
Abstract
Orchids are renowned for their spectacular flowers and ecological adaptations. After the sequencing of the genome of the tropical epiphytic orchid Phalaenopsis equestris, we combined Illumina HiSeq2000 for RNA-Seq and Trinity for de novo assembly to characterize the transcriptomes for 11 diverse P. equestris tissues representing the root, stem, leaf, flower buds, column, lip, petal, sepal and three developmental stages of seeds. Our aims were to contribute to a better understanding of the molecular mechanisms driving the analysed tissue characteristics and to enrich the available data for P. equestris. Here, we present three databases. The first dataset is the RNA-Seq raw reads, which can be used to execute new experiments with different analysis approaches. The other two datasets allow different types of searches for candidate homologues. The second dataset includes the sets of assembled unigenes and predicted coding sequences and proteins, enabling a sequence-based search. The third dataset consists of the annotation results of the aligned unigenes versus the Nonredundant (Nr) protein database, Kyoto Encyclopaedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG) databases with low e-values, enabling a name-based search.
Design Type(s) | transcription profiling by high throughput sequencing design • organism part comparison design |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | RNA sequencing |
Factor Type(s) | tissue |
Sample Characteristic(s) | Phalaenopsis equestris • root • stem • leaf • flower bud • column • plant lip • petal • sepal • seed |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Orchidaceae is the most diverse family of angiosperms, including approximately 25,000 species (i.e., approximately 8% of all vascular plant species), more than mammals, birds and reptiles combined1. Orchids are known for their very diverse and specialized reproductive and ecological strategies. The specific development of the labellum (the ‘lip’) and gynostemium (a fused structure of the stamens and pistils) to trick pollinators and to facilitate pollination is well documented2,3. In addition to the highly sophisticated floral structure, crassulacean acid metabolism (CAM), symbiosis with fungi, and epiphytism might also be linked to the adaptive radiation of orchids4–6, which might be related to their high diversification. Since the publication of Charles Darwin’s book On the Origin of Species, evolutionary biologists have been fascinated by orchids. Biologists have proposed various explanations for the extraordinary diversity of orchids but have been unable to identify its root causes.
The genome sequence of the tropical epiphytic orchid Phalaenopsis equestris, which represents the first sequenced genome for a plant with CAM, contains 29,431 predicted protein-coding genes3. The genomic sequence shows evidence of an orchid-specific polyploidy event that preceded the radiation of most orchid clades and suggests that gene duplication might have contributed to the evolution of CAM photosynthesis in P. equestris3. In addition, this species possesses expanded and diversified families of MADS-box C/D-class, B-class AP3, and AGL6-class genes, which might contribute to the highly specialized morphology of orchid flowers3. Furthermore, the P. equestris genome does not contain the β group of type I MADS-box genes (type I Mβ), although these genes do exist in Arabidopsis thaliana, Populus trichocarpa, and Oryza sativa. Interactions among type I MADS-box genes are important for the initiation of endosperm development7.
Some cDNA libraries have been constructed to examine the gene expression in Phalaenopsis mature flower buds8 and floral scent products by comparing their expression patterns in P. bellina and in the scentless species, P. equestris9, for which expressed sequence tags (ESTs) were sequenced and assembled into unigenes. Phalaenopsis ESTs are derived from cDNA-amplified fragment length polymorphism (cDNA-AFLP) and randomly amplified polymorphic cDNAs (cDNA-RAPD)10,11. These methods were used to systematically screen many differentially expressed cDNA fragments in the wild-type strain and somaclonal variants10,11. Several differentially expressed transcripts related to flower development and flower colour were identified10,11.
Two orchid transcriptomic databases have been established. One is OrchidBase, which contains the transcriptome sequences derived from 11 Phalaenopsis orchid cDNA libraries. OrchidBase was constructed from different species, including P. Aphrodite subsp. formosana, P. equestris and P. bellina, and from different tissues, including the developing seed, protocorm, vegetative tissue, leaf, cold-treated plantlet, pathogen-treated plantlet, inflorescence and flower buds12,13. The other database, Orchidstra, was constructed from the 233,924 unique contigs of the transcriptome sequences of P. aphrodite subsp. formosana. In Orchidstra, genes with tissue-specific expression were categorized by profiling analysis with RNA-Seq14.
