
Abstract
MicroRNAs (miRNAs) are endogenous, small non-coding RNAs that function in regulation of gene expression. Compelling evidences have demonstrated that miRNA expression is dysregulated in human cancer through various mechanisms, including amplification or deletion of miRNA genes, abnormal transcriptional control of miRNAs, dysregulated epigenetic changes and defects in the miRNA biogenesis machinery. MiRNAs may function as either oncogenes or tumor suppressors under certain conditions. The dysregulated miRNAs have been shown to affect the hallmarks of cancer, including sustaining proliferative signaling, evading growth suppressors, resisting cell death, activating invasion and metastasis, and inducing angiogenesis. An increasing number of studies have identified miRNAs as potential biomarkers for human cancer diagnosis, prognosis and therapeutic targets or tools, which needs further investigation and validation. In this review, we focus on how miRNAs regulate the development of human tumors by acting as tumor suppressors or oncogenes.
Similar content being viewed by others

Introduction
MicroRNAs (miRNAs) are a family of small non-coding RNAs that regulate a wide array of biological processes including carcinogenesis. In cancer cells, miRNAs have been found to be heavily dysregulated.
The first miRNA, lin-4, was discovered in Caenorhabditis elegans (C. elegans) by Ambros and colleagues.1 It was identified as a small non-protein-coding RNA affecting development through regulating the expression of the protein lin-14. After 7 years, Reinhart et al.2 reported another miRNA in C. elegans, let-7, which negatively regulates the expression of the heterochronic gene lin-41 through sequence-specific RNA–RNA interactions with the 3′-untranslated regions of its mRNA. Subsequently, miRNAs were found to be abundant in both invertebrates and vertebrates by three independent groups in 2001, and some of the miRNAs are highly conserved, which suggest that miRNA-mediated post-transcriptional regulation is a general regulatory function across species.3–5 Currently, there are total 1872 annotated human miRNA precursor genes that are processed into ~2578 mature miRNA sequences (http://www.mirbase.org), while functions of many miRNAs are still unknown.
The earliest evidence of miRNA involvement in human cancer was provided by Dr Croce’s group from studies attempting to identify tumor suppressors at chromosome 13q14 region in B-cell chronic lymphocytic leukemia cells.6 They found this region, frequently deleted in B-cell chronic lymphocytic leukemia, actually contains two miRNA genes, miR-15a and miR-16-1. Both genes are deleted or downregulated in the majority of clinical chronic lymphocytic leukemia cases. Further study revealed that miR-15 and miR-16-1 acts as tumor suppressors to induce apoptosis by repressing Bcl-2, an anti-apoptotic protein overexpressed in malignant nondividing B cells and many solid malignancies.7,8 Most importantly, the deletion of miR-15 and miR-16-1 cluster in mice recapitulated chronic lymphocytic leukemia-associated phenotypes observed in humans, which convincingly demonstrated the critical role of these two miRNAs in tumor suppression.9 In the following years, miRNA profiling and deep sequencing provided direct evidences that miRNA expression is dysregulated in cancer and its signatures could be used for tumor classification, diagnosis and prognosis. In this review, we briefly described miRNA biogenesis and regulation. Furthermore, we elucidated the mechanisms by which miRNA expression is dysregulated in human cancer. We dissected how miRNAs are involved in traits that are the hallmarks of cancer, either as oncogenes or tumor suppressors. Finally we stated the miRNA potentials as biomarkers for cancer diagnosis, prognosis and treatment, and the challenges in miRNA researches and applications.
MiRNA biogenesis and regulation
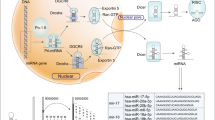
The miRNA biogenesis begins with transcribing gene into large primary transcript (pri-miRNA), which is 5′ capped and 3′ polyadenylated in structure. The transcription is typically mediated by RNA polymerase II, although some pre-miRNAs are generated by RNA polymerase III.10,11 The pri-miRNAs are then cleaved by a microprocessor complex, composed of RNA-binding protein DGCR8 and type III RNase Drosha, into a ~85-nucleotide stem–loop structure called precursor miRNA (pre-miRNA). Following transportation by Ran/GTP/Exportin 5 complex from nucleus to cytoplasm, the pre-miRNAs are processed by another RNase III enzyme Dicer to a ~20–22-nucleotide miRNA/miRNA* duplex (note: * indicates the passenger strand, while the other complementary stand is referred to as the mature or guide strand). After the duplex is unwound, the mature miRNA is incorporated into a protein complex termed RNA-induced silencing complex (RISC) and guides RISC to target mRNA (Figure 1).12

microRNA biogenesis. miRNA genes are usually transcribed by RNA polymerase II to produce the large primary transcripts termed pri-miRNAs, which are cleaved by a microprocessor complex, composed of RNA-binding protein DGCR8 and type III RNase Drosha, into an ~85-nucleotide stem–loop structure called pre-miRNA. Following transportation by Ran/GTP/Exportin 5 complex from nucleus to cytoplasm, the pre-miRNAs are processed by another RNase III enzyme Dicer to a ~20–22-nucleotide miRNA/miRNA* duplex. After the duplex is unwound, the mature miRNA is incorporated into a protein complex termed RISC. A miRNA-loaded RISC mediates gene silencing via mRNA cleavage and degradation, or translational repression depending on the complementarity between the miRNA and the targeted mRNA transcript. In addition, miRNAs may function as ligands to directly binding with Toll-like receptor (TLR), triggering downstream signaling pathways. Methyltransferase-like 3 (METTL3) is recently discovered to methylate pri-miRNAs, marking them for recognition and processing by DGCR8 to yield mature miRNA.
