
Abstract
A major question in neuroscience is how diverse subsets of synaptic connections in neural circuits are affected by experience dependent plasticity to form the basis for behavioral learning and memory. Differences in protein expression patterns at individual synapses could constitute a key to understanding both synaptic diversity and the effects of plasticity at different synapse populations. Our approach to this question leverages the immunohistochemical multiplexing capability of array tomography (ATomo) and the columnar organization of mouse barrel cortex to create a dataset comprising high resolution volumetric images of spared and deprived cortical whisker barrels stained for over a dozen synaptic molecules each. These dataset has been made available through the Open Connectome Project for interactive online viewing, and may also be downloaded for offline analysis using web, Matlab, and other interfaces.
Design Type(s) | organism part comparison design • parallel group design • injury design • sensory deprivation |
Measurement Type(s) | immunohistochemistry assay |
Technology Type(s) | Fluorescence Microscopy |
Factor Type(s) | intervention |
Sample Characteristic(s) | Mus musculus • adult organism |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
Many hundreds of distinct proteins are involved in the development of synapses and the mechanics of synaptic signaling1–7, and this complex molecular architecture exhibits striking diversity between brain regions, cell types, and even individual synapses belonging to the same neuron8. This complexity at first seems daunting, but may in fact enable researchers to better characterize synaptic populations and their experience-dependent modification. In particular, the definition of different synapse types based on characteristic expression of distinct molecular constituents by may enable identification of subpopulations involved in learning, while identifiable patterns of molecular modifications at existing connections may be used to reveal functional changes in synaptic strength8.
Array tomography (ATomo) is uniquely well suited to proteomic mapping of synaptic circuits. Ultrathin sectioning of resin-embedded tissue samples enables immunohistochemical multiplexing and high-resolution imaging of millions of synapses in situ, followed by computational reconstruction into precisely aligned image volumes9,10. The ability to measure the molecular composition of many individual synapses in the context of the larger circuits they comprise should greatly enhance our understanding of the roles of diverse synapse subsets in neuronal information storage and plasticity. (See Figure 1 for a summary of the method.)

Top Row: array production. Fixed brain tissue is dissected into a small sample, in this case a tissue punch, and embedded in resin (generally LR White). This embedded sample is sectioned into ribbons of ultrathin (50–200 nm) serial sections, which are each affixed to a microscope coverglass to form a stable array. Middle Row: imaging and image processing. A ribbon array is stained with antibodies against selected antigens, and indirect immunofluorescence (IF) is imaged using a high-resolution objective. The antibodies can be removed from the ribbon using a high-pH elution solution, and the array can then be used again for multiple cycles of immunostaining and imaging. Image processing software improves the resolution of the resulting images. Bottom Row: volume reconstruction. Custom software is used for stitching, registration, and alignment of acquired images into volumetric reconstructions of the original tissue sample. ‘Mosaics’ comprising multiple microscope fields of view are stitched into individual images of the same location in each serial section across the ribbon. Images of each serial section from different imaging sessions (with different antibody stains applied) are then registered into the same 2-dimensional data space. Serial sections are then three-dimensionally aligned with one another across the ribbon. Software used for image processing and reconstruction can be found at http://smithlabsoftware.googlecode.com.
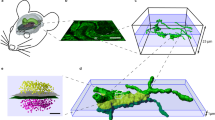
To enable this analysis, we have used ATomo to image the synaptic molecular architecture of neighboring whisker-associated columns of mouse somatosensory cortex (S1). The whisker region of S1 is organized as a grid-like spatial map, with rows of thalamically-innervated layer 4 (L4) ‘barrels’ corresponding to the layout of whiskers on the face11,12 (see Figure 2). The circuitry of this cortical barrel field is highly plastic, able to reorganize itself quickly if the whisker layout is altered13. The neurons immediately above and below a given barrel (the ‘barrel column’) normally respond primarily to stimulation of a single principal whisker (PW), but also exhibit weaker surround-whisker (SW) responses. Loss of input from the column’s PW depresses the column’s response to this whisker, while potentiating responses to spared SWs, which can come to dominate circuit activity in the deprived column14,15. This produces a remapping of the barrel field such that the representations of spared whiskers expand into neighboring deprived columns. Plasticity of many different components of the barrel column circuit have been implicated in this remapping, including excitatory and inhibitory synapses in thalamorecipient L4, superficial L2/3 and deep L5 (refs. 16, 17 and 18).