Recently, the first comprehensive analysis of the transcriptome and expression profiles during Phalaenopsis explant browning was reported using Illumina high-throughput technology. In this genome-wide level analysis, differentially expressed genes (DEGs) before and after Phalaenopsis explant browning were identified15. In addition, to study the regulation of Phalaenopsis flower organ development, RNA-Seq reads were generated with the Illumina platform for floral organs of the Phalaenopsis wild-type strain and a peloric mutant with a lip-like petal. In total, 43,552 contigs were obtained after de novo assembly. The comprehensive transcript profile and functional analysis suggest that PhAGL6a, PhAGL6b and PhMADS4 might play crucial roles in Phalaenopsis labellum development16.
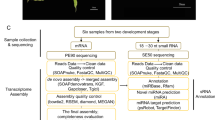
All this genomic and transcriptomic information will supply datasets for orchid molecular biology research. Here, we chose to focus on the transcriptomes of the root, stem, leaf, flower buds, column, lip, petal, sepal and three developmental stages of seeds from an individual plant of P. equestris used for genome sequencing. We provided high-quality transcriptome assemblies and annotated results, enabling comparisons with previously generated Phalaenopsis transcriptome data from the same or different tissues to further understand the highly specialized morphology of orchid flowers and the adaptive radiation of this highly diverse plant group. We also first presented the usage of these datasets using YABBY and NBS-encoding gene families as examples. All the experimental processes involved in the paper are shown in Fig. 1.

We collected one sample for each tissue type, including root, stem, leaf, flower bud, column, lip, petal, sepal and seeds from three developmental stages of P. equestris. Next, we sequenced cDNAs generated from the tissues on an Illumina HiSeq2000 in 90-bp paired-end (PE) reads, with 75-bp paired-end (PE) reads from the leaf tissue. The analysis started with assembling the short reads using the de novo assembly program Trinity and continued with functional analysis using BLASTX. Moreover, we performed quality control assessments at each step from the raw reads to the annotation datasets. Finally, we used YABBY and NBS-encoding gene families as examples of the usage of these datasets.
Methods
These methods are expanded from descriptions previously published in Nature Genetics3.
Plant sample collection and conditions
The experiments were performed on nine butterfly orchid P. equestris tissues: root, stem, leaf, flower buds, column, lip, petal, sepal and three developmental stages of seeds. All these tissues were obtained from the adult plant that was also used for genome sequencing and were grown at the National Orchid Conservation Centre of China and stored at −80 °C for further experiments.
Experimental design
One sample of each tissue of P. equestris—root, stem, leaf, flower buds, column, lip, petal, sepal and three developmental stages of seeds—was taken for RNA sequencing. The stem without the bud was from a three-year-old plant. The seeds we used for RNA sequencing were sown on 1/2 Murashige-Skoog (MS) medium for 4, 7 and 12 days, respectively.
RNA collection
Total RNA was extracted from each tissue using an RNAprep Pure Plant Kit (Qiagen). The quality and quantity of each RNA sample was assessed by agarose gel electrophoresis (Fig. 2).

CL, column; Fb, flower bud; L5, root; L6, stem; LP, lip; M, Marker DL2000; PHA, leaf; PT, petal; SP, sepal; 12, 12-day seed; 7, 7-day seed; 4, 4-day seed.
Library construction and illumina sequencing
A total of 3 μg RNA per sample was used to construct the cDNA library. Poly(A) mRNA was purified from total RNA using oligo(dT)-attached magnetic beads. Fragmentation buffer was used to cleave the mRNA into short fragments, which were then used as templates for the random hexamer-primed synthesis of first-strand cDNA. Second-strand cDNA was synthesized using buffer, dNTPs, RNase H, and DNA polymerase I. From this cDNA, a paired-end library was synthesized using a Genomic DNA Sample Preparation Kit (Illumina), according to the manufacturer’s instructions. Short fragments were purified with a QIAquick Gel Extraction Kit (Qiagen) and were then resolved with EB buffer for end repair and the addition of poly(A) tails. The short fragments were then connected with sequencing adapters, and suitable fragments were separated by agarose gel electrophoresis. Finally, the sequencing library was constructed by PCR amplification, and eleven cDNA libraries were generated. Sequencing using the Illumina HiSeq2000 system was performed to generate 90-bp paired-end (PE) reads, except in the leaf, for which 75-bp paired-end reads were generated.