In many cases, miRNA–target interactions are mediated by the seed region, a 6- to 8-nucleotide-long fragment at the 5′-end of the miRNA that forms Watson–Crick pairs with the cognate target.13 However, Helwak et al.14 recently identified additional non-canonical binding clusters independent of seed region using an unbiased technique CLASH. Regardless of the interaction complexity, once binding to their target mRNAs, miRNAs cause translational repression if imperfect complementarity, or target mRNA degradation in case of perfect complementarity.15
Besides regulating gene expression via base pairing with cognate mRNA, miRNA was recently discovered to function as ligand to activate signaling pathways. Fabbri et al.16 first discovered that tumor cell-secreted miR-21/miR-29a directly bind to murine Toll-like receptor 7 or human Toll-like receptor 8 when transferred into immune cells, resulting in a Toll-like receptor-mediated prometastatic inflammatory response that may ultimately lead to tumor growth and metastasis. Similarly, miRNA was found to affect nuclear factor κB signaling pathway in natural killer cells through directly interaction with Toll-like receptor 1 as ligand.17
The biogenesis of miRNAs is under tight control at multiple levels, including the levels of miRNA transcription, processing by Drosha and Dicer, transportation, RISC binding and miRNA decay. For example, DEAD-box RNA helicases and SMAD protein are reported to be involved in Drosha-mediated miRNA maturation.18,19 KSRP, the KH-type splicing regulatory protein, serves as a component of both Drosha and Dicer complexes to regulate the biogenesis of a subset of miRNAs in mammalian cells.20 Methyltransferase-like 3 is recently discovered to function as miRNA biogenesis regulator through methylating pri-miRNAs, marking them for recognition and processing by DGCR8 to yield mature miRNA.21
Mechanisms of miRNA dysregulation in cancer
Over the past decade it has become clear that miRNA expression is dysregulated in human malignancies. The underling mechanisms include chromosomal abnormalities, transcriptional control changes, epigenetic changes and defects in the miRNA biogenesis machinery.
Amplification or deletion of miRNA genes
Abnormal miRNA expression in malignant cells compared with normal cells are often attributed to alterations in genomic miRNA copy numbers and gene locations (amplification, deletion or translocation). The earliest discovery of miRNA gene location change is the loss of miR-15a/16-1 cluster gene at chromosome 13q14, which is frequently observed in B-cell chronic lymphocytic leukemia patients.6 In lung cancer, the 5q33 region harboring miR-143 and miR-145 is often deleted, resulting in decreased expression of both miRNAs.22 Conversely, amplification of miR-17–92 cluster gene was observed in B-cell lymphomas23 and lung cancers,24 and translocation of this cluster gene was also observed in T-cell acute lymphoblastic leukemia,25 leading to overexpression of these miRNAs in these malignancies. The high frequency of genomic alterations in miRNA loci was confirmed by high-resolution array-based comparative genomic hybridization in 227 specimens from human ovarian cancer, breast cancer and melanoma.26 Further genome-wide investigations revealed that many miRNA genes are located in cancer-associated genomic regions. These regions could be a minimal region of loss of heterozygosity, which could harbor tumor suppressor gene; a minimal region of amplification, which might contain oncogenes; or fragile sites or common breakpoint regions.27 Overall, these findings suggest that abnormal miRNA expression in malignant cells could arise from amplification or deletion of specific genomic regions encompassing miRNA genes.
Transcriptional control of miRNAs
MiRNA expression is tightly controlled by different transcription factors, so abnormal expression of miRNA in cancer could be due to dysregulation of some key transcription factors, such as c-Myc and p53.
O’Donnell et al.28 discovered that c-Myc, frequently upregulated in many malignancies to regulate cell proliferation and apoptosis, activates the transcription of oncogenic miR-17–92 cluster through binding to E-box elements in miR-17–92 promoter. Consistent with its oncogenic role, c-Myc also represses transcriptional activity of tumor suppressive miRNAs such as mir-15a, miR-26, miR-29, mir-30 and let-7 families.29 Ghoshal’s group found the reciprocal regulation of c-Myc and tumor suppressor miR-122 in hepatocellular cancer. c-Myc represses miR-122 expression by associating with its promoter and miR-122 indirectly inhibits c-Myc transcription by targeting Tfdp2 and E2f1. Therefore, the disruption of this feedback loop between miR-122 and c-Myc is essential for hepatocellular cancer development.30 Another functional c-Myc-miRNA feedback loop is also dysregulated in hepatocellular cancer. c-Myc directly binds to the promoters of miR-148a-5p/miR-363-3p genes and represses their expression, inducing hepatocellular tumorigenesis by promoting G1 to S phase progression. In turn, miR-148a-5p directly targets and inhibits c-Myc expression, whereas miR-363-3p destabilizes c-Myc by directly targeting ubiquitin-specific protease 28.31
The p53-miR-34 regulatory axis is another example of how transcriptional factor regulates miRNA expression to mediate tumor suppressive function.32 The p53 is a tumor suppressor encoded by the gene TP53, one of the most commonly mutated genes in human cancers. p53-regulated expression of many genes, including miRNA genes, forming a complex p53 network to regulate cell-cycle progression and apoptosis. Similar to p53-mediated phenotypes, miR-34 family including miR-34a/b/c promotes cell-cycle arrest, cell senescence and apoptosis in cancer,33 implying p53 and miR-34 are in the same regulatory pathway. The hypothesis was verified by Oren lab34 and Mendell lab35, which demonstrated that p53 can induce the expression of miR-34a to trigger apoptosis through direct binding to the promoter of mir-34a gene. In turn, miR-34a promotes p53 expression by targeting SIRT1, a negative regulator of p53 via deacetylation.36 Further studies indicated that p53 performs its function through regulating the expression of a range of miRNAs, such as miR-605,37 miR-1246,38 and miR-107.39
In addition to c-Myc and p53, two most intensively studied transcriptional factors, more transcriptional factors have been found to regulate miRNA expression. For example, miR-223 is preferentially expressed in the hematopoietic system with crucial functions in myeloid lineage development, and its expression is repressed in multiple tumors including hepatocellular cancer and acute myeloid leukemia (AML).40–43 Fukao et al.44 found that the expression of miR-223 gene is driven by the myeloid transcription factors PU.1 and C/EBPs. Fazi et al.45 discovered that miR-223 and transcription factors NFI-A and C/EBPα form a minicircuitry to control human granulocytic differentiation. These two transcription factors compete for binding to the miR-223 promoter: NFI-A maintains miR-223 at low levels, whereas the retinoic acid-induced C/EBPα replaces NFI-A to upregulate miR-223 expression. Therefore, miRNA expression is finely tuned by multiple factors to maintain normal transcription, and its dysregulation leads to tumorigenesis.
