
Abstract
Wild populations of the house mouse (Mus musculus) represent the raw genetic material for the classical inbred strains in biomedical research and are a major model system for evolutionary biology. We provide whole genome sequencing data of individuals representing natural populations of M. m. domesticus (24 individuals from 3 populations), M. m. helgolandicus (3 individuals), M. m. musculus (22 individuals from 3 populations) and M. spretus (8 individuals from one population). We use a single pipeline to map and call variants for these individuals and also include 10 additional individuals of M. m. castaneus for which genomic data are publically available. In addition, RNAseq data were obtained from 10 tissues of up to eight adult individuals from each of the three M. m. domesticus populations for which genomic data were collected. Data and analyses are presented via tracks viewable in the UCSC or IGV genome browsers. We also provide information on available outbred stocks and instructions on how to keep them in the laboratory.
Design Type(s) | parallel group design • species comparison design |
Measurement Type(s) | genetic sequence variation analysis • transcription profiling assay |
Technology Type(s) | whole genome sequencing • RNA sequencing |
Factor Type(s) | Subspecies • geographic location • tissue |
Sample Characteristic(s) | Mus musculus domesticus • Mus musculus musculus • Mus musculus castaneus • Mus spretus • brain • gut wall • heart • kidney • liver • lung • muscle structure • testis • spleen • thyroid gland • Massif Central • Bonn Urban District • Heligoland Islands • Ahvaz • Municipality of Studenec • Almaty City • Mazar-I-Sharif District • Himachal Pradesh State • Madrid |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

Background & Summary
The house mouse (Mus musculus) has a long-standing history as a model system in genetics and biomedical research, with many classical inbred strains available for purchase worldwide. By comparing the genetic make-up of classical inbred strains with those of mice collected in the wild, it became clear that classical inbred strains represent complex genomic mixtures with contributions from different subspecies and species of Mus1–3. While some of this genomic mixture stems from captive breeding of mice from different parts of the world during the early establishment of inbred strains, admixture4,5 and introgression of genomic material across subspecies and species6–9 also occurs in the wild and is thus likely to contribute to the genomic complexity observed in inbred strains. Classical inbred strains were found to exhibit a much reduced amount of genetic variation compared to their wild mice ancestors10. For example, all classical inbred strains share a single mitochondrial lineage derived from M. m. domesticus11, indicating that they all descend from the same female lineage of the wild ancestor.
An appreciation of the genetic diversity found in wild mice came with the advent of molecular mapping techniques that required crosses between lines with informative polymorphisms12,13. This has led to renewed interest in studying the evolutionary history of natural house mouse populations worldwide, with a main focus on clarifying its taxonomy and catalogue genetic variations found in the wild14–16. Currently, three major lineages of Mus musculus, classified as subspecies, are distinguished: the western house mouse Mus musculus domesticus, the eastern house mouse Mus musculus musculus and the southeast-Asian house mouse Mus musculus castaneus. All three lineages have their origin in Southern Asia and diverged roughly 0.5 million years ago, but still share haplotypes and appear to exchange genomic material6,8,9. Hybrid zones have been detected at areas of secondary contact between the subspecies17–19 and these serve for tracing genes involved in hybrid incompatibility20–22 as well as quantitative trait mapping23.
During the past 10,000 years house mice have developed commensalism with humans, which allowed them to a spread across the world. Among the recognized subspecies, M. m. domesticus seems to be the most successful colonizer of new continents during the past few hundred years12,13, partly revealing historical shipping routes of humans24, and has repeatedly colonized small islands25. One such recent island colonization occurred about 400 years ago and resulted in the naming of a new subspecies, i.e., M. m. helgolandicus26. Because of its molecular proximity to M. m. domesticus, we treat the M. musculus population from Heligoland, a small German archipelago in the North Sea, as a member of the subspecies M. m. domesticus in some further analyses.
One of the closest relatives to the Mus musculus subspecies complex is the Algerian mouse Mus spretus27, with populations inhabiting areas around the western Mediterranean Sea. With a divergence time of roughly 2 million years28,29 the Mus spretus lineage serves as an ideal outgroup to Mus musculus. Although viable offspring can be produced from crosses between Mus musculus and Mus spretus in the laboratory, it is morphologically and behaviorally rather distinct, justifying its status as separate species. Nevertheless, some exchange of genomic regions is still possible between these species in the wild7,9,30. A few individuals of Mus spretus are also represented among classical inbred strains31.
The unique combination of genetic and molecular knowledge derived from the classical inbred strains and the profound knowledge of the evolutionary history of wild mouse populations make Mus musculus a prime model for the study of evolution and molecular biology of natural populations12,13,32. We have previously used the Mus musculus model system to analyze patterns of positive and negative selection in the genome8,33–37, hybrid sterility21,22, evolution of copy number variation38, mapping of craniofacial traits23 and the composition and turnover of the microbiota39–41.
Here we describe the genomic resources that we have generated using mice collected in the wild over the past 10 years. These data can serve as a basis for in depth studies at a population level and to inform biomedical research projects on natural polymorphisms that are present in inbred strains. We provide information on a) genomic data for a total of nine populations (Fig. 1), covering the three major house mouse subspecies and one outgroup, b) tissue-specific RNAseq data from three M. m. domesticus populations, and c) details on animal husbandry of wild house mice in a Supplementary File and d) some general analyses and browser tracks for visualization. The genomic dataset is summarized using basic descriptive statistics, such as FST (ref. 42), π (nucleotide diversity43) and Tajima’s D44 as measures of population differentiation and selection, as well as a description of copy-number variation based on sequencing read depth. The RNAseq data are summarized as normalized RNAseq read coverage for each base pair of the genome. All statistics are made available as genome browser tracks (bed and bigWig files) to allow close visual inspection of any genomic region of interest in the UCSC browser45 or other visualization software such as IGV46,47.

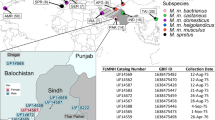
The map is modified from refs 3,13. The blue area depicts M. m. domesticus territory (includes M. m. helgolandicus (3), because of its close molecular proximity to M. m. domesticus), the red area depicts M. m. musculus territory, and the green area depicts M. m. castaneus territory. Mus spretus co-occurs with M. m. domesticus in Spain. The grey area harbors further lineages and possible additional subspecies16. Red arrows symbolize possible migration routes, mostly in post-glacial times during the spread of agriculture. Locations (year caught): 1, Massif Central/France (2005); 2, Cologne-Bonn/Germany (2006); 3, Heligoland/Germany (2012); 4, Ahvaz/Iran (2006); 5, Studenec/Czech Republic (2003); 6, Almaty/Kazakhstan (2002); 7, Afghanistan (2012); 8, Himachal Pradesh/India (2003); S, Madrid/Spain (2004).