De novo assembly and dataset annotation
De novo transcriptome reconstruction was performed using Trinity (version trinityrnaseq-r2013-02-25)17. Trinity was applied using the inchworm method with a minimum contig length of 200 nucleotides. The default settings for Trinity paired-end assembly were used for the remaining parameters. The assembly was further spliced and assembled to acquire non-redundant unigenes that were as long as possible. BLASTX (e-value≤1e−5) was performed to annotate the unigenes based on protein databases, including Nr, KEGG, and COG. The CDSs (coding DNA sequences) and protein sequences of all unigenes were predicted using BLASTX, ESTScan18, and the fifth-order Markov model. First, we utilized protein databases such as Nonredundant (Nr), Kyoto Encyclopaedia of Genes and Genomes (KEGG), and Clusters of Orthologous Groups (COG) to align against the unigenes using BLASTX with an E-value cutoff of 1e−5. The best alignment results were used to determine the sequence directions of the unigenes. Unigenes with sequences that produced matches in only one database were not searched further. When a unigene would not align to any database, ESTScan was used to predict coding regions and to determine the sequence direction. If the above two methods still could not predict the CDSs of the unigenes, we used a fifth-order Markov model to predict the CDSs.
HSP90, HSP70 and YABBY gene family identifications
We used hmmsearch of the Hidden Markov Model (HMM)-based HMMER program (3.3.2)19 to identify all HSP90, HSP70 and YABBY genes. HMM profiles of the HSP90, HSP70 and YABBY gene families (PF00183, PF00012 and PF04690.8 in pfam database20) were used in local searches of the P. equestris (PEQU) database, and deposited to Dryad Digital Repository (Data Citation 1). Subsequently, we used the Blastp program to search for the HSP90, HSP70 and YABBY genes in these transcriptomic protein datasets using the protein sequences of individual putative P. equestris HSP90, HSP70 and YABBY as queries, respectively. To maximize the confidence, only the HSP90 and HSP70 genes with E-values of 0.0 and YABBY genes with E-values ≤1e−20 were chosen, filtered for perfect matches, and aligned using MAFFT21 with an E-INS-I alignment strategy for sequence integrity analysis.
Identification of NBS-encoding genes
The complete set of NBS-encoding sequences was identified from the P. equestris genome3 in a reiterative process. First, all predicted proteins from the annotation of the P. equestris genome were screened using HMMER V.3 (ref. 19) analysis against the raw HMM corresponding to the Pfam NBS (NB-ARC) family (PF00931). The raw NB-ARC HMM was downloaded from the Pfam home page (http://pfam.xfam.org/)20. A set of 58 genes with the NBS motif was selected from the HMM search results with E-values ≤1e−10. In the second analytical step, selected protein sequences were aligned based only on the NBS domain using Muscle22. Next, the alignment was used to construct a P. equestris-specific HMM model. The refined HMM was compared against all predicted proteins from the P. equestris genome, and 65 genes were identified. In the third step, the NBS domains of the 65 identified genes were incorporated into the refined HMM model, which was used to search for related sequences. We also identified the same 65 genes in this step, which indicated that those 65 genes were reliable NBS-encoding gene candidates. Then, we used the Blastp program to search for NBS-encoding genes in these transcriptomic datasets against those 65 genes. To maximize the confidence, the NBS-encoding genes were further confirmed using SMART (http://smart.embl-heidelberg.de/).
Genome annotation methods
The methods for P. equestris genome assembly and annotation (Table 1) were presented in the previous publication3.
Data Records
For this study, we deposited six datasets. The first dataset consists of the genome annotation, constructed library reads and assembly sequences of P. equestris (Data Citations 1 and 2 and Tables 1–3). The genome annotation and scaffolds were deposited to the Dryad Digital Repository (Data Citation 1 and Table 1), while the 37 DNA paired-end library data, contigs and scaffolds were submitted to the NCBI database (Data Citation 2 and Tables 2 and 3). The second dataset consists of the RNA-Seq raw reads. This dataset contains a total of eleven samples (Data Citation 3 and Table 4). The third dataset contains the unigenes of the eleven samples (Data Citation 1 and Table 5). The fourth dataset is the annotation file, which contains the results annotated using all three databases and the predicted CDSs and protein files from the results from the eleven tissues (Data Citation 1 and Table 6 (available online only)). The fifth dataset includes the aligned full-length sequences of the HSP90 and HSP70 gene families, showing the integrity of the assembly (Data Citation 1 and Table 7). The sixth dataset contains the alignment results from 100 randomly selected conserved core eukaryotic genes (CEGs) among Arabidopsis thaliana, P. equestris and eleven transcriptomes for examining the transcript assembly completeness (Data Citation 1). The first dataset described above (Data Citations 1 and 2 and Tables 1–3) was previously published in our related work in the journal Nature Genetics3. The second dataset (Data Citation 3 and Table 4), the third dataset (Data Citation 1 and Table 5) and the fourth dataset (Data Citation 1 and Table 6 (available online only)) are the core of this work and have not been published previously.
