
Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease presenting major health and economic challenges that continue to grow. Mechanisms of disease are poorly understood but significant data point to metabolic defects that might contribute to disease pathogenesis. The Alzheimer Disease Metabolomics Consortium (ADMC) in partnership with Alzheimer Disease Neuroimaging Initiative (ADNI) is creating a comprehensive biochemical database for AD. Using targeted and non- targeted metabolomics and lipidomics platforms we are mapping metabolic pathway and network failures across the trajectory of disease. In this report we present quantitative metabolomics data generated on serum from 199 control, 356 mild cognitive impairment and 175 AD subjects enrolled in ADNI1 using AbsoluteIDQ-p180 platform, along with the pipeline for data preprocessing and medication classification for confound correction. The dataset presented here is the first of eight metabolomics datasets being generated for broad biochemical investigation of the AD metabolome. We expect that these collective metabolomics datasets will provide valuable resources for researchers to identify novel molecular mechanisms contributing to AD pathogenesis and disease phenotypes.
Design Type(s) | clinical history design • observation design |
Measurement Type(s) | Metabolomics |
Technology Type(s) | mass spectrometry assay |
Factor Type(s) | diagnosis |
Sample Characteristic(s) | Homo sapiens • blood serum |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Alzheimer’s disease is a degenerative brain disorder and the most common cause of dementia, presenting as the most common neurodegenerative disease in the United States1,2. It is characterized by a decline in memory, language, problem-solving and other cognitive skills that affects a person’s ability to perform everyday activities3. Data suggests that pathophysiological changes associated with AD begin decades before the emergence of clinical symptoms4,5. AD is becoming an increasing health burden in the United States and globally due to population aging6.
The disease is defined by the presence of tau neurofibrillary tangles and Aβ plaques, but coincident pathologies like Lewy body disease, vascular pathology and TDP-43 (transactive response DNA-binding protein 43) deposits are commonly found in AD patients. Current symptomatic therapeutic treatments have modest effects and do not modify the disease course. Researchers hope to develop therapies targeting specific genetic, molecular, and cellular mechanisms so that the actual underlying cause of the disease can be slowed or prevented but currently our understanding of disease mechanisms remains limited. While the majority of AD clinical trials to date have focused on Aβ treatments, other therapeutic approaches are necessary. Understanding biochemical trajectory of disease and metabolic changes related to Aβ and Tau pathology and cognitive decline is essential to advance our understanding of AD etiology as well as for developing novel approaches for drug development.
Recent advances in analytical chemistry led to the emergence of a new field called metabolomics. Metabolomics allows simultaneous measurement of 100’s to 1,000’s of metabolites for mapping perturbations in interconnected pathways and in metabolic networks enabling a systems approach to the study of AD7,8. An emerging body of evidence data supports the potential of metabolomics to provide added information for the prediction of AD and has identified a number of potentially important biochemical pathways in AD9–15. Though promising, in many cases to this point cohorts are quite small with little replication, and therefore do not allow the effects of confounds such as medications to be addressed. Larger and more comprehensive cohorts are needed to provide the statistical power to detect and develop robust predictive metabolomics models, while also accounting for confounding effects related to medication intake, gender and aging. The Alzheimer’s Disease Neuroimaging Initiative (ADNI) unites researchers around the globe to define the progression of Alzheimer’s disease. ADNI researchers collect, validate and utilize data such as MRI and PET images, genetics, cognitive tests, CSF and blood biomarkers from thousands of subjects as predictors for the disease16,17. The Alzheimer’s Disease Metabolomics Consortium (ADMC) has the goal of building a comprehensive metabolomics database for AD in partnership with ADNI. This will be a national resource for the Alzheimer’s community that enables interrogation of global metabolic changes within a pathway and network context and where metabolomics data can be used to compliment and inform genomics and imaging data. Eight targeted and non-targeted metabolomics platforms that have their own strengths and limitations are being used to profile thousands of ADNI subjects.
For this dataset, we used a targeted, widely utilized and cross-validated metabolomics platform AbsoluteIDQ®-p180 (Biocrates AG) to profile baseline serum samples from ADNI1 cohort where vast data exist on each patient including cognitive decline and imaging changes over many years, information on CSF markers, genetics and other omics data. The goal of this data generation is to aid in the discovery of metabolic failures correlated with disease and progression and biomarkers for a range of important physiological processes in AD. We describe a foundation for automated curation of metabolomics data, including R scripts for removing analytes with poor precision or with significant missing values, and samples that have missing clinical data or are metabolic outliers. We address approaches within this cohort for dealing with confounds that impact metabolomics data and findings including medications, broadly applicable to pharmacometabolomic investigations18–20.