Methods
Sampling procedure and sampling locations
The location of populations used for re-sequencing in this study are depicted in Fig. 1. They include three M. m. domesticus populations from Western Europe and Iran, the island subspecies M. m. helgolandicus, three M. m. musculus populations from the Czech Republic, Kazakhstan and Afghanistan and a M. spretus population from Spain. Populations were sampled between 2003 and 2012. The DNA samples used for genome sequencing were obtained either directly from wild caught animals, or from the first or second generation of out-breeding in our animal facility, i.e., they are expected to represent full wild type variation. Some aspects of the genome sequences obtained from the three M. m. domesticus populations, as well as the island subspecies M. m. helgolandicus, have previously been described26,38.
House mice form naturally extended family groups at a given location including breeding among relatives48,49. To obtain an unbiased population sample from a given region ideally requires sampling mice in a way to avoid catching related animals. Therefore, we aimed to collect only a single mouse per trapping location (or, for the purpose of setting up breeding colonies, one male and one female per location) and selected the next trapping location 500 m to 1 km apart. The whole area sampled ideally comprises a diameter of about 50 km. However, depending on the local conditions, following this sampling regime precisely was not always possible. Moreover, some samples were provided by collaborators who have only recorded the general area for trapping, but not the exact location (e.g., the mice from Kazakhstan). Other trapping locations had regional limitations. For example, the island of Heligoland is only 1.7 km2 in size, or the military Camp Marmal, Mazar-e-Sharif (Afghanistan) is only 8.7 km2 in size. In both cases, mice were collected in different localities on the island or military base respectively50. In Supplementary Table 1 we provide exact location information, as much as it is available for all animals involved in the study, as either directly having been sequenced, or as having been parent to one of the animal facility-born offspring of wild mice (see below).
To complement the genomic resources generated in our laboratory, we also re-analyzed previously published genome sequencing data from M. m. castaneus that were collected in the northwest Indian state of Himachal Pradesh51.
Mice were either caught in snap traps, or live traps (‘Mäusewippfalle’ No. 3451002, Firma Ehlert & Partner, 53859 Niederkassel, Germany) or were found dead after rodenticide-based pest management. Cervical dislocation was used to sacrifice mice caught in live traps in the field. Transportation of live mice to the animal facility, maintenance and handling were conducted in accordance with German animal welfare law (Tierschutzgesetz) and FELASA guidelines. Permits for keeping mice were obtained from the local veterinary office ‘Veterinäramt Kreis Plön’ (permit number: 1401-144/PLÖ-004697).
M. m. domesticus breeding scheme
For the M. m. domesticus populations our aim was to generate an RNAseq dataset from the same individuals for which we generated the DNAseq data. In order to standardize mice to the same sex, age and environmental conditions, we did not use wild caught mice but bred wild caught mice for one to two generations in our animal facility. Supplementary Table 2 shows the breeding scheme for the M. m. domesticus mice in the study. In one case, we caught a pregnant female in the wild and used its male offspring born in the facility for DNA and RNA sequencing. For the Iranian mice, we included two wild caught individuals in the DNAseq study (AH15 and AH23), for which we did not generate RNAseq data. For two additional Iranian individuals in the DNA sequencing study, tissue samples were lost and thus no corresponding RNAseq data exist. To compensate for this, we included four individuals representing male offspring from male AH15 and male AH23 respectively (both individuals are part of the DNAseq dataset; see Table 1 (available online only)). Two male offspring from each of these two males are represented as biological replicates in the RNAseq dataset.
Procedures for wild mouse handling
We established wild-derived outbred populations for M. m. domesticus (France, Germany) and M. m. musculus (Kazakhstan and Czech Republic). For the first 11–14 generations live mice obtained from the wild were set up in a cyclical breeding scheme aiming at maintaining maximum genetic variability over time. They were then partly refreshed with newly collected mice from the same original area and a HAN rotational breeding scheme52 was established. Individuals from each of the 4 outbred populations are maintained at the Max-Planck Institute in Plön, Germany, and are available from the authors upon request.
Wild mice are considerably more agile than classical inbred strain mice. Environmental enrichment is necessary and strongly reduces agitated stereotype behavior in wild mice kept under laboratory conditions. Standard mouse chow is provided ad libitum (e.g., Altromin 1,324 from ALTROMIN, 32,791 Lage, Germany). Further details on mouse handling and breeding are provided in Supplementary Material Text 1.
Molecular methods
DNA and RNA extraction
DNA was extracted from liver, spleen, or ear samples using salt extraction53 or DNeasy kits (Qiagen, Hilden, Germany). RNA was extracted only for M. m. domesticus mice from Germany (8 individuals), France (8 individuals) and Iran (8 individuals) using mostly the same individuals, which are included in the whole genome sequencing study (see details above and Supplementary Table 2).
Mice designated for RNA extraction were housed alone after weaning and were routinely visually inspected for health and vigor. Only male mice were included in the study, sacrificed at 12 weeks. All mice were fed standard mouse chow ad libitum (Altromin 1,324 from ALTROMIN, 32,791 Lage, Germany). Mice were sacrificed by CO2 asphyxiation. The coat was sprayed with 75% EtOH to reduce loose hair contaminating the organs during dissection of the animal. Organs were extracted in a specific order to improve comparability. Organs were shock frozen in liquid nitrogen and stored at −80° until RNA was extracted.
The Trizol reagent (Life Technologies, Carlsbad, California, USA) was used according to manufacturers instructions to extract RNA from each organ (Table 2 (available online only)). With the exception of the liver, for which we used only right and left medial lobe, whole organs were processed to minimize heterogeneity of the sample. The extracted RNA was quantified on a Nanodrop and analyzed for integrity on the Agilent Bioanalyzer.
DNA sequencing library preparation
All populations apart of the Afghanistan population were sequenced using the same protocol (see below) in the following batches: Batch 1 included the M. m. domesticus populations from France, Germany and Iran (locations 1, 2 and 4 in Fig. 1). Batch 2 included the 3 mice from Heligoland (location 3 in Fig. 1). Batch 3 included the M. m. musculus populations from the Czech Republic and Kazakhstan (locations 5 and 6 in Fig. 1) as well as the M. spretus population from Spain (location S in Fig. 1). Batch 4 included the M. m. musculus mice from the Afghanistan population (location 7 in Fig. 1), which were sequenced using a more recent Illumina technology (see below).