Dysregulated epigenetics change
The epigenetic alteration is a well-known feature in cancer, including global genomic DNA hypomethylation, aberrant DNA hypermethylation of tumor suppressor genes and disruption of the histone modification patterns. It is believed that miRNAs, similar to protein-coding genes, are also susceptible to epigenetic modulation.46,47 For instance, Fazi et al.48 has discovered that miR-223 expression was epigenetically silenced by AML1/ETO, a most common AML-associated fusion protein, through CpG methylation. The investigation by Saito et al.49 showed that 17 out of 313 human miRNAs are upregulated more than threefolds in T24 bladder cancer cells after simultaneous treatment with DNA methylation and histone acetylation inhibitors. Among these miRNAs, miR-127, embedded in a CpG island and lacks expression in cancer cells, had significantly elevated expression after the treatment, which was accompanied by the downregulation of proto-oncogene BCL6. These results indicate that DNA demethylation and histone deacetylase inhibition can activate the expression of miRNAs that may act as tumor suppressors. Using similar approach, Lujambio et al.50 discovered miR-148a, and miR-34b/c cluster is subject to specific hypermethylation-associated silencing in cancer cells. Moreover, restoration of these miRNAs in cancer cells inhibited their motility, reduced tumor growth and inhibited metastasis formation in vivo. Similarly, decreased expressions of miR-9-1, miR-124a and miR-145-5p are attributed to DNA hypermethylation in breast, lung and colon carcinomas, respectively.51–53 The above evidences highlighted the role of epigenetic regulation in miRNA expression during tumorigenesis, implying that aberrant DNA methylation and histone acetylation of miRNA genes could be served as useful biomarkers for cancer diagnosis and prognosis.
Defects in miRNA biogenesis machinery
As described above, miRNA biogenesis is elaborately controlled by several enzymes and regulatory proteins, such as Drosha, Dicer, DGCR8, Argonaute proteins and exprotin 5, allowing correct miRNA maturation from primary miRNA precursors. Therefore, mutation or aberrant expression of any component of the miRNA biogenesis machinery could lead to abnormal expression of miRNAs.
Drosha and Dicer are two key RNase III endonucleases in miRNA maturation, responsible for forming pre-miRNA and miRNA duplex. Recent researches showed that both enzymes are dysregulated in certain tumors. Thomson et al.54 found that a large fraction of miRNAs is regulated at the Drosha-processing step, and this regulation has a major impact on miRNA expression during embryonic development and in cancer. Walz et al.55 reported that DGCR8 and Drosha have single-nucleotide substitution/deletion mutations in 15% of 534 Wilms’ tumors, leading to significantly decreased expression of mature Let-7a and miR-200 family. Regarding to Dicer dysregulation, it was observed that Dicer1 impairment in colorectal cancer (CRC) cells induces the acquisition of a greater capacity for tumor initiation and metastasis.56 Moreover, high Dicer and Drosha mRNA levels in ovarian cancer are associated with increased median survival,57 and reversely, decreased Dicer expression significantly correlates with reduced patient survival.58,59 The positive correlation between lower Dicer mRNA levels and reduced let-7 expression with unfavorable postoperative survival was also discovered by Karube et al.60 in lung cancer patients.
Argonaute proteins are essential catalytic components of RISC and have a central role in RNA-silencing processes. Similar to Dicer and Drosha, dysregulation of Argonaute proteins also occurs in cancer. For example, human EIF2C1/hAgo1 gene is often lost in Wilms’ tumors of the kidney.61 The expression of human argonaute proteins (AGO) is regulated in a cell-dependent manner. For instance, AGO2 expression levels in primary gastric cancer and corresponding lymph node metastases are significantly higher than that in healthy controls,62 whereas AGO2 expression is lower, corresponding reduced RNAi efficiency, in melanoma compared with primary melanocytes.63
Exportin 5 (XPO5) is a double-stranded RNA-binding protein that mediates nuclear export of pre-miRNA into the cytoplasm. Melo et al. found that XPO5 gene has inactivating mutations in a subset of human tumors with microsatellite instability. In CRC cells HCT-15 and DLD-1, the insertion of an ‘‘A’’ in exon 32 generates a premature termination codon, resulting in frameshift mutation and production of truncated version of the protein. This truncated XPO5 loses the function to export pre-miRNAs. Pre-miRNAs are therefore trapped in the nucleus, resulting in reduced miRNA processing. Most importantly, the restoration of XPO5 functions reverses the impaired export of pre-miRNAs and has tumor suppressor features.64 In hepatocellular carcinoma, we also observed the failure of XPO5 to transport pre-miRNAs from nucleus to cytoplasm, which was caused by ERK kinase to phosphorylate XPO5 (unpublished data).
Significance of the altered miRNA expression in tumors
Hanahan and Weinberg65 have proposed that the hallmarks of human cancer comprise six biological capabilities acquired during tumor development, including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, activating invasion and metastasis and inducing angiogenesis. Given that abnormal miRNA expression in tumors, it is believed that the dysregulated miRNAs could affect one or several of the cancer hallmarks for tumor initiation and progression. Depending on their target genes, miRNA could function as either oncogene or tumor suppressor under certain circumstances.
Evading growth suppressors and sustaining proliferative signaling
Cell proliferation is the most important hallmark of cancer and its abnormality is the leading cause of tumorigenesis. In details, cell-cycle progression is controlled by intracellular programs and extracellular signal molecules, to reach the balance between promoting cell proliferation and suppressing it. Cells become cancerous when cell growth or division is out of control. Over the years of studies, it becomes apparent that some miRNAs functionally integrate into multiple critical cell proliferation pathways, and the dysregulation of these miRNAs is responsible for evading growth suppressors and sustaining proliferative signaling in cancer cells.