Methods
Alzheimer’s disease neuroimaging initiative (ADNI) cohort
Clinical and demographic data used for this study were obtained through the ADNI data repository (http://adni.loni.ucla.edu/). Written informed consent was obtained for all participants and prior Institutional Review Board approval was obtained at each participating institution. All demographic information, neuropsychological and clinical assessment data, and diagnostic information used in this study are available from the ADNI clinical data repository (http://adni.loni.ucla.edu/). Information about ADNI can be found in Petersen et al.16 and at http://www.adni-info.org/16. Key clinical and demographic variables for the ADNI1 participants as of May 2016 are summarized in Table 1 and available through Synapse (see Table 2). Note that ADNI data collection is ongoing, so variables on LONI may have been updated since that subset was downloaded. We have included this snapshot in order to enable analytic reproducibility despite a dynamic source of truth.
Serum collection and sample management
Morning fasting blood samples from the baseline visit were included in the study (all but 69 were fasting). Samples were collected in two bar-coded 10 ml red-top plastic Vacutainer blood tubes, blood was allowed to clot for 30 min followed by a 15 min centrifugation at 3,000 rpm (1,500 rcf) as described in the ADNI standard operating procedures (www.adni-info.org). Then the serum was transferred into a bar-coded 13 ml polypropylene transfer tube and capped and allowed to freeze in dry ice. Samples were shipped overnight to the ADNI biomarker core laboratory at the University of Pennsylvania Medical Center. Samples were thawed once in the core facility and aliquoted to 0.5 ml samples, then subsequently aliquoted once more for individual laboratory analyses. A 20 μl sample aliquot was delivered to the Duke Proteomics and Metabolomics Shared Resource for analysis with the p180 platform.
Metabolomics analysis and QC using the AbsoluteIDQ p180 kit
Sample preparation
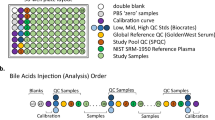
Samples were prepared and analyzed in the Duke Proteomics and Metabolomics Shared Resource using the AbsoluteIDQ p180 kit (Biocrates Life Sciences AG, Innsbruck, Austria) in accordance with the user manual. In brief, after the addition of 10 μl of the supplied internal standard solution to each well on a filterspot of the 96-well extraction plate, 10 μl of each serum sample, quality control (QC) samples, blank, zero sample, or calibration standard were added to the appropriate wells (Fig. 1). The plate was then dried under a gentle stream of nitrogen. The samples were derivatized with phenyl isothiocyanate (PITC) for the amino acids and biogenic amines, and dried again. Sample extract elution was performed with 5 mM ammonium acetate in methanol. Sample extracts were diluted with either 40% methanol in water for the UPLC-MS/MS analysis (15:1) or kit running solvent (Biocrates Life Sciences AG) for flow injection analysis (FIA)-MS/MS (20:1).

(a) 96-well plate layout used for sample preparation and data collection for the Absolute IDQ p180 metabolomics analysis. Each of the eleven plates (n=833 study samples) analyzed in the study used the same lot of calibrators, Biocrates QCs, study pool QC (SPQC), GoldenWest Serum and NIST SRM-1950 plasma. (b) Analysis order for each plate, showing how the calibration curve and QC samples bracket the actual sample analyses in order to decrease the likelihood of intraplate bias. LC-MS/MS and FIA-MS/MS use the same analysis order, but FIA-MS/MS excludes the calibration curve.
Quality control samples
The analysis of the samples using the AbsoluteIDQ p180 kit was performed using four specific sets of quality controls. First, low/mid/high level QC samples provided by Biocrates Life Sciences AG were prepared and analyzed on each plate as recommended by the manufacturer. These QC samples were used for a technical validation of each kit plate. Second, to allow appropriate inter-plate abundance scaling based specifically on this cohort of samples, we generated a Study Pool QC (SPQC) by combining approximately 10 μl from the first 76 samples for analysis. This sample was frozen in aliquots of 25 ul then prepared and analyzed twice on each plate. Third, there were 20 blinded analytical duplicates obtained from the same serum draw scattered throughout the study in a manner blinded to the investigators until data was sent to the ADNI informatics core for unblinding. The commonly used reference materials NIST SRM-1950 plasma (n=3 per plate) and GoldenWest serum pool (n=1 per plate) were also analyzed on each plate to allow cross-comparison against other sample cohorts in the future.