For whole genome sequencing of batch 1–3 we fragmented 1 μg of DNA of each individual using the 250 bp sonication protocol (Bioruptor, Diagenode, Liège, Belgium). The fragments were end-repaired and adaptor-ligated, including incorporation of sample index barcodes. The products were then purified and amplified (10 PCR cycles) to create the final libraries. The TruSeq DNA LT Sample Prep Kit v2 was used for all steps. After validation (Agilent 2,200 TapeStation), all libraries were quantified using the Peqlab KAPA Library Quantification Kit and the Applied Biosystems 7900HT Sequence Detection System. One library was loaded on two lanes of a Hiseq2000 sequencer and sequenced with a 2×100 bp v3 protocol.
The DNA quality of the Afghanistan mice (batch 4) proved problematic. Therefore, to recover high molecular weight genomic DNA, we ran a 0.7% agarose gel over night and extracted the genomic DNA from the gel using the Zymoclean Large Fragment DNA Recovery Kit (Zymo Research Europe, Freiburg im Breisgau, Germany). For whole genome sequencing we used the Nextera DNA library Prep Kit (Illumina) following manufacturer’s instructions and 50 ng of genomic DNA as starting material. Each sample was run on Agilent Bioanalyzer using the Agilent DNA7500 kit to verify that the fragment sizes were in the 500 bp range. To calculate the final concentration for the sequencing run, the samples were measured with the Quant-iT dsDNA BR Assay Kit on a Nanodrop 3,300 fluorometer. The samples were paired-end sequenced (76 bp) independently on a single flow cell on a NextSeq 500 using the NextSeq 500 High Output v2 150 cycles chemistry.
RNA sequencing library preparation
We used the NEBNext Ultra RNA Library Prep Kit for Illumina with the Poly(A) mRNA Magnetic Isolation module to generate the RNAseq libraries. We used a fragmentation time of 15 min (yielding ~180 bp fragments) and 15 PCR cycles for library enrichment. After validation (Agilent 2,200 TapeStation), all libraries were quantified using the Peqlab KAPA Library Quantification Kit and the Applied Biosystems 7900HT Sequence Detection System. Samples were pooled such that we generated about 12 million paired reads/sample, which were loaded on one lane each of a Hiseq2000 sequencer and sequenced with a 2×100 bp v3 protocol.
Data analysis
Mapping genomic reads and Single Nucleotide Polymorphism (SNP) calling
All sequencing reads (including those of the 10 previously published M. m. castaneus51 genomes) were processed according to a single standardized pipeline, which is outlined in Fig. 2a and described in detail (including commands) in Supplementary Material Text 2. In brief, reads were mapped against the mouse mm10 genome reference sequence54 (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/mouse/) using bwa-mem55. The Picard tools software suite (http://broadinstitute.github.io/picard/) was used for sorting, marking and removing duplicates. Raw SNP and indel calls were obtained from the alignment files following precisely the GATK56 ‘Best Practice’ instructions on joint genotyping of all samples together. The raw .vcf files were subjected to the GATK VSQR SNP filtering step, which uses known variants as training data to predict whether a new variant is likely a true positive, or a false positive. As training data we used the file ‘mgp.v5.merged.snps_all.dbSNP142.vcf’ downloaded from ftp://ftp-mouse.sanger.ac.uk/current_snps/57 which was filtered for ‘PASS’ SNPs. In addition, we used very stringent hard filtering criteria on our own dataset, and included these SNPs as training sets as well (see details in Supplementary Material Text 2). Due to an absence of a reliable indel reference dataset we did not generate VSQR calls for indels. Thus, all indels called in the .vcf file should be considered ‘raw’.

See Supplementary Material Text 2 for full details. Analysis steps for which files are provided are marked with ‘ftp’.
On our ftp website we provide a .vcf file with all SNPs and indels where we flag all SNPs within the 90% VSQR tranche as ‘PASS’ SNPs (this means that we accept all variants until we reach 90% of our known ‘truth’ set). This is a rather conservative filter on SNP quality, but users are free to perform their own filtering using their own training set and parameters for filtering on the raw .vcf file.
Transcriptome sequencing of M. m. domesticus populations
Transcriptome sequencing reads were processed according to the pipeline depicted in Fig. 2b. In short, they were trimmed according to quality values using Trimmomatic58, removing bases below Q20 and maintaining an average read quality above Q25. Pairs with one read below the quality thresholds were removed from the analyses. Quality-checked (QC) reads were mapped against the mouse mm10 genome reference using TopHat2 (ref. 59) and using the default settings for paired-end samples. The output alignments were sorted and indexed with samtools60. The sorted alignments were assigned to the version 82 of Ensemble Mouse gtf annotation61 using featureCounts from the subreads suite62 in paired-end (-p) exon mode (default). The gtf file contained only linear complete chromosomes (no scaffolds, no mitochondria). The unmapped pairs were counted with featureCounts from the ‘<unmapped.bam>’ file TopHat2 generates. Percentages are reported relative to the total number of QC reads. On average (across all tissues and all samples) 93% of the total number of QC reads could be mapped to the mm10 genome (range 86.5–96%, Table 2 (available online only), Supplementary Table 3). This number also includes spliced reads, which Tophat2 detects. This number dropped to 57% (range 33–66%) for reads uniquely mapping to ENS 82 annotated features (i.e., exons, Supplementary Table 3).
Copy number variation (CNV) analyses
We used the sequencing read depth approach implemented in the CNVnator software63 to predict CNV calls relative to the mouse mm10 reference assembly. We have previously experimentally confirmed that this is a reliable approach38. Optimal bin size for each individual was chosen such that the ratio of the average read depth signal to its standard deviation was between 4 and 5. Bin size ranged from 100–1,500 bp and was inversely proportional to genome coverage. Only linear complete chromosomes were considered. Calls intersecting annotated gaps in the reference genome were not considered. The CNV detection statistics are provided in Table 3 (available online only). Haploid copy numbers for each detected CNV, either per population or per individual, are included in the bed files for the UCSC browser tracks (available at the ftp site).