The E2F proteins, a family of transcription factors, are critical regulators of cell proliferation in a cell-cycle-dependent manner. A series of studies have demonstrated that miRNAs participate in regulation of E2F expression. The E2F member E2F1 induces target gene transcription during the G1 to S transition,66 and is defined as a tumor suppressor because the E2F1-deficient mice developed a wide variety of cancers. O’Donnell et al.28 showed that miR-17–92 inhibits E2F1 translation after being activated by c-Myc. Considering that c-Myc also directly induces E2F1 expression, miR-17–92 cluster may act as a brake on this possible positive feedback loop to ensure that E2F1 protein levels do not rise precipitously in response to c-myc activation.67 The miR-17–92 cluster was also found to regulate E2F2 and E2F3 translation,68 and the E2F transcription factors can in turn induce the expression of the miR-17–92 cluster.69 Therefore, the feedback system between miR-17–92 cluster and E2F provides a mechanism to keep regular cell-cycle progression under normal conditions. However, overexpression of miR-17–92, which is common among several tumors, disrupts the feedback loop to promote cell proliferation.70
Cell-cycle progression depends on different cyclins, cyclin-dependent kinases (Cdks) and their inhibitors, which are widely regulated by miRNAs. Hatfield et al.71 provided the first evidence that Drosophila germline stem cells with Dicer-1 knockout are blocked in the G1/S transition, suggesting that miRNAs are required for germline stem cells to pass the normal G1/S checkpoint. Moreover, Dicer-deficient germline stem cells exhibited increased expression of Dacapo, a member of the p21/p27 family of Cdk inhibitors, implying that this protein is negatively regulated by miRNAs to promote cell-cycle progression. Indeed, miR-221/222 has been identified to directly target the Cdk inhibitor p27Kip1 in glioblastoma cells,72 which was further confirmed in other cancer cell lines and primary tumor samples.73–75 Ectopic expression of miR-221/222 accelerated cell proliferation, whereas their suppression induced G1 cell-cycle arrest in cancer cells. Moreover, miR-221/222 expression has been found to be upregulated in a variety of human tumors, demonstrating that miR-221/222 regulation of p27Kip1 is a bona fide oncogenic pathway. Similar to p27Kip1, p21CIP1 and p16INK4a are also regulated by miRNAs such as miR-663, miR-302 family and miR-24.76,77 miR-663 was found to be upregulated in nasopharyngeal carcinoma, and acts as oncogene to promote the cellular G1/S transition in vitro and in vivo by directly targeting p21CIP1. Therefore, the miR-663/p21CIP1 axis clarifies the molecular mechanism of nasopharyngeal carcinoma cell proliferation.78 In addition to affect the expression of Cdk inhibitors, miRNAs are also regulators for expression of Cdk and cyclin. For examples, miRNA-545 leads to cell-cycle arrest in lung cancer cells by repressing expression of cyclin D1 and CDK4.79
MiRNAs take part in cell proliferation, not only through targeting cell-cycle components but also by extensively regulating multiple signaling pathways. For example, miR-486, significantly downregulated in non-small-cell lung cancer, was found to affect cell proliferation and migration through insulin growth receptor (IGF) and PI3K signaling pathways by targeting IGF1, IGF1R and p85α.80
Resisting cell death
Evasion of apoptosis is another significant hallmark of tumor progression, which is believed to be regulated by miRNAs.81,82 Tumor cells evolve a variety of strategies to limit or circumvent apoptosis. Among them, the loss of p53 tumor suppressor function is most common. The alternative ways to evade apoptosis include upregulation of anti-apoptotic regulators, suppression of proapoptotic factors and inhibition of death pathway induced by extrinsic ligands. The components involved in anti-apoptosis are broadly inhibited or activated by miRNAs.
A number of p53-regulated miRNAs have been identified to be involved in p53 functions, and some of these miRNAs can modulate p53 level and activity in a feedback fashion. For example, Pichiorri et al.83 identified that in multiple myeloma, three miRNAs (miR-192, miR-194 and miR-215) are transcriptionally activated by p53 to suppress Mdm2 expression via directly binding to its mRNA, thereby protecting p53 from degradation. These miRNAs are positive regulators of p53 and their downregulation has a key role in multiple myeloma development. There is another negative feedback regulation, which occurs between miR-122 and p53. MiR-122 promotes p53 activity via targeting cyclin G184 and cytoplasmic polyadenylation element-binding protein,85 which increases cell sensitivity to the drug doxorubicin, establishing a basis toward the development of combined chemo- and miRNA-based therapy for hepatocellular carcinoma.
Dysregulation of other p53-regulated miRNAs also confers cancer cells resistant to apoptosis. For example, miR-17–92 cluster is a novel target for p53-mediated transcriptional repression under hypoxia. Its downregulation sensitizes cells to hypoxia-induced apoptosis, whereas its overexpression inhibits apoptosis. Therefore, tumor cells with increased miR-17–92 expression may escape hypoxia-induced apoptosis.86 All the above results showed that p53 and its regulated miRNAs form a network to elaborately determine cell fate under normal conditions. However, cancer cells with dysregulated p53 or its target miRNAs could have capacity to resisting cell death.
Anti-apoptotic regulators (Bcl-2 and Bcl-xL) and proapoptotic factors (Bax, Bim and Puma) are potential targets of some miRNAs, which have important role in cell death. As discussed above, miR-15a and miR-16-1 are significantly downregulated in chronic lymphocytic leukemia and their expression inversely correlates with Bcl-2 expression. A further study demonstrated that these two miRNAs repress Bcl-2 expression and induce apoptosis. Bcl-2 was also regulated by other miRNAs, such as miR-204,87 miR-148a88 and miR-365.89 Denoyelle et al.90 found that miR-491-5p efficiently induces apoptosis in ovarian cancer cells by directly inhibiting Bcl-xL expression and by inducing Bim accumulation. MiR-221/222 inhibit cell apoptosis by targeting the proapoptotic gene PUMA in human glioma cells. And the knockdown of miR-221/222 induces PUMA expression and cell apoptosis, suggesting that miR-221/222 could be potential therapeutic targets for glioblastoma intervention.91
MiRNAs are also involved in resisting cell death by regulating components of extrinsic apoptotic pathway, such as the Fas ligand/Fas receptor. MiR-21, frequently upregulated in a variety of cancers, exerts an anti-apoptotic function in k-Ras-dependent lung tumors by inhibiting expression of Apaf-1, an important component of the intrinsic mitochondrial apoptotic pathway, and decreasing protein levels of Fas ligand, a key initiator of the extrinsic apoptotic pathway.92 The function of miR-21 was further confirmed by the observation that ectopic expression of miR-21 protected cancer cells from gemcitabine-induced apoptosis.93 Shaffiey et al.94 identified that miR-590 suppresses Fas ligand expression in AML to promote cell survival. Besides modulating ligand expression, dysregulated miRNAs also resist cell death via regulating the expression of death receptors. For example, Razumilava et al.95 found that miR-25, overexpressed in malignant cholangiocarcinoma cells, is able to protect cells against TNF-related apoptosis-inducing ligand-induced apoptosis by targeting death receptor-4 (DR4).