Figure 1a shows the preparation layout for the 96-well plates as utilized in this study. In total, eleven plates were prepared in order to analyze 831 serum samples. The blank, zero sample, calibration standards, and Low/Mid/High QC samples provided with the kit were arranged as recommended by Biocrates. In order to improve the ability to compare results with other metabolomics studies and reduce plate-to-plate batch effects, six additional wells were used for the additional QC samples as described above: two wells for the study pool QC (SPQC), one well for the GoldenWest Pooled Serum Standard21, and three wells for the NIST SRM-1950 Standard Reference Plasma. The remaining 76 wells were used for cohort samples. The analysis order of each plate is summarized in Fig. 1b, and was arranged to maximize quantitative accuracy and precision within a plate and limit the potential for batch effects. The analysis order included running the standard curve twice, once at the beginning and end of the samples (LC-MS/MS only). For both LC-MS/MS and FIA-MS/MS analysis, the Biocrates QC’s and Goldenwest Serum QC were prepared once but injected in technical triplicate, once before, in the middle (after 38 samples) and at the end of the sample set. The SPQC samples (n=2) were each analyzed once, with one analysis before and one after all samples on the plate. The NIST SRM-1950 plasma (n=3) were also analyzed once each at the beginning, middle, and end of the cohort samples. Bracketing the standard curves and nesting the analytical samples between the QCs offers the best chance of observing any system drift and assuring optimal instrument performance across the sample set.
Quantitative UPLC-MS/MS and FIA-MS/MS analysis
Mass spectrometry analysis was performed based on Standard Operating Procedures (SOP #8114) provided by Biocrates for the AbsoluteIDQ p180 kit. Chromatographic separation of amino acids and biogenic amines was performed using an ACQUITY UPLC System (Waters Corporation) using an ACQUITY 2.1 mm×50 mm 1.7 μm BEH C18 column fitted with an ACQUITY BEH C18 1.7 μm VanGuard guard column, and quantified by calibration curve plotting ratio of analyte to internal standard versus standard concentration, fitted using a linear regression with 1/x weighting. All amino acids and biogenic amines utilize either deuterated or 13C stable-isotope labeled internal standard of the exact analyte or closely-eluting compound of similar class. Acylcarnitines, sphingolipids, and glycerophospholipids were analyzed by flow injection analysis tandem mass spectrometry (FIA-MS/MS) and quantified by internal standard calibration; eight separate internal standards are used to quantify the various acylcarnitines, while a single internal standard is used for each of the other lipid classes. Thus, FIA-MS/MS analytes are reported as semi-quantitative values except where a stable-isotope labeled internal standard of that exact analyte was used. Samples for both UPLC and FIA analyzed using a Xevo TQ-S mass spectrometer (Waters Corporation) using positive electrospray ionization operating in the Multiple Reaction Monitoring (MRM) mode. MRM transitions (compound-specific precursor to product ion transitions) for each analyte and internal standard were collected over a scheduled retention time window using tune files and acquisition methods provided in the AbsoluteIDQ p180 kit. The UPLC data were imported into TargetLynx (Waters Corporation) for peak integration, calibration and concentration calculations. The UPLC data from TargetLynx and FIA data were analyzed using Biocrates’ MetIDQ v5.4.8 software. The kit data are reported in detail in the Supplementary Information on LONI, along with a color-coded key denoting samples that were below the limit of detection (<LOD), below the lowest calibration standard (<LLOQ), or quantified based on a ratio to a class-based internal standard (semi-quantitative). The data generated for the study samples, SPQC samples, as well as NIST-SRM 1950 from each plate can be downloaded at http://adni.loni.usc.edu/data-samples/access-data/ (See below).
Treatment of isobaric lipids
The p180 kit quantifies individual lipid species the ‘sum lipid composition’ level; that is, the quantity of the lipid reported as PC (36:3) (for example) is the sum of all possible phosphatidylcholine lipids in the sample which have a total of 36 carbons and 3 double bonds. Additionally, analyzed glycerophospholipids are differentiated according to the presence of ester and ether bonds in the glycerol moiety. Typically, the annotation ‘aa’ in lipid nomenclature indicates that both fatty acids at the sn-1 and sn-2 position are bound to the glycerol backbone via ester bonds, whereas ‘ae’ donates that fatty acid in the sn-1 or sn-2 position is bound via an ether bond. Total number of carbon atoms and double bonds present in lipid fatty acid chains are denoted as ‘C x:y’ where x indicates the number of carbons and y the number of double bonds. For the specific glycerophospholipid class sphingomyelins (SM), only the fatty acid bound to glycerol backbone at the sn-2 position are indicated under the standard assumption that sphingosine (d18:1) is bound at the sn-1 position. The FIA-MS/MS analysis with the Biocrates p180 kit is performed using mass spectrometry precursor/product transitions (‘MRMs’) with lipid species-specific precursor ions and class-specific fragment ions, to report quantitative values for a number of lipids at the sum lipid composition level.
