184, a MET, VEGFR-2 and RET kinase inhibitor for the treatment of thyroid cancer, glioblastoma multiforme and NSCLC. IDrugs 2010; 13: 112–121.
Knubel KH, Pernu BM, Sufit A, Nelson S, Pierce AM, Keating AK . MerTK inhibition is a novel therapeutic approach for glioblastoma multiforme. Oncotarget 2014; 5: 1338–1351.
Guessous F, Zhang Y, diPierro C, Marcinkiewicz L, Sarkaria J, Schiff D et al. An orally bioavailable c-Met kinase inhibitor potently inhibits brain tumor malignancy and growth. Anticancer Agents Med Chem 2010; 10: 28–35.
Zhou X, Zhao X, Li X, Ping G, Pei S, Chen M et al. PQ401, an IGF-1R inhibitor, induces apoptosis and inhibits growth, proliferation and migration of glioma cells. J Chemother 2016; 28: 44–49.
Zhou X, Shen F, Ma P, Hui H, Pei S, Chen M et al. GSK1838705A, an IGF-1R inhibitor, inhibits glioma cell proliferation and suppresses tumor growth in vivo. Mol Med Rep 2015; 12: 5641–5646.
Yin S, Girnita A, Stromberg T, Khan Z, Andersson S, Zheng H et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro Oncol 2010; 12: 19–27.
Premkumar DR, Jane EP, Pollack IF . Co-administration of NVP-AEW541 and dasatinib induces mitochondrial-mediated apoptosis through Bax activation in malignant human glioma cell lines. Int J Oncol 2010; 37: 633–643.
Zhou Q . BMS-536924, an ATP-competitive IGF-1R/IR inhibitor, decreases viability and migration of temozolomide-resistant glioma cells in vitro and suppresses tumor growth in vivo. Onco Targets Ther 2015; 8: 689–697.
Belda-Iniesta C, Carpeno Jde C, Saenz EC, Gutierrez M, Perona R, Baron MG . Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol Ther 2006; 5: 912–914.
Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol 2009; 20: 1596–1603.
Martens T, Schmidt NO, Eckerich C, Fillbrandt R, Merchant M, Schwall R et al. A novel one-armed anti-c-Met antibody inhibits glioblastoma growth in vivo. Clin Cancer Res 2006; 12 (20 Pt 1): 6144–6152.
Singh P, Alex JM, Bast F . Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol 2014; 31.
Yu YJ, Watts RJ . Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013; 10: 459–472.
Yu YJ, Atwal JK, Zhang Y, Tong RK, Wildsmith KR, Tan C et al. Therapeutic bispecific antibodies cross the blood-brain barrier in nonhuman primates. Sci Transl Med 2014; 6: 261ra154.
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014; 370: 699–708.
de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K et al. Phase II study of aflibercept in recurrent malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol 2011; 29: 2689–2695.
Giordano S . Rilotumumab, a mAb against human hepatocyte growth factor for the treatment of cancer. Curr Opin Mol Ther 2009; 11: 448–455.
Wen PY, Schiff D, Cloughesy TF, Raizer JJ, Laterra J, Smitt M et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro Oncol 2011; 13: 437–446.
Polivka Jr J, Janku F . Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther 2014; 142: 164–175.
LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA . Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat 2008; 11: 32–50.
Carracedo A, Pandolfi PP . The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 2008; 27: 5527–5541.
Fang X, Yu SX, Lu Y, Bast RC Jr, Woodgett JR, Mills GB . Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci USA 2000; 97: 11960–11965.
Bai D, Ueno L, Vogt PK . Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 2009; 125: 2863–2870.
Burris HA 3rd . Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol 2013; 71: 829–842.
Memmott RM, Dennis PA . Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal 2009; 21: 656–664.
Li X, Gao T . mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep 2014; 15: 191–198.
Yang G, Murashige DS, Humphrey SJ, James DE . A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep 2015; 12: 937–943.
Land SC, Tee AR . Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem 2007; 282: 20534–20543.
Kolch W . Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J 2000; 351 (Pt 2): 289–305.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 2007; 1773: 1263–1284.
Lim JH, Lee ES, You HJ, Lee JW, Park JW, Chun YS . Ras-dependent induction of HIF-1alpha785 via the Raf/MEK/ERK pathway: a novel mechanism of Ras-mediated tumor promotion. Oncogene 2004; 23: 9427–9431.
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN . Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 2000; 25: 55–57.
Sonoda Y, Ozawa T, Aldape KD, Deen DF, Berger MS, Pieper RO . Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res 2001; 61: 6674–6678.
Gallia GL, Tyler BM, Hann CL, Siu IM, Giranda VL, Vescovi AL et al. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol Cancer Ther 2009; 8: 386–393.
Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D et al. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 1997; 57: 4183–4186.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007; 13: 84–88.
Dasgupta B, Gutmann DH . Neurofibromatosis 1: closing the GAP between mice and men. Curr Opin Genet Dev 2003; 13: 20–27.