(a) Chessboard pattern of whisker deprivation. (b) Spared whiskers (magenta) surrounding the C2 whisker (cyan) were stimulated in anesthetized mice. Intrinsic optical signal (IOS) imaged transcranially over left somatosensory cortex (S1). (c) In vivo images of D1-whisker stimulation-evoked IOS peak (pseudocolor) and cerebral surface vasculature (grayscale). (d) Fixed and dissected left S1 cortex with tissue paint and registered IOS peaks (pseudocolor). (e) Remainder tissue after removal of tissue punch centered on the C2 whisker column. (f) Cytochrome oxidase (CO)-stained 80 micron-thick section of remainder tissue registered to the intact remainder tissue, with barrel field traced to confirm the correct punch localization (gray overlay). (g) Estimated barrel column positions within embedded tissue punch (yellow outlines) based on vascular and tissue paint features (cf. red traced blood vessels in panel f) with estimate of optimal cross-section through the C1 and C2 barrel columns parallel to the C-row axis (dashed cyan line). (h) Portion of ATomo ribbon imaged for DAPI at 10x magnification. (i) Maximum-intensity z-projection (MIP) of volume reconstruction of 25x magnification images of ribbon in h. Left: DAPI (gray). Right: DAPI (gray) and YFP (green). High-resolution imaging is targeted to C1 & C2, L3-L5a (red outlines). (j) MIP of multi-session volume reconstruction of 63x magnification images of region shown in (i), with YFP (grey) vGluT1 (green) and vGluT2 (red).
In our experiments, we trimmed alternating facial whiskers of adult mice in a ‘chessboard’ pattern, every 2-3 days for 7 days, which has been shown to produce significant functional and structural plasticity in barrel cortex well into adulthood19–21. We developed a method to precisely dissect pairs of neighboring barrel columns for ATomo imaging, and produced volume reconstructions of stains against many functionally important synaptic molecules in these adjacent spared and deprived columns. Additionally, this data contains cell-type specific transgenic labeling of layer 5 pyramidal neurons with YFP22,23, as well as immunohistochemical labeling of PV+ interneurons24–26. Thus, synaptic subpopulations may be classified based on characteristic molecular signatures or association with YFP+ pyramidal neurons or PV+ interneurons, and the prevalence of these populations as well as changes in expression of molecules thought to play a role in experience-dependent plasticity may be compared between spared and deprived columns.
The Chessboard Dataset described here comprises over 6 million cubic microns of neocortical tissue within a well-defined microanatomical structure, stained for over a dozen different synaptic proteins and imaged at synaptic resolution. As such, it is likely to be a valuable resource for researchers interested in statistical characterization of the diversity of protein expression patterns in mouse neocortical synapses and the effects of experience-dependent plasticity on these patterns8. We make this dataset publicly available alongside two supplementary datasets (see Data Records) in the hopes that other researchers will apply their own creative approaches to discovering patterns of synapse molecular diversity and novel signatures of experience dependent cortical learning.
Methods
Animals
All procedures related to the care and treatment of animals were approved by the Administrative Panel on Laboratory Animal Care at Stanford University. Animals used to generate these datasets were derived from hemizygous transgenic founder mice of the thy1-yfp H line22 purchased from Jackson Labs (Strain Name: B6.Cg-Tg(Thy1-YFP)HJrs/J, Stock number: 003782). Mouse Ex1 was excluded from the Chessboard Dataset (see Data Records) because it was found not to express YFP. Thereafter, we used the Transnetyx genotyping service to restrict experiments to YFP-positive individuals based on the results of automated rtPCR comparison with JAX gene sequences.