Due to the relatively low mass resolution of triple quadrupole MS instruments, the detected flow-injection based MRM signal is a sum of several isobaric lipids within the same class. For example, according to the LIPID MAPS database (www.lipidmaps.org), the signal of PC aa C36:6 can arise from at least 15 different lipid species that have different fatty acid composition (e.g., PC 16:1/20:5 versus PC 18:4/18:2), various positioning of fatty acids sn-1 and sn-2 position (e.g., PC 18:4/18:2 versus PC 18:2/18:4) and different double bond positions in those fatty acid chains (e.g., PC(18:4(6Z,9Z,12Z,15Z)/18:2(9Z,12Z)) versus PC(18:4(9E,11E,13E,15E)/18:2(9Z,12Z))). The Biocrates MetIDQ software applies an isotopologue correction for the obtained lipid data in order to increase accuracy, but due to limitations of FIA-MS/MS data on triple quadrupoles, this correction is incomplete for some lipids22. To assist with clarity in the interpretation of the results reported herein using the p180 kit, we have included a list of measured lipids. Their common names and possible isobars and isomers are summarized in Supplementary Table 1: The list of lipids measured with the AbsoluteIDQ p180 kit. Within this table, possible isobars are given within ±0.5 Da range due to the typical conditions under which triple quadrupole mass spectrometers are operated for the kit, and the isobars are reported as sum compositions23. The position of acyl chains (sn-1, sn-2) and double bonds are not indicated for the possible isomers. The possible isotopologue interferences are indicated in cursive. For each isomer, examples of LIPID MAPS Structure Database (http://www.lipidmaps.org/data/databases.html) entries are listed, where applicable. The table comprises examples of possible isobars and isomers to the best of our current knowledge, but does not prioritize the subspecies based on likelihood of existence or relative abundance in any particular biological system.
Data processing
Statistical preprocessing was performed using the open-source, statistical software, R v3.2.4 (www.r-project.org), with scripts available for download at https://doi.org/10.7303/syn7354353. The processing included the steps briefly described herein, and graphically depicted as a flowchart in Fig. 2.

The use of Levels (shown at left) breaks the workflow into discrete steps which can be applied to multiple metabolomics data types. The workflow executed in R is described on the right. *Subjects flagged for exclusion in Level 4 are not physically excluded from the table until Level 5.
In the first step we excluded four samples due to erroneous inclusion in the cohort (thawed during shipment), and the values of each analyte were scaled across the different plates using the Study Pool QC (SPQC) duplicates analyzed twice on each plate. Given SPQC duplicates, the correction factor for each analyte in a specific plate was obtained by dividing its global average by its average within the plate to adjust for the batch effects, yielding the Intermediate Data Level 2.
The second set of steps include filtering of analytes based on quality metrics. We applied two criteria to filter individual metabolites as part of our data quality control (QC) evaluation process based on the 20 blinded ADNI duplicates: 1) a coefficient of variation (CV) <20% across plates, and 2) an intraclass correlation coefficient (ICC) >0.65. ICC compared the two measurements for each of the blinded duplicates. Additionally, analytes with >40% of measurements below the lower limit of detection (<LOD) were excluded from the analysis. Combined, these three steps allow only the most robust analytes from the panel through to the Level 3 data matrix, and reduced the total number of analytes reported in the dataset from 182 analytes (Intermediate Data—Level 2) to 138 analytes (Intermediate Data—Level 3). The QC Results for all analytes including LOD percentage, CV of the blinded replicates, and ICC values are reported in the Supplementary Table 2 (FIA-MS/MS) and 3 (UPLC-MS/MS).
The steps between Level 3 and Level 4 in the pipeline (Fig. 2) perform missing value replacement and allow for exclusion of samples due to other missing data that may vary from study to study. Remaining samples with values reported as ‘<LOD’ were imputed using LOD/2 value for each specific analyte. Also, there were 73 samples determined to be pre-analytical outliers for one or more of the following reasons which were flagged and removed from the dataset. These included a total of 69 samples identified as non-fasting, 2 samples lacking corresponding body mass index (BMI) values, and 1 for which no baseline medication record was reported. After these steps, the Intermediate Data—Level 4 contained n=754 samples (734 subjects) and n=138 analytes.