Technical Validation
Sequencing quality control
We used two steps for testing sequencing quality. The first step included counts of the total reads and total bases for each sample to ensure that the amounts were approximately of the same order of magnitude. These amounts were 16–70 million reads (Table 8). As a second step, we tested samples using FastQC23 for Q20 and GC content (Table 8).
Assembly quality control
To ensure that the produced contigs were correct following the use of Trinity, we compared our transcriptome model to the published Phalaenopsis transcriptomes. We compared basic statistics, such as the average contig length (Table 9), which was longer than the average transcript size from OrchidBase13 (approximately 350 bp, http://orchidbase.itps.ncku.edu.tw/), and shorter than those from leaves of Phalaenopsis sp. (ref. 15) and Phalaenopsis Brother Spring Dancer ‘KHM190’ (ref. 16), 1,108.07 and 2,094, respectively. We also compared the total transcripts with the number of those mapped to the P. equestris genome3, which has a similar number. We subsequently tested full-length transcripts against the HSP90 gene family24 to examine the completeness of the data. We found only one gene (Unigene017669_ORF) in the leaf (PHA), one gene (Unigene037471_ORF) in the root (L5), and one gene (Unigene029033_ORF) in the flower bud (fb) that were almost full-length; the others were reconstructed perfectly (the fifth dataset in Data Citation 4). We also found that there was partial sequencing missed in the PEQU_19561 gene of P. equestris genome. We also tested the HSP70 gene family, which is constitutively expressed and up-regulated in response to various stressors, such as heat, cold, anoxia, and heavy metal exposure25,26. Only six pairs of unigenes should be merged based on the sequence analysis: Unigene019149_ORF and Unigene019150_ORF in the fb, Unigene052632_ORF and Unigene052633_ORF in the stem (L6), and Unigene020433_ORF and Unigene020432_ORF in the PHA, sepal_c24932_g1_i1_7684 and sepal_c24932_g2_i1_6884 in the sepal, petal_c31129_g2_i1_17690 and petal_c31129_g1_i1_15744 in the petal, column_c50529_g2_i1_17726 and column_c50529_g1_i1_29153 in the column. In addition, two genes of the P. equestris genome had missed sequences: PEQU_21700 and PEQU_20114 (the fifth dataset in Data Citation 1). The HSP70 sequences from the root, lip and three developmental stages of seeds were perfectly reconstructed (the fifth dataset in Data Citation 1). Next, we used Bowtie to map the reads back to the unigenes to test the mapping rate (Table 10)17; more than 85% of the reads were proper pairs, showing a high read utilization rate. Finally, the 248 conserved CEGs were used to assess transcript assembly completeness using CEGMA software27 (Table 10). The completeness of PHA was likely low because fewer reads were returned or because some conserved CEGs are not expressed. The transcript assembly completeness of all other tissues had high values (i.e., greater than 80%). We manually examined 100 randomly selected CEG sequences from A. thaliana to align with PEQU genome sequences and eleven tissue transcriptome homologous genes (the sixth dataset in Data Citation 1). Of these, 82 CEG sequences (82%) were perfectly reconstructed, showing high consistency, although some sequences suggested that partial sequencing was missed in the PEQU genome, such as sequences from At2g36880.1 homologous genes, and some sequences in transcriptomes should be merged, such as sequences from At4g39280.1 homologous genes.
Annotation quality control
We estimated the functional annotation results based on the aforementioned database and detailed information from the Nr database (Table 11 and Fig. 3), which revealed 18,787–32,996 unigenes with low e-values that were aligned versus the Nr database showing similar annotation gene numbers with the P. equestris genome3. Additionally, the statistical results of the predicted CDSs are shown in Table 12.