Visualization of data within and between populations
For the genomic data we set up UCSC45 genome browser tracks for 10 kb windows of nucleotide diversity π, Tajima´s D and FST, calculated using vcftools64, and CNV tracks. The 90% truth VSQR-filtered ‘PASS’ data were used for all vcftools calculations, allowing only bi-allelic SNPs and a maximum of 20% of missing data per SNP. The output tables were converted to bigWig format using the BigWig utilities65. For the CNV tracks we used bedtools66 to intersect calls from all individuals belonging to the same population. Within each population average copy number across individuals was calculated for every given interval and transformed to log2 values. The genomic browser tracks provided at the ftp site allow a visualization of differences between the populations and species for genomic regions of interest.
For RNAseq data, the coverage (number of reads at a given base pair) is expected to be proportional to the expression level of the gene from which the read originated. In order to compare RNAseq coverage (and thus gene expression) across individuals with slightly varying total numbers of mapped reads, we normalized the data by proportionally sub-sampling reads from the alignment file (*.bam) for the individuals with higher numbers of total mapped reads. Specifically, for each given tissue, we first determined the individual with the lowest number of total mapped reads. This individual is assigned the normalization factor of 1. For each additional individual we calculated the factor c, total number of mapped reads in the individual with the lowest number / total number of reads in sampled individual. Values of c range from 0 to <1. Thus, the individual with the highest number of mapped reads will have a value of c closest to zero. This normalization factor is then used with the samtools view –s x.y command, where x=0 and y=the decimal part of the normalization factor c to generate the normalized alignment file.
To visualize the data as number of reads covering a particular location (i.e., base pair) in the genome, we converted normalized alignment files into bedgraph files, which were further compressed into bigWig format (available on the ftp site). The RNAseq based read coverage for each basepair in the genome can then be visualized in the UCSC (available as public session under ‘wildmouse’) or IGV browsers46,47. Figure 3 shows two examples of screen shots from IGV sessions. The first is a general overview across all tissues from a section of chromosome 10. The second shows only a single tissue (brain), but with read coverage information for each individual.

(a) Region chr10:61,200,000–63,600,000 in the mouse mm10 genome displaying results for all tissues with the data combined from all individuals of the three M. m. domesticus populations. The population order is for each tissue Germany-Iran-France from top. (b) Region chr7:59,165,000–60,177,000 in the mm10 mouse genome, displaying results for brain normalized RNAseq read coverage with all individuals displayed for each of the three M. m. domesticus populations. Note that there is RNAseq read coverage (i.e., ‘gene expression’) between the annotated coding genes, a region corresponding to known non-coding snoRNAs located within tandem repeats. This expression is limited to the brain among the tissues sampled. Expression differences between the populations are evident.
Data Records
The primary read files for the genome sequences are available at the European Nucleotide Archive (ENA) under project accession number PRJEB9450 (Data Citation 1) for the M. m. domesticus genomes, under project accession number PRJEB11742 (Data Citation 2) and PRJEB14167 (Data Citation 3) for the M. m. musculus and the M. spretus genomes and under project accession number PRJEB2176 (Data Citation 4) for the M. m. castaneus genomes processed in this study. All genome samples and their associated sample designations are listed in Table 1 (available online only). The transcriptome read files are available at ENA under project accession number PRJEB11897 (Data Citation 5). The samples and their sample designation are described in Table 2 (available online only).
The files with the mapped reads (bam), variant calling (vcf) and browser tracks (bigWig) for the genomes and transcriptomes, as well as the IGV session files for the transcriptomes are available at:
http://wwwuser.gwdg.de/~evolbio/evolgen/wildmouse/ where they can be accessed via ftp.
Technical Validation
We used the software angsd67 and its -doDepth 1 command (with options -minMapQ 30 -minQ 20) to assess the quality of each DNA sequencing library with respect to good quality coverage (both mapping and base quality) of the genome. The angsd depth analysis is based on all sequenced and mapped bases, rather than only on called genotypes at variable sites. Thus, this is the most comprehensive way to assess coverage across the whole genome. The average per-base coverage for each sequenced genome is given in Table 1 (available online only) for autosomes, the X-chromosome and the Y chromosome separately. It was calculated as: , where ni is number of bases sequenced at depth i and genome size is 2,395,908,738 for autosomes, 163,487,995 for the X chromosome and 88,124,698 for the Y chromosome.
The autosomal coverage is variable across individuals (both within and between sequencing batch) but should be high enough to obtain good quality SNP calls. For males, the X-chromosome coverage is expected to be half of the autosomal coverage, while for females, the coverage should be similar for X chromosome and autosomes. We can use this fact to obtain independent confirmation of the animal’s sex recorded in the field. For all but one case, the genomic sex (based on X/autosome coverage ratio) matched the sex determined in the field. Individual AL42 was recorded as male in the field, but its genomic data clearly suggest it was a female. For juvenile wild mice it can sometimes be difficult to accurately determine their sex. The lower X-chromosomal coverage for males indicates that some caution should be taken when using called genotypes for this chromosome for population genetic inferences. Approaches that take the genotype likelihoods into account to estimate parameters may be better suited for the X-chromosomal data (i.e., ref. 67). The Y chromosome is extremely poorly covered (see below).
We also assessed the uniformity of coverage across the genome, aiming at identifying specific regions, where few or no reads could be mapped. We ran the angsd -doDepth 1 command for non-overlapping 100 kb windows recording the average (across individuals within subspecies) % bases covered at >10 reads/individual in 100 kb windows. As shown in Supplementary Fig. 4 using all M. m. domesticus individuals as an example, there are some regions in the genome where coverage is low or absent. This pattern was highly correlated in the other subspecies/species (data not shown), suggesting that features of the reference genome (possibly presence of repetitive elements or un-sequenced parts of the reference genome) limit mapping of reads in these regions. Regional variation in coverage is especially striking on the Y-chromosome, where coverage is limited to several short regions within the proximal 10 Mb of the chromosome. This region roughly corresponds to the male-specific short arm of the Y chromosome and its centromere.
Confirmation that mice are naturally inbred
The natural inbreeding status of wild caught mice is expected to be influenced by population history, as well as their tendency to form extended family structures with breeding among relatives48,49. The degree of inbreeding is expected to be higher for small populations and also for populations that recently colonized new habitats, a process which often involves a bottleneck. For the M. musculus mice included in this dataset, Iranian, Afghanistan and Indian mice are closest to the center of origin of this species and thus are expected to be least inbred, as such populations are expected to have been large and stable over time. On the other hand, M. m. helgolandicus inhabits a very small island and experienced a strong founder event during colonization26. For Mus spretus, we do not have a good expectation, as wild populations of this species have never been studied. It is generally assumed that M. spretus does not form extended family structures, as these mice are not human commensals but live in fields, hedges and boundaries to forests.