The last steps in the statistical pretreatment pipeline serve to combine replicate measurements to give one value per biological sample, filter out any statistical outlier subjects, and perform log-transformation if necessary. To obtain a single value for the 20 subjects with blinded duplicates we calculated the average of the duplicates for each subject and reported this single value. We checked for the presence of outlier subjects by performing principal components analysis, and evaluating the subject distance from the centroid in the K-dimensional space based on principal components that explained >90% cumulative variance. Subjects located more than 7 s.d. from the mean were flagged as outliers. This procedure identified two additional samples that were excluded from the final data matrix. Finally, log2 transformation was performed for those analytes which show P-value for D’Agostino <0.05 and Skewness test >2. The final preprocessed data matrix (Final Data Matrix—Fig. 2, Level 5) contained data for 732 subjects and 138 analytes.
It should be noted that the statistical curation process described above is very stringent, leaving behind only the most robust analytes but in the process potentially excluding some good measurements. One weakness in our pipeline is that by filtering on intraclass correlation coefficient (ICC) it is possible that we filtered out some robust measurements which simply had very narrow biological measurement range over the blinded samples, precluding the observation of a correlational trend. Examples of this potentially include histamine (17.2% missing values, 5.4% CV, 0.09 ICC) and methionine (0% missing values, 8.1% CV, 0.65 ICC), which almost certainly would have been left in the dataset using many commonly-used and less stringent filtering criteria. To address this shortcoming, future studies are being designed with at least three measurements of each blinded replicate sample and triplicate preps of the SPQC on each plate instead of the NIST SRM-1950, to allow more robust filtering based on imprecision across a wider dynamic range. Metabolites excluded from the analysis suggested here may also be recovered when reanalyzing data in the context of comparative data from other cohorts.
Collection and curation of medication data
Many classes of medications have been shown to affect metabolism and change levels of certain metabolites18–20,24. It is thus necessary to take drug information into account as a potential confounder for metabolomics analysis. In order to convert free text medication information into computable drug data, we applied a pipeline described previously25. In brief, as shown in Fig. 3, we employed the National Library of Medicine’s (NLM) RxNorm API (application programming interface) to match drug names extracted from patient medication information containing lexical variations and misspellings to standardized drug concept identifiers. Corresponding concept identifiers are returned along with confidence scores. Low scoring terms were reviewed manually and adjusted as appropriate.

Drug names are parsed and passed to the RxNorm API to determine approximate string matches. Low scoring matches are reviewed manually. Once the drug, whether brand name or generic, has been mapped to an RxNorm ingredient, corresponding classes are ascertained.
We mapped all versions of a drug, whether brand name or generic, to its respective ingredients, then mapped those ingredients to corresponding drug classes. A subset of drug categories were selected from 3 standardized drug classification systems (NDF-RT, ATC, and MeSH) based on input from experts in Alzheimer’s disease and metabolomics (see Table 3 (available online only)). Criteria for selection of classes to be included in analysis were classes of drugs known to impact metabolomics pathways and/or those likely to be taken by a cognitively impaired population. In this way, each patient was assigned a Boolean flag for whether or not he or she was taking any drug in each respective class. Binary variables can then be used to address potential confounding in subsequent association analyses. The code for this pipeline, including R scripts and API configuration files, is available in Synapse: https://doi.org/10.7303/syn7477310. The final table showing which ADNI1 participants were taking which classes of drugs at their first visit is available at https://doi.org/10.7303/syn7440367.1.
Important note
The method described here is the first in a series of iterative approaches to tackle the complex challenge of medications as confounding variables in metabolomic profiling. Medication terminology and software tools continue to evolve. We recommend that those performing future analyses related to medication effects revisit this site and the ADNI data repository for updated curation of medication data.
Data Records
The primary access site for this dataset is through Sage Bionetworks’ Synapse platform (Data Citation 1). ADNI’s data use agreement prohibits redistribution of ADNI data outside of LONI, so actual data files are hosted by the University of Southern California’s Laboratory of Neuroimaging (LONI). The scripts used for data processing and medication mapping, however, reside in the Synapse platform. Core data files along with associated metadata files, scripts, and Supplementary Files are listed in Table 2. Note that ADNI requires registration to access the data. Researchers may apply for data access at https://ida.loni.usc.edu/collaboration/access/appLicense.jsp.