Yunoue S, Tokuo H, Fukunaga K, Feng L, Ozawa T, Nishi T et al. Neurofibromatosis type I tumor suppressor neurofibromin regulates neuronal differentiation via its GTPase-activating protein function toward Ras. J Biol Chem 2003; 278: 26958–26969.
Jeong TS, Yee GT . Glioblastoma in a patient with neurofibromatosis type 1: a case report and review of the literature. Brain Tumor Res Treat 2014; 2: 36–38.
Koul D, Shen R, Kim YW, Kondo Y, Lu Y, Bankson J et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol 2010; 12: 559–569.
Pitz MW, Eisenhauer EA, MacNeil MV, Thiessen B, Easaw JC, Macdonald DR et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro Oncol 2015; 17: 1270–1274.
Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA et al. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol 2011; 13: 384–392.
Heffron TP, Ndubaku CO, Salphati L, Alicke B, Cheong J, Drobnick J et al. Discovery of clinical development candidate GDC-0084, a brain penetrant inhibitor of PI3K and mTOR. ACS Med Chem Lett 2016; 7: 351–356.
Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs 2005; 23: 357–361.
Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE 2nd et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol 2010; 96: 219–230.
Ma DJ, Galanis E, Anderson SK, Schiff D, Kaufmann TJ, Peller PJ et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro Oncol 2015; 17: 1261–1269.
Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K et al. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol 2014; 16: 29–37.
Mortensen DS, Fultz KE, Hickman M, Khambatta G, Xu W, Perrin-Ninkovic SM et al. 337 The discovery and preclinical characterization of CC-223, a novel mTOR kinase inhibitor under clinical investigation. European Journal of Cancer 2012; 48 (suppl6): 103.
Lin F, Buil L, Sherris D, Beijnen JH, van Tellingen O . Dual mTORC1 and mTORC2 inhibitor Palomid 529 penetrates the blood-brain barrier without restriction by ABCB1 and ABCG2. Int J Cancer 2013; 133: 1222–1233.
Caraglia M, Santini D, Marra M, Vincenzi B, Tonini G, Budillon A . Emerging anti-cancer molecular mechanisms of aminobisphosphonates. Endocr Relat Cancer 2006; 13: 7–26.
Marra M, Salzano G, Leonetti C, Tassone P, Scarsella M, Zappavigna S et al. Nanotechnologies to use bisphosphonates as potent anticancer agents: the effects of zoledronic acid encapsulated into liposomes. Nanomedicine 2011; 7: 955–964.
Salzano G, Zappavigna S, Luce A, D'Onofrio N, Balestrieri ML, Grimaldi A et al. Transferrin-targeted nanoparticles containing zoledronic acid as a potential tool to inhibit glioblastoma growth. J Biomed Nanotechnol 2016; 12: 811–830.
Porru M, Zappavigna S, Salzano G, Luce A, Stoppacciaro A, Balestrieri ML et al. Medical treatment of orthotopic glioblastoma with transferrin-conjugated nanoparticles encapsulating zoledronic acid. Oncotarget 2014; 5: 10446–10459.
Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S et al. NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol 2013; 15: 490–496.
Muanza T, Shenouda G, Souhami L, Leblanc R, Mohr G, Corns R et al. High dose tamoxifen and radiotherapy in patients with glioblastoma multiforme: a phase IB study. Can J Neurol Sci 2000; 27: 302–306.
Robins HI, Won M, Seiferheld WF, Schultz CJ, Choucair AK, Brachman DG et al. Phase 2 trial of radiation plus high-dose tamoxifen for glioblastoma multiforme: RTOG protocol BR-0021. Neuro Oncol 2006; 8: 47–52.
Puchner MJ, Giese A, Lohmann F, Cristante L . High-dose tamoxifen treatment increases the incidence of multifocal tumor recurrences in glioblastoma patients. Anticancer Res 2004; 24: 4195–4203.
Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol 2010; 28: 1168–1174.
Harris SL, Levine AJ . The p53 pathway: positive and negative feedback loops. Oncogene 2005; 24: 2899–2908.
Levine AJ, Feng Z, Mak TW, You H, Jin S . Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev 2006; 20: 267–275.
He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene 2005; 24: 2929–2943.
Krause K, Wasner M, Reinhard W, Haugwitz U, Dohna CL, Mossner J et al. The tumor suppressor protein p53 can repress transcription of cyclin B. Nucleic Acids Res 2000; 28: 4410–4418.
Kim SS, Rait A, Kim E, Pirollo KF, Chang EH . A tumor-targeting p53 nanodelivery system limits chemoresistance to temozolomide prolonging survival in a mouse model of glioblastoma multiforme. Nanomedicine 2015; 11: 301–311.
Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol 2003; 21: 2508–2518.
Nevins JR . The Rb/E2F pathway and cancer. Hum Mol Genet 2001; 10: 699–703.
Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV et al. Aberrant signaling pathways in glioma. Cancers (Basel) 2011; 3: 3242–3278.
Munro S, Carr SM, La Thangue NB . Diversity within the pRb pathway: is there a code of conduct? Oncogene 2012; 31: 4343–4352.
Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 2010; 70: 3228–3238.
Gerson SL . MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 2004; 4: 296–307.
Kaina B, Christmann M, Naumann S, Roos WP . MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007; 6: 1079–1099.
Wedge SR, Newlands ES . O6-benzylguanine enhances the sensitivity of a glioma xenograft with low O6-alkylguanine-DNA alkyltransferase activity to temozolomide and BCNU. Br J Cancer 1996; 73: 1049–1052.
Quinn JA, Pluda J, Dolan ME, Delaney S, Kaplan R, Rich JN et al. Phase II trial of carmustine plus O(6)-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol 2002; 20: 2277–2283.
Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol 2009; 27: 1262–1267.
Rooke HM, Crosier KE . The smad proteins and TGFbeta signaling: uncovering a pathway critical in cancer. Pathology 2001; 33: 73–84.
Han J, Alvarez-Breckenridge CA, Wang QE, Yu J . TGF-beta signaling and its targeting for glioma treatment. Am J Cancer Res 2015; 5: 945–955.
Zhang YE . Non-Smad pathways in TGF-beta signaling. Cell Res 2009; 19: 128–139.
Lamouille S, Derynck R . Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178: 437–451.
Kuppner MC, Hamou MF, Bodmer S, Fontana A, de Tribolet N . The glioblastoma-derived T-cell suppressor factor/transforming growth factor beta 2 inhibits the generation of lymphokine-activated killer (LAK) cells. Int J Cancer 1988; 42: 562–567.
Hau P, Jachimczak P, Schlingensiepen R, Schulmeyer F, Jauch T, Steinbrecher A et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides 2007, Summer 17: 201–212.
Hjelmeland MD, Hjelmeland AB, Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ et al. SB-431542, a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther 2004; 3: 737–745.
Zhang M, Kleber S, Rohrich M, Timke C, Han N, Tuettenberg J et al. Blockade of TGF-beta signaling by the TGFbetaR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res 2011; 71: 7155–7167.
Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res 2012; 72: 4119–4129.
Baik CS, Chamberlain MC, Chow LQ . Targeted therapy for brain metastases in EGFR-mutated and ALK-rearranged non-small-cell lung cancer. J Thorac Oncol 2015; 10: 1268–1278.
de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O . Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs 2012; 30: 443–449.
Steiniger SC, Kreuter J, Khalansky AS, Skidan IN, Bobruskin AI, Smirnova ZS et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int J Cancer 2004; 109: 759–767.
Ren J, Shen S, Wang D, Xi Z, Guo L, Pang Z et al. The targeted delivery of anticancer drugs to brain glioma by PEGylated oxidized multi-walled carbon nanotubes modified with angiopep-2. Biomaterials 2012; 33: 3324–3333.
Liu HL, Hua MY, Chen PY, Chu PC, Pan CH, Yang HW et al. Blood-brain barrier disruption with focused ultrasound enhances delivery of chemotherapeutic drugs for glioblastoma treatment. Radiology 2010; 255: 415–425.
Ting CY, Fan CH, Liu HL, Huang CY, Hsieh HY, Yen TC et al. Concurrent blood-brain barrier opening and local drug delivery using drug-carrying microbubbles and focused ultrasound for brain glioma treatment. Biomaterials 2012; 33: 704–712.
Fan CH, Ting CY, Liu HL, Huang CY, Hsieh HY, Yen TC et al. Antiangiogenic-targeting drug-loaded microbubbles combined with focused ultrasound for glioma treatment. Biomaterials 2013; 34: 2142–2155.
Izumoto S, Tsuboi A, Oka Y, Suzuki T, Hashiba T, Kagawa N et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J Neurosurg 2008; 108: 963–971.
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 2015; 17: 854–861.
Cho DY, Yang WK, Lee HC, Hsu DM, Lin HL, Lin SZ et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial. World Neurosurg 2012; 77: 736–744.
Acknowledgements
This work was supported by the John and Lucille van Geest Foundation, the John van Geest Cancer Research Centre (Nottingham Trent University) and the Headcase cancer trust.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pearson, J., Regad, T. Targeting cellular pathways in glioblastoma multiforme. Sig Transduct Target Ther 2, 17040 (2017). https://doi.org/10.1038/sigtrans.2017.40
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/sigtrans.2017.40
This article is cited by
-
Targeting of REST with rationally-designed small molecule compounds exhibits synergetic therapeutic potential in human glioblastoma cells
BMC Biology (2024)
-
CD133 significance in glioblastoma development: in silico and in vitro study
European Journal of Medical Research (2024)
-
Decoding cancer insights: recent progress and strategies in proteomics for biomarker discovery
Journal of Proteins and Proteomics (2024)
-
Let-7b-5p promotes triptolide-induced growth-inhibiting effects in glioma by targeting IGF1R
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Dysregulated lipid metabolism in TMZ-resistant glioblastoma: pathways, proteins, metabolites and therapeutic opportunities
Lipids in Health and Disease (2023)