Activating invasion and metastasis
Metastasis is a complex, multistep and dynamic biological event. Epithelial–mesenchymal transition (EMT) is considered an early and key step in the metastatic cascade, characterized by loss of cell adhesion through repression of E-cadherin and activation of genes associated with motility and invasion. EMT is thought to be regulated by a variety of signaling pathways such as transforming growth factor (TGF)-β, all of which converge on the key transcription factors such as ZEB, SNAIL and TWIST.96
Growing evidences show that miRNAs have an important role in EMT and cancer metastasis. TGF-β-regulated miRNAs were found to engage in TGF-β signaling to induce EMT and facilitate metastasis in advanced malignancy. MiR-155 is one of the miRNAs involved in this regulation process. It is overexpressed in several malignancies and transcriptionally activated by TGF-β/SMAD4 signaling. Mechanistic studies revealed that miR-155 promote EMT by targeting RhoA GTPase, an important regulator of cellular polarity and tight junction formation and stability. The knockdown of miR-155 suppresses TGF-β-induced EMT and tight junction dissolution, as well as cell migration and invasion.97 In contrast to miR-155, miR-200 and miR-203 are inhibited by TGF-β. The miR-200 family was shown to affect EMT by inhibiting the expression of E-cadherin transcriptional repressors ZEB1 and ZEB2.98 In turn, the miR-200 primary transcript is also repressed by ZEB1 and ZEB2,99 forming a double-negative feedback loop between ZEB1/ZEB2 and miR-200 family. This loop was proposed to explain a central dilemma in our understanding of the metastatic cascade: miR-200 expression is significantly downregulated in invasive breast cancer cells with increased metastatic potential that convey a mesenchymal phenotype. Therefore, enforced overexpression of miR-200c in the mesenchymal cells increases expression of E-cadherin and promotes an epithelial phenotype by inducing MET.100,101 In addition, p53-regulated miRNAs miR-200 and miR-192 are critical mediators of p53-regulated EMT, supported by the observation that these miRNAs are transactivated by p53 and modulate EMT program via repressing ZEB1/2 expression.102,103
TWIST and SNAIL are the other two key transcription factors to promote epithelial motility, invasiveness and metastasis through regulating the expression of certain miRNAs. For example, miR-10b is highly expressed in metastatic breast cancer cells and positively regulates cell migration and invasion, which is induced by direct binding of TWIST to the putative promoter of miR-10b gene. Moreover, ectopic expression of miR-10b in non-metastatic SUM149 and SUM159 human breast cancer cell lines does induce aggressive invasion and micrometastasis formation in severe combined immunodeficiency mouse models, providing experimental validation that overexpression of individual miRNAs can contribute to metastasis formation in vivo.104 In addition, miRNAs regulating the expression of these EMT factors are also critical for controlling metastasis. For instance, miR-203 is significantly downregulated because of hypermethylation of its promoter in highly metastatic breast cancer cells. The restoration of miR-203 in breast cancer cells inhibits tumor cell invasion in vitro and lung metastatic colonization in vivo by repressing SNAI2, suggesting that the SNAI2 and miR-203 regulatory loop has an important role in EMT and tumor metastasis.105,106
Other important miRNAs involved in regulating metastasis include miR-9 and miR-212. MiR-9 expression is activated by c-Myc and n-Myc, both of which directly bind to the miR-9-3 locus. The expression level of miR-9 closely correlates with MYCN amplification, tumor grade and metastatic status in neuroblastoma tumors. In primary breast tumors of patients with metastatic disease, miR-9 expression is much higher than that in metastasis-free patients, implying that miR-9 is a potential regulator of the metastatic process. Ma et al. identified that miR-9 reduces the expression of E-cadherin in breast cancer cells via directly binding to its 3′-untranslate region. The consequence of the E-cadherin downregulation by miR-9 is the activation of β-catenin signaling to trigger the expression of downstream oncogenic genes, which leads to increased cell motility and invasiveness. The function of miR-9 is further confirmed by the fact that inhibition of miR-9 using a miRNA ‘sponge’ suppresses metastasis formation in animal model, implying that miR-9 silencing may represent a new therapeutic approach in advanced breast cancers to prevent metastasis formation.107,108 MiR-212 is significantly downregulated in human CRC tissues due to both promoter hypermethylation and loss of heterozygosity. Overexpression of miR-212 inhibits CRC cell migration and invasion in vitro and pulmonary metastasis in vivo by targeting expression of MnSOD, which is required for downregulation of epithelial markers and upregulation of mesenchymal markers in CRC cells. Therefore, miR-212 could be a prognostic marker for CRC patients to predict their survival, and both miR-212 and MnSOD might also be therapeutic targets for cancer.109
Inducing angiogenesis
Angiogenesis is a highly coordinated process to develop new blood vessels from pre-existing ones to satisfy the needs for food and oxygen in tumor growth and metastasis.110 As tumor tissues have significantly lower oxygen concentration than the surrounding normal tissues, hypoxia has a critical role in the tumor microenvironment by allowing the development and maintenance of cancer cells. Hypoxia-inducible factor (HIF) is a key transcription factor in response to hypoxia, which influences the expression of a number of genes, including miRNAs. Vascular endothelial growth factor (VEGF) is a pivotal angiogenic factor, directing endothelial cells to build new vessels upon binding to its receptors.111 Therefore, miRNAs that target HIF or VEGF signaling pathways are likely to have significant impact on the angiogenesis. It is now well documented that the process of angiogenesis is elaborately regulated by miRNAs, some of which are described in detail below.