We used a combination of angsd67 to calculate genotype likelihoods and ngsF68 to calculate inbreeding coefficients for each individual based on randomly selected 1,000 autosomal 10 kb fragments (see Supplementary Material for scripts). As shown in Fig. 4, our expectations are mostly met, with ancestral populations from India and Iran showing the lowest estimated inbreeding coefficient, while Heligoland individuals are close to representing inbred lines. Surprisingly, the Mus spretus individuals from Spain are highly inbred, suggesting that they may have experienced a recent bottleneck.

M. m. domesticus and M. m. helgolandicus populations are highlighted in blue, M. m. musculus populations are highlighted in red, the M. m. castaneus population is highlighted in green and Mus spretus is highlighted in purple.
Confirmation that animals cluster with their respective population
The aim of this analysis was to confirm that each sequenced individual is assigned to its respective population, based on its SNP genotypes. We used the software NgsAdmix69 on the same randomly selected 1,000 autosomal 10 kb fragments as used in the previous analysis. As before, genotype likelihoods were generated by angsd. We ran the NgsAdmix software for K=1 to K=9 (Fig. 5). The likelihood of the data increases dramatically from K=1 to K=4 and then plateaus (data not shown). Such a pattern (see ref. 70) is usually taken as evidence that K=4 fits the data best. K=4 clusters all individuals with their correct subspecies (M. m. helgolandicus correctly clusters with M. m. domesticus26) and species respectively. At K=7, we can subdivide the M. m. domesticus subspecies into its respective populations (with M. m. helgolandicus clustering with the German individuals, confirming the previous results based on microsatellites26). For the M. m. musculus subspecies we find the individuals from the Czech Republic to split off from those from Afghanistan and Kazakhstan. The latter two populations seem to be more closely related. Generally, the genetic clustering analysis suggests that the populations are well defined and differentiated and that there are no recent immigrants from other areas among the sequenced individuals (note that the seeming admixture in K=9 is an artifact of too high K).

Plots for each individual in the sequencing study, for K=4 to K=9 (number of assumed populations). Individuals are sorted by population and subspecies. M. m. domesticus populations are highlighted in blue, M. m. musculus populations are highlighted in red, the M. m. castaneus population is highlighted in green and Mus spretus is highlighted in purple.
Confirmation that VSQR 90 tranche filtering yields expected levels of polymorphism
We assessed the GATK VSQR 90% tranche PASS-filtered SNPs for levels of polymorphism (Watterson’s θ71) within each population and compared the estimates to several reference data sets, which have previously been generated using Sanger sequencing on smaller number of loci. The Indian population is especially informative, as it has been extensively sequenced51,72. For each chromosome and each population we determined the number of segregating sites using the software PopGenome73. Values were summarized over the autosomal genome and converted into Watterson’s θ in % by dividing by ai71 and the number of sites sequenced (Supplementary Table 4). The resulting θW/bp in % for the Indian population (0.74) lies in between the value obtained by ref. 51 (0.91, for 4-fold degenerated sites and 0.83 for intronic sites) and the one obtained by ref. 72 for the same Indian population (0.664). θW for the Western European M. m. domesticus individuals was 0.213 in ref. 72, 0.18 for our German population and 0.2 for our French population. Thus, overall, the VSQR 90% PASS-filtered SNPs dataset seem to reflect the previously inferred levels of polymorphism of house mouse populations quite well.
Analysis of relatedness in the sample
We used the —relatedness2 option of vcftools to assess pairwise individual relatedness among all mice in the dataset, using the KING method74. This analysis is based on GATK called genotypes and the 90% tranche PASS-filtered SNPs. We restricted the dataset to only include autosomal SNPs, thinned to 1 SNP every 1 Mb. We also removed sites that had more than 20% missing data and only included bi-allelic markers in the analysis. As described in Table 1 of ref. 74, expected ranges of kinship coefficients (‘Phi’) are >0.354 for duplicate samples/monozygotic twins, [0.177–0.354] for 1st degree relatives, [0.0884–0.177] for 2nd degree relative, [0.0442–0.0884] for 3rd degree relatives and <0.0442 for unrelated samples. Out of 2,211 pairwise individual relatedness estimates, 35 indicated first (10 pairwise comparisons), second (three pairwise comparisons) and third degree (22 pairwise comparisons) relatives (Supplementary Table 5). However, since we detected third degree relationship also among animals that were unequivocally caught far apart (e.g., SP39-SP68), we only consider first and second degree relatedness relevant here. No duplicate samples were detected (expected Phi=0.5). Relatedness was only detected within populations, and was absent between them. No first or second-degree relatedness was found for the German M. m. domesticus, the Afghanistan M. m. musculus and the Indian M. m. castaneus populations. Most related animals were found in the populations from Iran and Kazakhstan. In the case of the Iranian population the increased relatedness within the sample can be explained by the fact that some breeding adults were used in multiple crosses (see Supplementary Table 2). The relatedness observed in the population from Kazakhstan is best explained by the fact that mice were collected in close proximity, rather than over a larger regional scale.
We can use the known breeding setup of the Iranian mice to confirm the inferred relatedness categories in this population. The KING method detected 2 first-degree relationships in the Iranian population. Male AH15 is indeed the father of JR5-F1C. However, JR-7F1C is the brother of the mother (i.e., uncle) of JR11 and thus a second-degree relative. The second-degree relative identified by the KING method in the Iranian sample is consistent with the breeding scheme, with JR11 and JR15 being half siblings (they have the same father). Two third degree relationships are incorrectly identified and should be second-degree relationships instead (i.e., JR5-F1C is the uncle of JR15 and AH15 is grandfather of JR15) and one third-degree relationship does not have any known breeding history confirming it. The KING method assumes Hardy-Weinberg equilibrium among SNPs with the same underlying allele frequencies. This assumption is most likely being violated in our dataset (given that wild mice are generally inbred, see above), and could explain some of the miss-assignments between categories.