Chessboard whisker deprivation
We induced experience dependent map plasticity in 8 adult male mice (P63-70) by unilaterally trimming facial whiskers on the right side of the face in a chessboard pattern (that is, alternating spared and trimmed whiskers) every 2-3 days for 7 days (Figure 2a). We trimmed whiskers to within 1 mm of the face, and did not observe significant regrowth between trimming sessions. Of the 7 mice included in the Chessboard Dataset (see Data Records), 4 had the chessboard pattern centered on a spared C2 whisker (C2 spared), and 3 had the chessboard pattern centered on a deprived C2 whisker (C2 deprived). Animals were group-housed and supplied with cardboard tubes for mild enrichment and to discourage whisker barbering by cagemates27. Mice were excluded from these dataset if any whisker loss was observed in addition to the chessboard trimming.
Rodent surgery
Following 7 days of chessboard deprivation, we imaged whisker-evoked intrinsic optical signals (IOS)28 to localize barrel columns C1 and C2 for dissection. Mice were sedated with intraperitoneal injections of 0.05 ml chlorprothixene and anesthetized with isoflurane (0.8% in O2). Mice were kept on a heating pad, and we ensured that mice remained at a stable level of anesthesia by monitoring their core temperature and tracking their respiration rate. We also locally anesthetized the scalp with subcutaneous injections of ~0.01 ml lidocaine. We covered the eyes with petroleum jelly and provided hydration with a pre-operative subcutaneous injection of 0.5 ml physiological saline.
We stabilized the heads of the mice with ear-bars and surgically exposed a region of the skull overlying the left somatosensory cortex. We then affixed a custom metal head-plate with a 2 mm round imaging window to the exposed skull approximately centered on the location of left barrel cortex, and clamped this head plate to a metal post attached to the imaging apparatus. Once the mice were thus head-fixed, we removed the ear-bars, filled the imaging window with warm low-melting-point agarose, and covered the window with a round glass coverslip.
Intrinsic Optical Signal (IOS) imaging
To identify the location of barrel-columns C1 and C2 without producing additional plasticity in these columns by direct stimulation, we used IOS imaging to map the representations of adjacent spared whiskers (Figure 2b). In C2-spared mice, we mapped the locations of the spared A2, B1, D1 and E2 whisker representations, while in C1-spared mice, we mapped the spared A1, B2, D2, and E1 whiskers. We used a custom piezo-electric actuator (PiezoSystems) to stimulate individual right facial whiskers at 10 Hz in 5-second bouts with an inter-stimulus interval of 15 s. The imaging window was illuminated with a 630 nm red/orange LED light (LumiLEDs), and a Pantera 1M30 camera (Dalsa) equipped with a nose-to-nose macro-lens was used to record changes in reflectance through the skull29. We measured IOS with the camera focused 500 microns beneath the cortical surface to maximize signals originating L4 barrels.
IOS peaks associated with the 0.05 Hz period of whisker stimulation bouts were obtained using LabView software designed by David Ferster, which calculated the real-time fast fourier transform (FFT) of changes in reflectance at this frequency. This FFT-based method allows fast and robust detection of stimulus-associated IOS peaks with little interference from other cardiovascular rhythms, which occur at much higher frequencies30. Clear IOS peaks were generally detected within 15 bouts (~5 min) of stimulation. For each whisker, once an IOS peak was established, we photographed the vascular pattern at the surface of the cortex with contrast enhanced by 530 nm green LED illumination (LumiLEDs). Because the position of the camera relative to the imaging window could shift slightly as we switched whiskers, we registered the vascular images and their paired IOS peaks to map the location of each whisker-associated peak relative to the surface vasculature (Figure 2c).
Barrel column dissection
Following IOS imaging, animals were deeply anesthetized with 5% isoflurane, their brains extracted and the left somatosensory cortex dissected and immersed in fixative (4% PFA, 2.5% sucrose in 0.01 M PBS)31. We estimated the locations of the C-row barrel columns by registering in vivo images of the surface vasculature collected under 530 nm illumination to the visible vasculature of the fixed somatosensory cortex tissue. We used tissue paint (Polysciences) to help reveal additional vascular features in the fixed tissue and to highlight the estimated position of the C-row of the barrel field (Figure 2d). We embedded this painted tissue in agarose gel for stability, and used a custom guide-chamber and sharpened oval tube (inner diameter ~1×0.75 mm) to precisely target extraction of a tissue punch centered on the border between barrels C1 and C2 (Figure 2e), as estimated by vascular and tissue-paint features registered to the functionally measured ISI peaks. The punched tissue was then embedded in LR-white resin for ultra-thin sectioning into ATomo ribbons (see ATomo Tissue Processing, below).