MiR-210 is the most consistently and significantly induced miRNA during hypoxia.112 Two independent studies demonstrated that miR-210 overexpression in normoxic human umbilical vein endothelial cells stimulates the formation of capillary-like structures and VEGF-dependent cell migration. In contrast, miR-210 blockade antagonizes these processes.113,114 Furthermore, miR-210 promotes angiogenesis not only by targeting the receptor tyrosine kinase ligand ephrin-A3, which is an anti-angiogenic factor,113 but also by enhancing the expression of VEGF and VEGF receptor-2 (VEGFR2).115
MiR-424 is induced by hypoxia in endothelial cells to promote angiogenesis in vitro and in vivo by targeting cullin 2, a scaffold protein for ubiquitin ligase. This process stabilizes HIF1α and allows it to transcriptionally activate VEGF expression.116 Another miRNA that induces angiogenesis is miR-21. It targets PTEN to activate the downstream Akt/ERK signaling pathways, leading to high expression of HIF1α and VEGF.117 In contrast, miR-20b and miR-519c negatively regulate angiogenesis by targeting VEGF and/or HIF1α.118,119 Besides regulating HIF1α, miR-107 was able to inhibit the expression of HIF1β, so downregulation of miR-107 promotes tumor angiogenesis under hypoxic conditions.120
Recent researches have demonstrated that exosomal miRNA from cancer cells could help modulate the tumor microenvironment. One of the evidences was provided by Umezu et al. They observed that miR-135b, which is overexpressed in exosomes from hypoxia-resistant multiple myeloma cells, suppresses factor-inhibiting HIF1 (FIH-1) in endothelial cells, thus promoting endothelial tube formation via the HIF–FIH signaling pathway. Therefore, exosomal miR-135b may be a target for controlling multiple myeloma angiogenesis.121
Conclusions and future challenges
Since the discovery of miR-15a and miR-16-1 deletions in chronic lymphocytic leukemia, many laboratories around the world have demonstrated the expression of miRNAs is dysregulated in different tumors. Such dysregulation could be caused by multiple mechanisms, including amplification or deletion of miRNA genes, abnormal transcriptional control of miRNAs, dysregulated epigenetic changes and defects in the miRNA biogenesis machinery. Cancer cells with abnormal miRNA expression evolve the capability to sustain proliferative signaling, evade growth suppressors, resist cell death, activate invasion and metastasis and induce angiogenesis. MiRNA may function as either tumor suppressor or oncogene under certain circumstances. Although miRNAs have multiple targets, their function in tumorigenesis could be due to their regulation of a few specific targets. Therefore, a future challenge will be to identify the critical targets of the miRNAs involved in cancer and establish their contribution to malignant transformation.
Genome-wide profiling demonstrates that miRNA expression signatures are associated with tumor type, tumor grade and clinical outcomes, so miRNAs could be potential candidates for diagnostic biomarkers, prognostic biomarkers, therapeutic targets or tools. However, more efforts are still needed to screen miRNA candidates by deep sequencing and validate them as diagnostic and prognostic biomarkers in a large cohort of patient samples. For the development of miRNA therapeutic strategies, the following issues should be addressed: the validation of the targets and the accurate prevision of the putative unwanted off-target effects; and the development of efficient and specific miRNA delivery system.
Recent researches focus on identification of miRNAs in the secreted exosomes of the clinical samples. Owing to their good stability, higher specificity and sensitivity, exosomal miRNAs could be potential biomarkers for clinical applications. However, exosomal miRNA biomarkers are still in the early discovery/development stage and their potential value in clinical diagnostics waits to be fully explored. It is for sure that further researches into identifying the novel miRNAs, their biological functions and their target genes will boost our knowledge of the miRNA roles in tumorigenesis and warrant the development of miRNA-related cancer prognosis, diagnosis and treatment.
References
Lee RC, Feinbaum RL, Ambros V . The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993; 75: 843–854.
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000; 403: 901–906.
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T . Identification of novel genes coding for small expressed RNAs. Science 2001; 294: 853–858.
Lau NC, Lim LP, Weinstein EG, Bartel DP . An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001; 294: 858–862.
Lee RC, Ambros V . An extensive class of small RNAs in Caenorhabditis elegans. Science 2001; 294: 862–864.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA 2002; 99: 15524–15529.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102: 13944–13949.
Calin GA, Cimmino A, Fabbri M, Ferracin M, Wojcik SE, Shimizu M et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc Natl Acad Sci USA 2008; 105: 5166–5171.
Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010; 17: 28–40.
Borchert GM, Lanier W, Davidson BL . RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 2006; 13: 1097–1101.
Lee Y, Kim M, Han J, Yeom KH, Lee S et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23: 4051–4060.
Macfarlane LA, Murphy PR . MicroRNA: biogenesis, function and role in cancer. Curr Genomics 2010; 11: 537–561.
Bartel DP . MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215–233.
Helwak A, Kudla G, Dudnakova T, Tollervey D . Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013; 153: 654–665.
Vasudevan S, Tong Y, Steitz JA . Switching from repression to activation: microRNAs can up-regulate translation. Science 2007; 318: 1931–1934.
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA 2012; 109: E2110–E2116.
He S, Chu J, Wu LC, Mao H, Peng Y, Alvarez-Breckenridge CA et al. MicroRNAs activate natural killer cells through Toll-like receptor signaling. Blood 2013; 121: 4663–4671.
Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol 2007; 9: 604–611.
Davis BN, Hilyard AC, Lagna G, Hata A . SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008; 454: 56–61.
Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A et al. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 2009; 459: 1010–1014.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF . N6-methyladenosine marks primary microRNAs for processing. Nature 2015; 519: 482–485.
Calin GA, Croce CM . MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene 2006; 25: 6202–6210.
Tagawa H, Seto M . A microRNA cluster as a target of genomic amplification in malignant lymphoma. Leukemia 2005; 19: 2013–2016.
Hayashita Y, Osada H, Tatematsu Y, Yamada H, Yanagisawa K, Tomida S et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65: 9628–9632.
Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol 2010; 12: 372–379.
Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A et al. MicroRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA 2006; 103: 9136–9141.
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA 2004; 101: 2999–3004.
O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT . c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005; 435: 839–843.
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 2008; 40: 43–50.
Wang B, Hsu SH, Wang X, Kutay H, Bid HK, Yu J et al. Reciprocal regulation of microRNA-122 and c-Myc in hepatocellular cancer: role of E2F1 and transcription factor dimerization partner 2. Hepatology 2014; 59: 555–566.
Han H, Sun D, Li W, Shen H, Zhu Y, Li C et al. c-Myc-MicroRNA functional feedback loop affects hepatocarcinogenesis. Hepatology 2013; 57: 2378–2389.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134.
Hermeking H . The miR-34 family in cancer and apoptosis. Cell Death Differ 2010; 17: 193–199.
Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26: 731–743.
Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752.
Yamakuchi M, Lowenstein CJ . MiR-34, SIRT1, and p53: The feedback loop. Cell Cycle 2009; 8: 712–715.
Xiao J, Lin H, Luo X, Luo X, Wang Z . miR-605 joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J 2011; 30: 524–532.
Zhang Y, Liao JM, Zeng SX, Lu H . p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep 2011; 12: 811–817.
Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT et al. p53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci USA 2010; 107: 6334–6339.
Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008; 451: 1125–1129.