Confirmation of known t-haplotype carriers and identifying t-haplotype carriers in the total dataset
The t-haplotype is complex set of 4 inversions, comprising a 20 cM (30–40 Mbp) region of the proximal third of chromosome 17 in house mice75. It is a selfish genetic element, which causes transmission ratio distortion, with heterozygous t-haplotype carriers predominantly (sometimes up to 99% of times) transmitting the t-haplotype carrying chromosome to their offspring. Homozygous individuals for the t-haplotype, however, die in utero. Despite their massive transmission advantage, t-haplotype carrying individuals are rare in natural populations of mice, but have been found in all recognized subspecies.
We have previously used two published primer pairs76 to genotype individuals in our collections of mouse samples for presence/absence of the t-haplotype. Both primer pairs span t-haplotype diagnostic indels that can be analyzed on an agarose gel (the proximal locus Tcp1 spans a 175 bp indel and the distal locus Hba-4ps spans a 16 bp indel). Three individuals typed with those primers are identical to samples included in the whole genome sequencing study described here: male AL41, male CR16 and female H14. Of those, AL41 and H14 were found to be carriers (heterozygous) of the t-haplotype, while CR16 was wildtype. We use ENSEMBL Blast to determine the location of those primers in the mm10 reference sequence. All primers yielded unique hits. We then extracted all indels spanning the region between the primer locations for all samples from the .vcf-file, and searched for (combinations of) indels matching the sizes above. For the distal locus, we found a single 17 bp indel at position chr17:26,286,509 (‘ TACTACTATGCACTGAA’). For the proximal locus, we found one indel at position chr17:12,921,682 that was identified as ‘ GTTTTTTTTTTT’. Illumina sequencing is not capable to sequence through long homo-polymer stretches which is likely the reason why the identified indel is reported shorter than the expected 175 bp. However, it is the only indel >3 bp in the region and moreover, genotypes at this indel are in almost perfect linkage disequilibrium with genotypes at the distal locus (see Table 1 (available online only)), which is highly unexpected over a distance of 13.3 Mb. The two individuals that we previously found to be positive for the t-haplotype using the PCR primers are also heterozygous for the respective indels in the whole genome dataset, while CR16 was wildtype based on the whole genome sequence. Thus, indels at chr17:12,921,682 and chr17:26,286,509 were used to genotype the remaining individuals in the dataset for the presence/absence of t-haplotypes. All M. musculus individuals positive for the t-haplotype indels were heterozygous for that t-allele. Mus spretus, on the other hand, was homozygous for the proximal t-specific indel, which is consistent with a rather old origin of the t-allele (1–3 Myr77), despite not having any obvious transmission ratio distortion properties in a species outside the M. musculus complex. t+/wt individuals were found in every population apart from the Iranian population and among the three individuals from Heligoland. T-haplotype carriers reached frequencies of 50% in two M. musculus populations (Afghanistan and Czech Republic). 37,5% of the French individuals carried the t-haplotype, as did 30% of Indian individuals, 25% of individuals from Kazakhstan and 12.5% of the German population. The frequency of t-haplotypes in our data is somewhat higher than reported previously (reviewed in ref. 78), however well below the theoretical expectations based on transmission ratio distortion (see ref. 79).
K-mer distributions to determine complexity of RNAseq libraries
We analyzed the k-mer (DNA sequence stretch of length k) frequency spectrum to identify potential problems with the RNAseq libraries, such as DNA contamination and overabundance (potentially due to PCR amplification) of particular sequence stretches. Ideally, libraries are complex, meaning they exhibit great diversity in unique sequences and repeated structures80 and there should be no sign of DNA contamination among the RNA based reads generated. In total, we generated 224 RNAseq libraries from up to 10 tissues in 24 M. m. domesticus individuals. For each library, we ran the software jellyfish81 on all forward reads with a k-mer length of k=12. Using reverse reads yielded the same results (data not shown). For visualization in Supplementary Fig. 5 we randomly choose three individuals from each population and three tissues (brain, testis and liver). The complete set of all 224 RNAseq k-mer distributions are available on our ftp server. The k-mer distribution of the mouse mm10 DNA sequence (red in Supplementary Fig. 5) produces a characteristic line with 2 prominent humps when plotted on a log10–log10 scale. The k-mer distribution for annotated cDNAs in the mouse genome (ENSEMBL version 83 (ref. 61)) does not produce such humps and compared to the DNA mm10 sequence is more ‘complex’, i.e., shows proportionally more unique sequences (with low 12-mer occurrence, but high frequency, left side of plots in Supplementary Fig. 5). The k-mer distribution of the RNAseq forward reads (grey in Supplementary Fig. 5) falls in between the cDNA and genomic DNA profile. Since we generated our RNAseq libraries from poly-adenylated RNAs, comparing the RNAseq profile to annotated cDNA seems appropriate. Most notably, the RNAseq k-mer profile lacks the characteristic humps of the genomic DNA k-mer profile, suggesting that the RNA libraries are not contaminated with DNA. Moreover, the diversity of unique sequences matched the cDNA profile much better than the DNA profile. Very similar patterns have been observed for human RNAseq data, that have been deemed good quality (see Supplementary Fig. 6A in ref. 80).
Additional information
How to cite this article: Harr, B. et al. Genomic resources for wild populations of the house mouse, Mus musculus and its close relative Mus spretus. Sci. Data 3:160075 doi: 10.1038/sdata.2016.75 (2016).
References
References
Yang, H., Bell, T. A., Churchill, G. A. & Pardo-Manuel de Villena, F. On the subspecific origin of the laboratory mouse. Nat. Genet. 39, 1100–1107 (2007).
Yang, H. et al. A customized and versatile high-density genotyping array for the mouse. Nat. Methods 6, 663–666 (2009).
Didion, J. P. & de Villena, F. P.-M. Deconstructing Mus gemischus: advances in understanding ancestry, structure, and variation in the genome of the laboratory mouse. Mammalian Genome 24, 1–20 (2013).
Payseur, B. A. & Nachman, M. W. The genomics of speciation: investigating the molecular correlates of X chromosome introgression across the hybrid zone between Mus domesticus and Mus musculus. Biological Journal of the Linnean Society 84, 523–534 (2005).
Teeter, K. C. et al. The variable genomic architecture of isolation between hybridizing species of house mice. Evolution 64, 472–485 (2010).
Bonhomme, F. et al. Species-wide distribution of highly polymorphic minisatellite markers suggests past and present genetic exchanges among house mouse subspecies. Genome Biol 8, R80 (2007).
Song, Y. et al. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Curr. Biol. 21, 1296–1301 (2011).