The x-axis shows the eleven tissues, different colours outline the range of E-values, and the y-axis provides the percentages.
Usage Notes
The data provided in these experimental datasets can be used for the following two purposes. First, it is possible to use the raw reads to conduct new experiments using different analytical methods. Second, each analysis step can be performed differently because all the technical experimental information is publicly available.
De novo assembly
Using the unigenes generated with Trinity, a dataset search for genes of interest can be easily performed by searching for homologues using Blast or by performing a text-based search when using an annotation table. We can also identify the gene families that are expressed in specific tissues. For example, we demonstrated that the YABBY gene family plays a key role in determining leaf polarity28–30. The results indicated that the gene family does not exist in the root and seed (Table 13), a finding that is consistent with their function. Furthermore, we identified disease resistance (R) genes (Table 14), which play important roles in resistance to major plant pathogens31, and NBS domain sequences that are commonly used to identify R genes and to classify the genes into subgroups bearing different functions32. Among these tissues, at least 7 R genes were identified in the 7-day seeds, whereas 24, 21, and 22 genes were found in the flower bud, root and stem, respectively. These findings suggest that the flower bud, stem and root may be more susceptible than 7-day seeds to major diseases or that resistance to various orchid pathogens is related to not only the R gene numbers but also R gene expression. We also found that MADS-box genes mostly existed in flower tissue, suggesting a distinct role for these genes in orchid floral morphogenesis3.
Downstream analysis
Future downstream analyses could entail a comparison of the tissues sequenced in this work to other tissues to determine genes that are differentially expressed in other plant organs. Additionally, because orchids are divided into different ecotypes (epiphytic, lithophytic, and terrestrial plants)5,33, comparing transcriptomes from the same tissues, particularly the root, among different orchid ecotypes could provide new insights into the molecular mechanisms of orchid ecological differentiation.
Additional Information
How to cite this article: Niu, S.-C. et al. De novo transcriptome assembly databases for the butterfly orchid Phalaenopsis equestris. Sci. Data 3:160083 doi: 10.1038/sdata.2016.83 (2016).
References
References
Givnish, T. J. et al. Orchid phylogenomics and multiple drivers of their extraordinary diversification. Proc. Roy. Soc. B 282, 20151553 (2015).
Schiestl, F. P. et al. The chemistry of sexual deception in an orchid-wasp pollination system. Science 302, 437–438 (2003).
Cai, J. et al. The genome sequence of the orchid Phalaenopsis equestris . Nat Genet 47, 65–72 (2015).
Benzing, D. H. The evolution of epiphytism. Vascular plants as epiphytes [Ulrich Lüttge (Ed.)] [15–41] (Berlin, Springer, 1989).
Gravendeel, B., Smithson, A., Slik, F. J. & Schuiteman, A. Epiphytism and pollinator specialization: drivers for orchid diversity? Philosophical Transactions of the Royal Society B: Biological Sciences 359, 1523–1535 (2004).
Silvera, K., Santiago, L. S., Cushman, J. C. & Winter, K. Crassulacean acid metabolism and epiphytism linked to adaptive radiations in the Orchidaceae. Plant Physiol. 149, 1838–1847 (2009).
Masiero, S., Colombo, L., Grini, P. E., Schnittger, A. & Kater, M. M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23, 865–872 (2011).
Tsai, W.-C. et al. Expression analysis of the ESTs derived from the flower buds of Phalaenopsis equestris . Plant Science 170, 426–432 (2006).
Hsiao, Y. Y. et al. Comparison of transcripts in Phalaenopsis bellina and Phalaenopsis equestris (Orchidaceae) flowers to deduce monoterpene biosynthesis pathway. BMC Plant Biol 6, 14 (2006).
Chen, Y. H. et al. Transcription analysis of peloric mutants of Phalaenopsis orchids derived from tissue culture. Cell Research 15, 639–657 (2005).
Hsu, T.-W. et al. Differential gene expression analysis by cDNA-AFLP between flower buds of Phalaenopsis Hsiang Fei cv. H.F. and its somaclonal variant. Plant Science 175, 415–422 (2008).
Hsiao, Y.-Y. et al. Gene discovery using next-generation pyrosequencing to develop ESTs for Phalaenopsis orchids. BMC genomics 12, 360 (2011).