R Scripts for data processing can be found at https://doi.org/10.7303/syn7354353. Input files (Fig. 2, Level 0) are found in (Data Citation 2) and (Data Citation 3) for FIA and UPLC respectively. Processed data (Fig. 2, Level 5) are available at (Data Citation 4) and (Data Citation 5) for FIA and UPLC respectively. The latest version of clinical annotation data is in (Data Citation 6).
Also included in Supplementary Information on LONI (http://adni.loni.usc.edu/data-samples/access-data/) are the original Excel format exports from the MetIDQ software (Biocrates, Inc). These files include information on calibration ranges, limits of quantification and detection for the assays, and QC sample measurements. They use the original blinded identifiers.
Similarly, the code for the medication mapping pipeline and a link to the current medications file on LONI is available through (Data Citation 7). The medications file must be downloaded and placed in the same directory as the medication mapping R scripts in order to reproduce the medication mapping workflow. The output table showing which patients were taking which classes of drugs is available at (Data Citation 8). Note that, as described in the readme file that accompanies these scripts, manual intervention was used at three points in the pipeline: running RxMix using two different configuration files, and expert review of medication name mapping to accept, reject, or correct results for low-scoring matches.
Technical Validation
AbsoluteIDQ® p180 kit has been fully validated according to European Medicine Agency Guideline on bioanalytical method validation, and this kit has been utilized in over 200 peer-reviewed publications including a number in dementia and AD9,13,26. A recent ring trial showed that inter-laboratory precision was <20% for 82% of the analytes measured with the kit, and 83% of the analytes were accurate within <20%27. Additionally, each analyzed kit plate includes an automated technical validation to approve the validity of the run and to provide verification of the actual performance of the applied quantitative procedure including instrumental analysis. Interplate technical validation of each analyzed kit plate was performed using MetIDQ software based on results obtained and defined acceptance criteria for blank, zero samples, calibration standards and curves, low/medium/high level QC samples and measured signal intensity of internal standards over the plate. Technical validation for the Xevo TQS was performed according to the following criteria. For the Blank samples the signal intensity for all metabolites and internal standards had to be smaller than a defined minimum value. Signal intensities obtained for zero sample were used for the calculation of plate specific limit of detection (LOD) for FIA-MS/MS analysis. LOD value was defined as concentration that corresponded to three times level of the blank sample. A specific standard measurement (calibrators) was considered valid when calculated concentration was within +/− 30% range of the target concentration. For a specific analyte a minimum of 75% of all calibration standards had to be valid. Biocrates-provided QC samples were human plasma pool spiked with analytes at known concentrations at three different levels. The valid measured concentration range was set for each analyte separately and was within +/− 45% of a target concentration. For a specific analyte a minimum of 67% of all QCs had to be valid as well as a minimum of 50% of all QCs of a certain level had to be valid. Additionally signal intensity for internal standards had to be within valid minimum and maximum intensity value defined by kit manufacturer. All measured plates fulfilled the above described criteria hence confirming the quality and accuracy of the obtained quantitative metabolomics data according to manufacturer recommendations.
To ensure the quality and reproducibility of the quality control and analysis performed prior to data release, independent analysts completed the computational workflow, and achieved reproducibility out to three significant digits across all calculations.
Usage Notes
Details on how to apply for data access and usage rules can be found at the ADNI website: http://adni.loni.usc.edu/data-samples/access-data/. In brief, users agree to keep the data secure and not to attempt to re-identify research participants. Users also agree to acknowledge ADNI and the ADMC in any derivative publications as follows:
-
1
On the by-line of the manuscript, after the named authors, include the phrase ‘for the Alzheimer’s Disease Neuroimaging Initiative*’ with the asterisk referring to the following statement and list of names: *Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
-
2
On the by-line of the manuscript, after the named authors, include the phrase ‘for the Alzheimer’s Disease Metabolomics Consortium**’ with the double asterisk referring to the following statement and list of names: **Data used in preparation of this article were generated by the Alzheimer’s Disease Metabolomics Consortium (ADMC). As such, the investigators within the ADMC provided data but did not participate in analysis or writing of this report. A complete listing of ADMC investigators can be found at: https://sites.duke.edu/adnimetab/admc-team-directory/
-
3
The results published here are in whole or in part based on data obtained from the AMPAD Knowledge POrtal accessed at doi:10.7303/syn2580853
Access to scripts and other files described herein that are available through the https://www.synapse.org/#!Synapse:syn2580853/wiki/409840 AMPAD Knowledge Portal hosted on the Sage Bionetworks Synapse informatics data sharing platform, requires adherence to the terms of use described at http://docs.synapse.org/articles/governance.html. Users are required to sign an oath (http://docs.synapse.org/assets/other/oath.html) stating they will not re-identify participants, redistribute the data, or use for advertising and that they will keep data secure, protect privacy, support open access, report any breaches, credit participants, and follow privacy laws.