Staubach, F. et al. Genome patterns of selection and introgression of haplotypes in natural populations of the house mouse (Mus musculus). PLoS Genet. 8, e1002891 (2012).
Liu, K. J. et al. Interspecific introgressive origin of genomic diversity in the house mouse. Proc. Natl. Acad. Sci. USA 112, 196–201 (2015).
Salcedo, T., Geraldes, A. & Nachman, M. W. Nucleotide variation in wild and inbred mice. Genetics 177, 2277–2291 (2007).
Goios, A., Pereira, L., Bogue, M., Macaulay, V. & Amorim, A. mtDNA phylogeny and evolution of laboratory mouse strains. Genome Res. 17, 293–298 (2007).
Guenet, J. L. & Bonhomme, F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends in Genetics 19, 24–31 (2003).
Phifer-Rixey, M. & Nachman, M. W. Insights into mammalian biology from the wild house mouse Mus musculus. eLife 4, e05959 (2015).
Rajabi-Maham, H., Orth, A. & Bonhomme, F. Phylogeography and postglacial expansion of Mus musculus domesticus inferred from mitochondrial DNA coalescent, from Iran to Europe. Mol. Ecol. 17, 627–641 (2008).
Duvaux, L., Belkhir, K., Boulesteix, M. & Boursot, P. Isolation and gene flow: inferring the speciation history of European house mice. Mol. Ecol. 20, 5248–5264 (2011).
Hardouin, E. A. et al. Eurasian house mouse (Mus musculus L.) differentiation at microsatellite loci identifies the Iranian plateau as a phylogeographic hotspot. Bmc Evolutionary Biology 15, 26 (2015).
Sage, R. D., Heyneman, D., Lim, K. C. & Wilson, A. C. Wormy mice in a hybrid zone. Nature 324, 60–63 (1986).
Tucker, P. K., Sage, R. D., Warner, J., Wilson, A. C. & Eicher, E. M. Abrupt cline for sex-chromosomes in a hybrid zone between 2 species of mice. Evolution 46, 1146–1163 (1992).
Jing, M. et al. Phylogeography of Chinese house mice (Mus musculus musculus/castaneus): distribution, routes of colonization and geographic regions of hybridization. Mol. Ecol. 23, 4387–4405 (2014).
Janoušek, V. et al. Genome-wide architecture of reproductive isolation in a naturally occurring hybrid zone between Mus musculus musculus and M. m. domesticus. Mol. Ecol. 21, 3032–3047 (2012).
Turner, L. M., Schwahn, D. J. & Harr, B. Reduced male fertility is common but highly variable in form and severity in a natural house mouse hybrid zone. Evolution 66, 443–458 (2012).
Turner, L. M. & Harr, B. Genome-wide mapping in a house mouse hybrid zone reveals hybrid sterility loci and Dobzhansky-Muller interactions. eLife 3, doi:10.7554/eLife.02504 (2014).
Pallares, L. F., Harr, B., Turner, L. M. & Tautz, D. Use of a natural hybrid zone for genomewide association mapping of craniofacial traits in the house mouse. Mol. Ecol. 23, 5756–5770 (2014).
Jones, E. P., Eager, H. M., Gabriel, S. I., Johannesdottir, F. & Searle, J. B. Genetic tracking of mice and other bioproxies to infer human history. Trends in Genetics 29, 298–308 (2013).
Hardouin, E. A. et al. House mouse colonization patterns on the sub-Antarctic Kerguelen Archipelago suggest singular primary invasions and resilience against re-invasion. Bmc Evolutionary Biology 10, 325 (2010).
Babiker, H. & Tautz, D. Molecular and phenotypic distinction of the very recently evolved insular subspecies Mus musculus helgolandicus ZIMMERMANN, 1953. Bmc Evolutionary Biology 15, 160 (2015).
Lundrigan, B. L., Jansa, S. A. & Tucker, P. K. Phylogenetic relationships in the genus Mus, based on paternally, maternally, and biparentally inherited characters. Syst. Biol. 51, 410–431 (2002).
Suzuki, H., Shimada, T., Terashima, M., Tsuchiya, K. & Aplin, K. Temporal, spatial, and ecological modes of evolution of Eurasian Mus based on mitochondrial and nuclear gene sequences. Mol. Phylogenet. Evol. 33, 626–646 (2004).
Galtier, N. et al. Mouse biodiversity in the genomic era. Cytogenet Genome Res 105, 385–394 (2004).
Orth, A. et al. Natural hybridization between 2 sympatric species of mice, Mus musculus domesticus L. and Mus spretus Lataste. C. R. Biol 325, 89–97 (2002).
Dejager, L., Libert, C. & Montagutelli, X. Thirty years of Mus spretus: a promising future. Trends in Genetics 25, 234–241 (2009).
Macholán M., Baird S. J. E., Munclinger P. & Pialek J. (eds.) Evolution of the house mouse (Cambridge University Press, 2012).
Ihle, S., Ravaoarimanana, I., Thomas, M. & Tautz, D. An analysis of signatures of selective sweeps in natural populations of the house mouse. Mol. Biol. Evol. 23, 790–797 (2006).
Harr, B. et al. A change of expression in the conserved signaling gene MKK7 is associated with a selective sweep in the western house mouse Mus musculus domesticus. J Evol Biol 19, 1486–1496 (2006).
Teschke, M., Mukabayire, O., Wiehe, T. & Tautz, D. Identification of selective sweeps in closely related populations of the house mouse based on microsatellite scans. Genetics 180, 1537–1545 (2008).
Halligan, D. L. et al. Positive and negative selection in murine ultraconserved noncoding elements. Mol. Biol. Evol. 28, 2651–2660 (2011).
Halligan, D. L. et al. Contributions of protein-coding and regulatory change to adaptive molecular evolution in murid rodents. PLoS Genet. 9, e1003995 (2013).
Pezer, Ž., Harr, B., Teschke, M., Babiker, H. & Tautz, D. Divergence patterns of genic copy number variation in natural populations of the house mouse (Mus musculus domesticus) reveal three conserved genes with major population-specific expansions. Genome Res. 25, 1114–1124 (2015).
Linnenbrink, M. et al. The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol. Ecol. 22, 1904–1916 (2013).
Wang, J. et al. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc. Natl. Acad. Sci. USA 111, E2703–E2710 (2014).