Wong QW, Lung RW, Law PT, Lai PB, Chan KY, To KF et al. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology 2008; 135: 257–269.
Eyholzer M, Schmid S, Schardt JA, Haefliger S, Mueller BU, Pabst T . Complexity of miR-223 regulation by CEBPA in human AML. Leuk Res 2010; 34: 672–676.
Stamatopoulos B, Meuleman N, Haibe-Kains B, Saussoy P, Van Den Neste E et al. microRNA-29c and microRNA-223 down-regulation has in vivo significance in chronic lymphocytic leukemia and improves disease risk stratification. Blood 2009; 113: 5237–5245.
Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y et al. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 2007; 129: 617–631.
Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 2005; 123: 819–831.
Han L, Witmer PD, Casey E, Valle D, Sukumar S . DNA methylation regulates MicroRNA expression. Cancer Biol Ther 2007; 6: 1284–1288.
Saito Y, Jones PA . Epigenetic activation of tumor suppressor microRNAs in human cancer cells. Cell Cycle 2006; 5: 2220–2222.
Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 2007; 12: 457–466.
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–443.
Lujambio A, Calin GA, Villanueva A, Ropero S, Sanchez-Cespedes M, Blanco D et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 2008; 105: 13556–13561.
Lehmann U, Hasemeier B, Christgen M, Müller M, Römermann D, Länger F et al. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol 2008; 214: 17–24.
Lujambio A, Esteller M . CpG island hypermethylation of tumor suppressor microRNAs in human cancer. Cell Cycle 2007; 6: 1455–1459.
Donzelli S, Mori F, Bellissimo T, Sacconi A, Casini B, Frixa T et al. Epigenetic silencing of miR-145-5p contributes to brain metastasis. Oncotarget 2015; 6: 35183–35201.
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM . Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 2006; 20: 2202–2207.
Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 2015; 27: 286–297.
Iliou MS, da Silva-Diz V, Carmona FJ, Ramalho-Carvalho J, Heyn H, Villanueva A et al. Impaired DICER1 function promotes stemness and metastasis in colon cancer. Oncogene 2014; 33: 4003–4015.
Merritt WM, Lin YG, Han LY, Kamat AA, Spannuth WA, Schmandt R et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N Engl J Med 2008; 359: 2641–2650.
Pampalakis G, Diamandis EP, Katsaros D, Sotiropoulou G . Down-regulation of dicer expression in ovarian cancer tissues. Clin Biochem 2010; 43: 324–327.
Faggad A, Budczies J, Tchernitsa O, Darb-Esfahani S, Sehouli J, Müller BM et al. Prognostic significance of Dicer expression in ovarian cancer-link to global microRNA changes and oestrogen receptor expression. J Pathol 2010; 220: 382–391.
Karube Y, Tanaka H, Osada H, Tomida S, Tatematsu Y, Yanagisawa K et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 2005; 96: 111–115.
Dome JS, Coppes MJ . Recent advances in Wilms tumor genetics. Curr Opin Pediatr 2002; 14: 5–11.
Zhang J, Fan XS, Wang CX, Liu B, Li Q, Zhou XJ . Up-regulation of Ago2 expression in gastric carcinoma. Med Oncol 2013; 30: 628.
Völler D, Reinders J, Meister G, Bosserhoff AK . Strong reduction of AGO2 expression in melanoma and cellular consequences. Br J Cancer 2013; 109: 3116–3124.
Melo SA, Moutinho C, Ropero S, Calin GA, Rossi S, Spizzo R et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010; 18: 303–315.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Trimarchi JM, Lees JA . Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 2002; 3: 11–20.
Coller HA, Forman JJ, Legesse-Miller A . ‘Myc’ed messages’: Myc induces transcription of E2F1 while inhibiting its translation via a microRNA polycistron. PLoS Genet 2007; 3: e146.
Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V et al. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem 2007; 282: 2135–2143.
Woods K, Thomson JM, Hammond SM . Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem 2007; 282: 2130–2134.
He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S et al. A microRNA polycistron as a potential human oncogene. Nature 2005; 435: 828–833.
Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW et al. Stem cell division is regulated by the microRNA pathway. Nature 2005; 435: 974–978.
Gillies JK, Lorimer IA . Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 2007; 6: 2005–2009.
Galardi S, Mercatelli N, Giorda E, Massalini S, Frajese GV, Ciafrè SA et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem 2007; 282: 23716–23724.
le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J 2007; 26: 3699–3708.
Visone R, Russo L, Pallante P, De Martino I, Ferraro A, Leone V et al. MicroRNAs (miR)-221 and miR-222, both overexpressed in human thyroid papillary carcinomas, regulate p27Kip1 protein levels and cell cycle. Endocr Relat Cancer 2007; 14: 791–798.
Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann Jr R, Srikantan S et al. p16(INK4a) translation suppressed by miR-24. PLoS One 2008; 3: e1864.
Dolezalova D, Mraz M, Barta T, Plevova K, Vinarsky V, Holubcova Z et al. MicroRNAs regulate p21(Waf1/Cip1) protein expression and the DNA damage response in human embryonic stem cells. Stem Cells 2012; 30: 1362–1372.
Yi C, Wang Q, Wang L, Huang Y, Li L, Liu L et al. MiR-663, a microRNA targeting p21WAF1/CIP1, promotes the proliferation and tumorigenesis of nasopharyngeal carcinoma. Oncogene 2012; 31: 4421–4433.
Du B, Wang Z, Zhang X, Feng S, Wang G, He J et al. MicroRNA-545 suppresses cell proliferation by targeting cyclin D1 and CDK4 in lung cancer cells. PLoS One 2014; 9: e88022.
Peng Y, Dai Y, Hitchcock C, Yang X, Kassis ES, Liu L et al. Insulin growth factor signaling is regulated by microRNA-486, an underexpressed microRNA in lung cancer. Proc Natl Acad Sci USA 2013; 110: 15043–15048.
Lima RT, Busacca S, Almeida GM, Gaudino G, Fennell DA, Vasconcelos MH . MicroRNA regulation of core apoptosis pathways in cancer. Eur J Cancer 2011; 47: 163–174.
Li C, Hashimi SM, Good DA, Cao S, Duan W, Plummer PN et al. Apoptosis and microRNA aberrations in cancer. Clin Exp Pharmacol Physiol 2012; 39: 739–746.
Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010; 18: 367–381.