Tsai, W. C. et al. OrchidBase 2.0: comprehensive collection of Orchidaceae floral transcriptomes. Plant Cell Physiol. 54, e7 (2013).
Su, C.-L. et al. De novo assembly of expressed transcripts and global analysis of the Phalaenopsis aphrodite transcriptome. Plant Cell Physiol. 52, 1501–1514 (2011).
Xu, C., Zeng, B., Huang, J., Huang, W. & Liu, Y. Genome-wide transcriptome and expression profile analysis of Phalaenopsis during explant browning. PLoS ONE 10, e0123356 (2015).
Huang, J. Z. et al. A de novo floral transcriptome reveals clues into Phalaenopsis orchid flower development. PLoS ONE 10, e0123474 (2015).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature protocols 8, 1494–1512 (2013).
Iseli, C., Jongeneel, C. V. & Bucher, P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 138–148 (1999).
Eddy, S. R. Profile hidden Markov models. Bioinformatics 14, 755–763 (1998).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Katoh, K. & Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9, 286–298 (2008).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Andrews, S. FASTQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Krishna, P. & Gloor, G. The Hsp90 family of proteins in Arabidopsis thaliana . Cell stress & chaperones 6, 238 (2001).
Sung, D. Y., Vierling, E. & Guy, C. L. Comprehensive expression profile analysis of the Arabidopsis Hsp70 gene family. Plant Physiol. 126, 789–800 (2001).
Li, Q.-B., Haskell, D. W. & Guy, C. L. Coordinate and non-coordinate expression of the stress 70 family and other molecular chaperones at high and low temperature in spinach and tomato. Plant molecular biology 39, 21–34 (1999).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Sarojam, R. et al. Differentiating Arabidopsis shoots from leaves by combined YABBY activities. The Plant Cell Online 22, 2113–2130 (2010).
Floyd, S. K. & Bowman, J. L. Gene expression patterns in seed plant shoot meristems and leaves: homoplasy or homology? Journal of plant research 123, 43–55 (2010).
Bowman, J. L. The YABBY gene family and abaxial cell fate. Current Opinion in Plant Biology 3, 17–22 (2000).
McHale, L., Tan, X., Koehl, P. & Michelmore, R. W. Plant NBS-LRR proteins: adaptable guards. Genome Biol 7, 212 (2006).
Traut, T. W. The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur. J. Biochem. 222, 9–19 (1994).
Roberts, D. L. & Dixon, K. W. Orchids. Curr Biol 18, R325–R329 (2008).
Data Citations
Liu, Z.-J. Dryad Digital Repository http://dx.doi.org/10.5061/dryad.8253q (2016)
Liu, Z.-J. NCBI BioProject PRJNA192198 (2015)
Liu, Z.-J. NCBI Sequence Read Archive SRP059975 (2016)
Acknowledgements
We thank Liang-Sheng Zhang for discussions regarding the YABBY gene family identification and Lorenzo Pecoraro and Ming-He Li for their revision of this manuscript. This work was funded by (Development Funds for Emerging Industries of Strategic Importance of Shenzhen) NYSW20140331010039, (Development Funds for Emerging Industries of Strategic Importance of Shenzhen) JCYJ20140402093332029, and (the Innovation Fund for Forestry Technology of Guangdong) 2014KJCX009.
Author information
Authors and Affiliations
Contributions
Z.-J.L. and Y.-B.L. conceived and implemented the study. G.-Q.Z. prepared the final datasets. S.-C.N., Q.X. and Y.-Q.Z. analysed and acquired the data. W.-C.T., J.-L.H. and C.-K.L. provided partial data. S.-C.N. and Z.-J.L. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Niu, SC., Xu, Q., Zhang, GQ. et al. De novo transcriptome assembly databases for the butterfly orchid Phalaenopsis equestris. Sci Data 3, 160083 (2016). https://doi.org/10.1038/sdata.2016.83
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.83
This article is cited by
-
Expression regulation of MALATE SYNTHASE involved in glyoxylate cycle during protocorm development in Phalaenopsis aphrodite (Orchidaceae)
Scientific Reports (2020)
-
Genome-wide identification and classification of MIKC-type MADS-box genes in Streptophyte lineages and expression analyses to reveal their role in seed germination of orchid
BMC Plant Biology (2019)
-
Post genomics era for orchid research
Botanical Studies (2017)
-
A perspective on orchid seed and protocorm development
Botanical Studies (2017)