Wang, J. et al. Analysis of intestinal microbiota in hybrid house mice reveals evolutionary divergence in a vertebrate hologenome. Nature Communications 6, 6440 (2015).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population-structure. Evolution 38, 1358–1370 (1984).
Nei, M. & LI, W. H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76, 5269–5273 (1979).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Robinson, J. T. et al. Integrative genomics viewer. Nature Biotechnology 29, 24–26 (2011).
Thorvaldsdottir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics 14, 178–192 (2013).
Berry, R. J. & Bronson, F. H. Life history and bioeconomy of the house mouse. Biological Reviews 67, 519–550 (1992).
Montero, I., Teschke, M. & Tautz, D. Paternal imprinting of mating preferences between natural populations of house mice (Mus musculus domesticus). Mol. Ecol. 22, 2549–2562 (2013).
Schlegel, M. et al. Spielen Nagetiere als Überträger von Zoonoseerregern im Einsatzgebiet der Bundeswehr in Afghanistan eine Rolle? Wehrmed. Mschr 8-9, 203–207 (2012).
Halligan, D. L., Oliver, F., Eyre-Walker, A., Harr, B. & Keightley, P. D. Evidence for pervasive adaptive protein evolution in wild mice. PLoS Genet. 6, e1000825 (2010).
Rapp, K. G. HAN-rotation, a new system for rigorous outbreeding. Zeitschrift für Versuchstierkunde 14, 133–142 (1972).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215 (1988).
Waterston, R. H. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Keane, T. M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 14, R36 (2013).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Cunningham, F. et al. Ensembl 2015. Nucleic Acids Res. 43, D662–D669 (2015).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Kent, W. J., Zweig, A. S., Barber, G., Hinrichs, A. S. & Karolchik, D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 (2010).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics 15 (2014).
Vieira, F. G., Fumagalli, M., Albrechtsen, A. & Nielsen, R. Estimating inbreeding coefficients from NGS data: Impact on genotype calling and allele frequency estimation. Genome Res. 23, 1852–1861 (2013).
Skotte, L., Korneliussen, T. S. & Albrechtsen, A. Estimating Individual Admixture Proportions from Next Generation Sequencing Data. Genetics 195, 693–69 (2013).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7, 256–276 (1975).
Geraldes, A., Basset, P., Smith, K. L. & Nachman, M. W. Higher differentiation among subspecies of the house mouse (Mus musculus) in genomic regions with low recombination. Mol. Ecol. 20, 4722–4736 (2011).
Pfeifer, B., Wittelsbürger, U., Ramos-Onsins, S. E. & Lercher, M. J. PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936 (2014).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Ardlie, K. G. & Silver, L. M. Low frequency of t haplotypes in natural populations of house mice (Mus musculus domesticus). Evolution 52, 1185–1196 (1998).
Planchart, A., You, Y. & Schimenti, J. C. Physical mapping of male fertility and meiotic drive quantitative trait loci in the mouse t complex using chromosome deficiencies. Genetics 155, 803–812 (2000).
Delarbre, C. et al. Phylogenetic distribution in the genus Mus of t-complex-specific DNA and protein markers: inferences on the origin of t-haplotypes. Mol. Biol. Evol. 5, 120–133 (1988).
Huang, S. W., Ardlie, K. G. & Yu, H. T. Frequency and distribution of t-haplotypes in the Southeast Asian house mouse (Mus musculus castaneus) in Taiwan. Mol. Ecol. 10, 2349–2354 (2001).
Manser, A., Lindholm, A. K., König, B. & Bagheri, H. C. Polyandry and the decrease of a selfish genetic element in a wild house mouse population. Evolution 65, 2435–2447 (2011).
Anvar, S. Y. et al. Determining the quality and complexity of next-generation sequencing data without a reference genome. Genome Biology 15, 555 (2014).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Data Citations
European Nucleotide Archive PRJEB9450 (2016)
European Nucleotide Archive PRJEB11742 (2016)
European Nucleotide Archive PRJEB14167 (2016)
European Nucleotide Archive PRJEB2176 (2016)
European Nucleotide Archive PRJEB11897 (2016)
Acknowledgements
This work was mostly financed by institutional resources of the Max-Planck Society, a DFG grant to B.H. and M.T. (HA 3139/4-1) and an ERC grant to D.T. (NewGenes, 322564). We thank Sonja Ihle, Susanne Krächter, Ruth Rottscheidt for contributing to collecting animals in the wild and our animal care takers for active involvement of optimizing the scheme for wild mouse keeping. The initial analysis of mice from Afghanistan was funded by contract-research-project for the Bundeswehr Medical Service M/SABX/005. We thank Bastian Pfeifer for help with software package PopGenome, Leslie Turner for discussion and Daniel M. Hooper and Trevor Price for helpful comments on the manuscript. D.T. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Contributions
B.H. contributed to study design, collected mice, prepared samples, established the genomic mapping pipeline, analyzed DNAseq data, and wrote the manuscript. E.K. contributed to and ran the genomic mapping pipelines. R.N. prepared samples, generated and ran the RNAseq mapping pipelines and analyzed RNAseq data. M.T. contributed to study design, collected mice and prepared samples. C.P. collected mice and developed the wild mouse keeping procedures. Z.P. conducted the copy number analyses. H.B. collected mice and prepared samples. M.L. collected mice and prepared samples. I.M. collected mice and prepared samples. R.S. collected mice and prepared samples. M.A. collected mice. M.P.M. collected mice. M.S. collected mice. A.B. prepared RNA samples. R.G.U. collected mice. J.A. conducted the sequencing. M.F. conducted the sequencing. S.K. conducted the sequencing. D.T. organized the study, wrote the manuscript, with input from the co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Harr, B., Karakoc, E., Neme, R. et al. Genomic resources for wild populations of the house mouse, Mus musculus and its close relative Mus spretus. Sci Data 3, 160075 (2016). https://doi.org/10.1038/sdata.2016.75
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2016.75
This article is cited by
-
Detecting haplotype-specific transcript variation in long reads with FLAIR2
Genome Biology (2024)
-
Competitive ability is a fast-evolving trait between house mouse populations (Mus musculus domesticus)
Frontiers in Zoology (2022)
-
Taxonomic assessment of two wild house mouse subspecies using whole-genome sequencing
Scientific Reports (2022)
-
Population structure and inbreeding in wild house mice (Mus musculus) at different geographic scales
Heredity (2022)
-
Selection shapes the landscape of functional variation in wild house mice
BMC Biology (2021)