Cytochrome oxidase staining
We confirmed that our punch contained the C1 and C2 barrel columns by making 80-micron sections of the somatosensory cortex tissue remaining after the tissue punch procedure (hereafter ‘remainder tissue’), and stained these for CO to reveal the pattern of the barrel field in layer 4. We then registered our standard barrel field map to the CO stain of remainder tissue sections to reveal which barrels overlapped with the punched-out hole in these sections (Figure 2f).
The CO-stained sections also allowed us to estimate the precise position and orientation of the columns contained within the embedded tissue-punch in order to plan the sectioning of ribbons for ATomo (Figure 2g). For this purpose, we registered images of the vascular features and tissue paint at the pia surface at each stage of the dissection procedure. Based on these common features, we were able to register images of 1) the fixed tissue prior to punch extraction, 2) the superficial sections of post-punch remainder tissue, and 3) the surface of the punched tissue before and after embedding. We aligned adjacent sections of the remainder tissue from the superficial sections containing vascular features down to the L4 sections containing the CO-stained barrel field based on vertically projecting capillaries visible from section to section. Following this alignment, the standard barrel field map and embedded punch images were also in registry, and so we could estimate the locations of the embedded barrel columns with great precision.
ATomo tissue processing
We dehydrated the tissue punches containing C1 and C2 columns in ascending alcohols up to 80% to retain YFP fluorescence, and embedded them in LR White resin according to previously published methods9,31,32.
The resin block was oriented based on the registration procedure described above (Figure 2g) and sectioned into ribbons of 70 nm serial sections (~50 sections/ribbon) parallel to the axis of the C-row such that each ribbon comprised ~3.5 micron cross-sections of the embedded C1 and C2 columns and contained all layers of cortex between the pia surface and the subcortical white matter33. We collected 40–100 ribbons from each embedded block, enough to contain all or most of the targeted pairs of barrel columns. Although we could in principle attempt to reconstruct the columns in their entirety, the scale of such a project was outside the capabilities of our current image acquisition and reconstruction pipeline. Instead, for each block we identified the ribbon that best approximated a plane passing through the centers of both neighboring barrels for imaging and reconstruction (Figure 2g). To aid in this selection, we took sequential pictures of the surface of the punch during sectioning to keep track of our progress through the columns, so that we could select ribbons from as near as possible to the C-row center (that is, equidistant from rows B and D).
Note that for most animals, ribbons were cut from the tissue punch starting from the E-side of the C-row (Ex1, Ex2, Ex3, Ex6, Ex10, Ex14) while others were cut from the B-side (Ex12, Ex13), producing different left-right orientations of the columns in the final data volumes (See Table 1).
ATomo ribbon imaging
For all imaging experiments, we first create a low-resolution overview of the ribbon to be imaged by making a mosaic image at low magnification (10x) of a DAPI stain for cell nuclei included in the mounting medium (molecular probes ref S36939) (Figure 2h). To ensure that we selected good quality ribbons for high-resolution immunohistochemical imaging, we imaged DAPI and intrinsic YFP fluorescence in several ribbons surrounding the estimated row center with a 25x magnification objective (Zeiss). These 25x images allowed us to reconstruct large cross-sections of barrel columns (Figure 2i; see ATomo Image Processing and Reconstruction, below), which aided in selecting ribbons taken from closest to the barrel centers, based on their larger barrel hollows visible as lower DAPI and YFP density. Once a ribbon was selected, the 25x DAPI/YFP images were also used to identify boundaries between columns and layers to guide higher magnification imaging of layers 3–5a in columns C1 and C2. Column boundaries could be detected by the higher density of YFP dendrites running between columns. The centers of L4 barrels were identified by their low DAPI density compared to higher density ‘septal’ regions between barrels, which is a characteristic feature of mouse barrel cortex15.