Because they are managed by different entities, users must register for separate user accounts for LONI (where data are stored) and https://www.synapse.org/#!Synapse:syn2580853/wiki/409840 AMPAD Knowledge Portal on Synapse (repository for scripts and additional information) respectively.
Consortia
The Alzheimer Disease Neuroimaging Initiative
A full list of members is available at http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
The Alzheimer Disease Metabolomics Consortium
A list of members is available at: https://sites.duke.edu/adnimetab/files/2017/06/ADMCTeamMembersJun14.pdf.
Additional information
How to cite this article: St John-Williams, L. et al. Targeted metabolomics and medication classification data from participants in the ADNI1 cohort. Sci. Data 4:170140 doi: 10.1038/sdata.2017.140 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Barker, W. W. et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 16, 203–212 (2002).
Wilson, R. S. et al. The natural history of cognitive decline in Alzheimer's disease. Psychol. Aging 27, 1008–1017 (2012).
Alzheimer's Association . Alzheimer's disease facts and figures. Alzheimers Dement. 12, 459–509 (2016).
Jack, C. R. Jr et al. Age-specific population frequencies of cerebral beta-amyloidosis and neurodegeneration among people with normal cognitive function aged 50-89 years: a cross-sectional study. Lancet Neurol. 13, 997–1005 (2014).
Toledo, J. B. et al. Alzheimer's disease cerebrospinal fluid biomarker in cognitively normal subjects. Brain 138, 2701–2715 (2015).
James, B. D. et al. Contribution of Alzheimer disease to mortality in the United States. Neurology 82, 1045–1050 (2014).
Kaddurah-Daouk, R. & Krishnan, K. R. Metabolomics: a global biochemical approach to the study of central nervous system diseases. Neuropsychopharmacology 34, 173–186 (2009).
Patti, G. J., Yanes, O. & Siuzdak, G. Innovation: Metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 13, 263–269 (2012).
Fiandaca, M. S. et al. Plasma 24-metabolite Panel Predicts Preclinical Transition to Clinical Stages of Alzheimer's Disease. Front Neurol 6, 237 (2015).
Han, X., Holtzman, D. M. & McKeel, D. W. Jr. Plasmalogen deficiency in early Alzheimer's disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 77, 1168–1180 (2001).
Han, X. et al. Metabolomics in early Alzheimer's disease: identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS ONE 6, e21643 (2011).
Klavins, K. et al. The ratio of phosphatidylcholines to lysophosphatidylcholines in plasma differentiates healthy controls from patients with Alzheimer's disease and mild cognitive impairment. Alzheimers Dement. 1, 295–302 (2015).
Mapstone, M. et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med 20, 415–418 (2014).
Simpson, B. N. et al. Blood metabolite markers of cognitive performance and brain function in aging. J. Cereb. Blood Flow Metab. 36, 1212–1223 (2016).
Wood, P. L. et al. Circulating plasmalogen levels and Alzheimer Disease Assessment Scale-Cognitive scores in Alzheimer patients. J Psychiatry Neurosci 35, 59–62 (2010).
Petersen, R. C. et al. Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74, 201–209 (2010).
Weiner, M. W. et al. Impact of the Alzheimer's Disease Neuroimaging Initiative, 2004 to 2014. Alzheimers Dement 11, 865–884 (2015).
Kaddurah-Daouk, R., Kristal, B. S. & Weinshilboum, R. M. Metabolomics: a global biochemical approach to drug response and disease. Annu. Rev. Pharmacol. Toxicol. 48, 653–683 (2008).
Kaddurah-Daouk, R., Weinshilboum, R. & Pharmacometabolomics Research Network. Metabolomic Signatures for Drug Response Phenotypes: Pharmacometabolomics Enables Precision Medicine. Clin. Pharmacol. Ther. 98, 71–75 (2015).
Kaddurah-Daouk, R., Weinshilboum, R. M. & Pharmacometabolomics Research Network. Pharmacometabolomics: implications for clinical pharmacology and systems pharmacology. Clin Pharmacol. Ther. 95, 154–167 (2014).