Fornari F, Gramantieri L, Giovannini C, Veronese A, Ferracin M, Sabbioni S et al. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res 2009; 69: 5761–5767.
Burns DM, D’Ambrogio A, Nottrott S, Richter JD . CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 2011; 473: 105–108.
Yan HL, Xue G, Mei Q, Wang YZ, Ding FX, Liu MF et al. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J 2009; 28: 2719–2732.
Sacconi A, Biagioni F, Canu V, Mori F, Di Benedetto A, Lorenzon L et al. miR-204 targets Bcl-2 expression and enhances responsiveness of gastric cancer. Cell Death Dis 2012; 3: e423.
Zhang H, Li Y, Huang Q, Ren X, Hu H, Sheng H et al. MiR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell Death Differ 2011; 18: 1702–1710.
Nie J, Liu L, Zheng W, Chen L, Wu X, Xu Y et al. microRNA-365, down-regulated in colon cancer, inhibits cell cycle progression and promotes apoptosis of colon cancer cells by probably targeting Cyclin D1 and Bcl-2. Carcinogenesis 2012; 33: 220–225.
Denoyelle C, Lambert B, Meryet-Figuière M, Vigneron N, Brotin E, Lecerf C et al. miR-491-5p-induced apoptosis in ovarian carcinoma depends on the direct inhibition of both BCL-XL and EGFR leading to BIM activation. Cell Death Dis 2014; 5: e1445.
Zhang CZ, Zhang JX, Zhang AL, Shi ZD, Han L, Jia ZF et al. MiR-221 and miR-222 target PUMA to induce cell survival in glioblastoma. Mol Cancer 2010; 9: 229.
Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E et al. Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21. Cancer Cell 2010; 18: 282–293.
Wang P, Zhuang L, Zhang J, Fan J, Luo J, Chen H et al. The serum miR-21 level serves as a predictor for the chemosensitivity of advanced pancreatic cancer, and miR-21 expression confers chemoresistance by targeting FasL. Mol Oncol 2013; 7: 334–345.
Shaffiey F, Cross E, Sathyanarayana P . Mir-590 is a novel STAT5 regulated oncogenic miRNA and targets FasL in acute myeloid leukemia. Blood 2013; 122: 3811–3811.
Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR et al. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 2012; 55: 465–475.
Kalluri R, Weinberg RA . The basics of epithelial mesenchymal transition. J Clin Invest 2009; 119: 1420–1428.
Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS et al. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 2008; 28: 6773–6784.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601.
Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008; 68: 7846–7854.
Hurteau GJ, Carlson JA, Spivack SD, Brock GJ . Over-expression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 2007; 67: 7972–7976.
Korpal M, Lee ES, Hu G, Kang Y . The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008; 283: 14910–14914.
Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 2011; 13: 317–323.
Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 2011; 208: 875–883.
Ma L, Teruya-Feldstein J, Weinberg RA . Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007; 449: 682–688.
Ding X, Park SI, McCauley LK, Wang CY . Signaling between transforming growth factor β (TGF-β) and transcription factor SNAI2 represses expression of microRNA miR-203 to promote epithelial-mesenchymal transition and tumor metastasis. J Biol Chem 2013; 88: 10241–10253.
Zhang Z, Zhang B, Li W, Fu L, Fu L, Zhu Z et al. Epigenetic silencing of miR-203 upregulates SNAI2 and contributes to the invasiveness of malignant breast cancer cells. Genes Cancer 2011; 2: 782–791.
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol 2010; 12: 247–256.
Almeida MI, Reis RM, Calin GA . MYC-microRNA-9-metastasis connection in breast cancer. Cell Res 2010; 20: 602–603.
Meng X, Wu J, Pan C, Wang H, Ying X, Zhou Y et al. Genetic and epigenetic down-regulation of microRNA-212 promotes colorectal tumor metastasis via dysregulation of MnSOD. Gastroenterology 2013; 145: 426–436.
Carmeliet P . Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000; 6: 389–595.
Ferrara N . VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002; 2: 795–803.
Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H et al. hsa-miR-210 is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res 2008; 14: 1340–1348.
Fasanaro P, D’Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G et al. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 2008; 283: 15878–15883.
Lou YL, Guo F, Liu FL, Gao FL, Zhang PQ, Niu X et al. MiR-210 activates notch signaling pathway in angiogenesis induced by cerebral ischemia. Mol Cell Biochem 2012; 370: 45–51.
Liu F, Lou YL, Wu J, Ruan QF, Xie A, Guo F et al. Upregulation of MicroRNA-210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro. Kidney Blood Press Res 2012; 35: 182–191.
Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest 2010; 120: 4141–4154.
Liu LZ, Li C, Chen Q, Jing Y, Carpenter R, Jiang Y et al. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1alpha expression. PLoS One 2011; 6: e19139.
Lei Z, Li B, Yang Z, Fang H, Zhang GM, Feng ZH et al. Regulation of HIF-1alpha and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS One 2009; 4: e7629.
Cha ST, Chen PS, Johansson G, Chu CY, Wang MY, Jeng YM et al. MicroRNA-519c suppresses hypoxia-inducible factor-1alpha expression and tumor angiogenesis. Cancer Res 2010; 70: 2675–2685.
Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT et al. p53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci USA 2010; 107: 6334–6339.
Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH . Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1. Blood 2014; 124: 3748–3757.
Acknowledgements
This work was supported by the US National Institutes of Health (U01CA166905 and P01CA081534 to CMC); National Natural Science Foundation of China (No. 81572739 to YP) and Science & Technology Foundation of Sichuan Province, China (No. 2014SZ0188 to YP).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Peng, Y., Croce, C. The role of MicroRNAs in human cancer. Sig Transduct Target Ther 1, 15004 (2016). https://doi.org/10.1038/sigtrans.2015.4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/sigtrans.2015.4
This article is cited by
-
Four microRNA gene polymorphisms are associated with Iraqi patients with colorectal cancer
Egyptian Journal of Medical Human Genetics (2024)
-
The potential of microRNAs in cancer diagnostic and therapeutic strategies: a narrative review
The Journal of Basic and Applied Zoology (2024)
-
The role of MicroRNAs in the diagnosis and treatment of oral premalignant disorders
Odontology (2024)
-
From diagnosis to resistance: a symphony of miRNAs in pheochromocytoma progression and treatment response
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Overexpression of miR-32 in Chinese hamster ovary cells increases production of Fc-fusion protein
AMB Express (2023)