For selected ribbons from each chessboard-deprived animal, we performed 6–8 rounds of triple or quadruple immunostaining, paired with high-resolution imaging with a 63x plan-apochromat objective (Zeiss)34,35. For all sections in each ribbon, we imaged a region of the section corresponding to layers 3-5a in all of the columns present (C1-C2 in 5 animals, C1-C3 in 1 animal). Automated mosaic imaging of the selected region was targeted using custom software (MosaicPlanner; available at https://smithlabsoftware.googlecode.com/). For several ribbons, to minimize focus artifacts resulting from deviations of the 70 nm sections from the objective’s limited focal plane, z-stacks of each mosaic position were imaged and then computationally merged using an extended depth of field algorithm (Zeiss Axiovision).
ATomo image processing and reconstruction
All custom code described in this section is available for download at https://smithlabsoftware.googlecode.com .
Image processing
Following imaging, we used plugins available in the FIJI image processing software as well as custom routines to create high-dimensional volume reconstructions of the imaged tissue with all of the antibody stains in registry. First, all images were background subtracted using a 20 pixel rolling ball filter (FIJI/ImageJ) and deconvolved (Matlab) based on empirical point-spread functions measured with fluorescent tetraspeck beads. Following deconvolution, the effective resolution of the images was 100×100×70 nm (ref. 36).
Mosaic stitching
We used a DAPI nuclear stain present in our mounting medium in all imaging sessions as a fiducial marker both for stitching together adjacent microscope fields of view and registering images of the same tissue sections taken in different imaging sessions (see below).
2D registration
Minor shifts in the position of the coverslip-affixed ribbon between imaging sessions can be simply modeled as rigid linear transformations to achieve appropriate session registration using custom FIJI/ImageJ plugins. These transformations were calculated based on SIFT features detected in the corresponding DAPI images from each imaging session37. These features were filtered using RANSAC38 to detect inliers specifying the model for rigid transformation between sessions, which could then be applied to images from the other channels of that session to bring them into registry with the first session.
3D alignment
Unlike the rigid transformations used for 2d registration between imaging sessions, mechanical deformations of the tissue during the process of sectioning make the transformations needed for 3d alignment of ultrathin serial-section images significantly nonlinear. Synapse-scale alignment was achieved with another custom FIJI/ImageJ plugin by applying the elastic algorithm developed by Saalfeld and colleagues to the Synapsin1 channel39. This channel was used for fine alignment because it densely and reliably stains synapses across multiple 70 nm sections. The alignment calculated from Synapsin1 was then applied to all of the other channels already in registry to produce full multi-channel reconstructed data volumes.
Data Records
There are 3 data records associated with this data descriptor, all derived from serial-section ribbons from 9 chessboard-deprived mice (Table 1), imaged across multiple rounds of staining with a panel of 21 antibodies (Table 2).
1: Chessboard dataset
The primary Chessboard Dataset consists of 12 reconstructed data volumes (Table 3 (available online only)) comprising 20–25 immunohistochemical channels each (Table 4 (available online only)). These data volumes were reconstructed from multiple rounds of imaging of eight ribbons of serial sections containing paired spared and deprived barrel columns from seven chessboard-deprived mice (Table 5 (available online only)).
The Chessboard Dataset is hosted for interactive browsing and download through the Open Connectome Project (OCP) at http://openconnecto.me. (For further instructions, see Data Access, below, as well as Usage Notes: OCP Data Access.) Individual data files for each channel in each volume reconstruction are also available through FigShare (Data Citation 1), as files 8–295 described in Supplementary Table 1.
Chessboard dataset caveats
Please note that the number of animals, imaged ribbons, and derived data volumes differ because (1) two ribbons from mouse Ex12 were imaged and (2) multiple barrel columns from ribbons Ex2R18, Ex3R43, and Ex6R15 were reconstructed separately into 2-3 columnar data volumes per ribbon. Also, note that animal ribbon Ex14-R58 was subject to several significant problems during antibody staining (see Table 5 (available online only)), particularly as relates to the staining for vGluT2. We include the derived data volumes in the present dataset with this caveat, and we have excluded this ribbon from the quantification of vGluT puncta density (See Usage Notes: Analysis Recommendations & Caveats).