Grant, R. P. & Hoofnagle, A. N. From lost in translation to paradise found: enabling protein biomarker method transfer by mass spectrometry. Clin. Chem. 60, 941–944 (2014).
Eibl, G. et al. Isotope correction of mass spectrometry profiles. Rapid Commun. Mass Spectrom. 22, 2248–2252 (2008).
Liebisch, G. et al. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 54, 1523–1530 (2013).
Neavin, D., Kaddurah-Daouk, R. & Weinshilboum, R. Pharmacometabolomics informs Pharmacogenomics. Metabolomics 12, 121 (2016).
Blach, C. et al. Use of RxNorm and NDF-RT to normalize and characterize participant-reported medications in an i2b2-based research repository. AMIA Jt. Summits Trans.l Sci. Proc 2014, 35–40 (2014).
Casanova, R. et al. Blood metabolite markers of preclinical Alzheimer's disease in two longitudinally followed cohorts of older individuals. Alzheimers Dement. 12, 815–822 (2016).
Siskos, A. P. et al. Inter-laboratory reproducibility of a targeted metabolomics platform for analysis of human serum and plasma. Anal. Chem. 89, 656–665 (2017).
Data Citations
ADMC Synapse https://www.synapse.org/#!Synapse:syn5592519 (2017)
Sage Bionetworks https://doi.org/10.7303/syn7440354.1 (2016)
Sage Bionetworks https://doi.org/10.7303/syn7440355.1 (2016)
Sage Bionetworks https://doi.org/10.7303/syn7440356.1 (2016)
Sage Bionetworks https://doi.org/10.7303/syn7440357.1 (2016)
Sage Bionetworks https://doi.org/10.7303/syn7477271.1 (2016)
Sage Bionetworks https://doi.org/10.7303/syn7477310 (2016)
Sage Bionetworks https://doi.org/10.7303/syn7440367.1 (2016)
Acknowledgements
We thank Lisa Howerton for her enthusiastic coordination and administrative support of this work. Funding for ADMC (Alzheimer’s Disease Metabolomics Consortium, led by Dr R.K.-D. at Duke University) was provided by National Institute on Aging grant R01AG046171, a component of the https://www.nia.nih.gov/research/dn/amp-ad-target-discovery-and-preclinical-validation-project Accelerated Medicines Partnership (AMP-AD) Target Discovery and Preclinical Validation Project, and the National Institute on Aging grant RF1 AG0151550, a component of the Molecular Mechanisms of Vascular Etiology (https://www.nia.nih.gov/news/decoding-molecular-ties-between-vascular-disease-and-alzheimers M2OVE-AD) Consortium. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (A.D.N.I.) (National Institutes of Health Grant U01 AG024904) and DOD A.D.N.I. (Department of Defense award number W81XWH-12-2-0012). A.D.N.I. is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support A.D.N.I. clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. A.D.N.I. data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. J.B.T. is supported by P01 AG032953, PO1 AG017586, P30 AG010124 and P50 NS053488.
Author information
Authors and Affiliations
Consortia
Contributions
R.K.-D. conceived the study and is the PI of the project. M.A.M. and T.K. provided expertise in experimental design. L.S.J.-W. and J.W.T. performed the assays. J.B.T., S.M., and A.M.R. performed statistical analysis. K.K. provided analysis required for the lipids table. J.D.T. was responsible for data coordination and medication mapping. C.B. built the medication mapping pipeline. J.W.T., L.S.J.-W., J.D.T., and K.K. wrote the manuscript. M.A.M., T.K., and R.K.-D. edited the manuscript. All authors reviewed and approved the manuscript for important scientific content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
St John-Williams, L., Blach, C., Toledo, J. et al. Targeted metabolomics and medication classification data from participants in the ADNI1 cohort. Sci Data 4, 170140 (2017). https://doi.org/10.1038/sdata.2017.140
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.140
This article is cited by
-
Multiomics analysis to explore blood metabolite biomarkers in an Alzheimer’s Disease Neuroimaging Initiative cohort
Scientific Reports (2024)
-
The associations of serum valine with mild cognitive impairment and Alzheimer’s disease
Aging Clinical and Experimental Research (2022)
-
Alterations in acylcarnitines, amines, and lipids inform about the mechanism of action of citalopram/escitalopram in major depression
Translational Psychiatry (2021)
-
Sex and APOE ε4 genotype modify the Alzheimer’s disease serum metabolome
Nature Communications (2020)
-
Bile acids targeted metabolomics and medication classification data in the ADNI1 and ADNIGO/2 cohorts
Scientific Data (2019)