Data access
There are two ways to access the Chessboard Dataset through OCP: 1) interactively browsing through entire data volumes over the web through OCP’s CATMAID interface, which is ideal for quick visual inspection of the data, or 2) by downloading specific data regions (called ‘cutouts’) through OCP’s HDF5 interface, which is ideal for handling the data for analysis. OCP also has its own API for those wishing to interact more deeply with the project’s data structures. For the most up-to-date descriptions of OCP’s services, visit http://openconnecto.me/#!services/chru.
Online data viewing
CATMAID stands for Collaborative Annotation Toolkit for Massive Amounts of Image Data (http://catmaid.org/). OCP uses this interface to enable fast, browsable online data viewing as well as collaborative data annotation. To view the data, visit http://openconnecto.me/catmaid and browse projects labeled ‘Array Tomography (Weiler et al.)’. You may view these projects only by selecting Home> Array Tomography in the upper left-hand corner of the browser window. We have curated several useful multi-channel volumes to explore, but users may also construct their own combinations of channels to visualize different molecular relationships using hdf5 data cutouts (see below).
Viewing/downloading data cutouts
To access data volumes for analysis, OCP provides a ‘cutout service’ which allows users to download arbitrary 3d data cubes using the HDF5 interface through Matlab, R, C, C++, or C# or by downloading the NumPy pickle for use with python. (See Usage Notes: OCP Data Access for a guide to using the hdf5 web interface to view or download cutouts.).
For additional documentation of the cutout service, see the OCP wiki page at the following address: http://tinyurl.com/OCP-cutout-wiki. We have prepared sample scripts for data access using MATLAB at https://gist.github.com/ncweiler. The cutout service also enables collaborative annotation of the data (labeling neuronal or synaptic structures, for instance). These metadata annotations are registered to the original data and can be either used privately or shared publicly. See more details on data annotations at http://tinyurl.com/OCP-annotation-wiki.
2: Chessboard dataset low-magnification overview images
Prior to the high-resolution, high-magnification imaging that generated the Chessboard Dataset image volumes, we first imaged intrinsic YFP and DAPI-stained cell bodies in each ribbon at low magnification. These images allowed us to create a roughly aligned reconstruction of the complete ribbons, which we then used to help identify anatomical layers and columns for the subsequent high-magnification imaging that generated the Chessboard Dataset image volumes. 2d projections of these whole-ribbon reconstructions, created using either a standard deviation projection or maximum intensity projection (FIJI), are also made available through FigShare as RGB tiff images to support the identification and analysis of specific anatomical regions (Data Citation 2).
3: Elution-test dataset
The supplementary Elution-Test Dataset consists of data volumes derived from seven short ribbon segments derived from mouse Ex1 (Table 6 (available online only)), each stained and imaged repeatedly with a different panel of antibodies (Tables 7 and 8 (available online only)). Analysis of stain repeatability can help estimate the signal to noise ratio of each antibody—the likelihood that a punctum of stain will be seen on repeated staining of the same tissue (see Technical Validation).
In the Elution-Test Dataset, each ribbon was divided into distinct segments, which were stained separately with different antibody panels. These ribbon segments are designated with the letters A through F. Unlike the Chessboard Dataset, only small fields of view from layer 4 were imaged in each of these ribbons, since the objective of this experiment was antibody testing, rather than analysis of differences between regions. Six of ribbon segments have been reconstructed into data volumes, but one other ribbon segment (Ex1-R02A) was imaged at four non-consecutive tissue sections, preventing volumetric reconstruction.
The Elution-Test Dataset may be downloaded from Figshare (Data Citation 1), as files 1–7 described in Supplementary Table 1. Unlike the Chessboard Dataset, each of these files is a zip file containing all channels for a given data volume.
OCP—FigShare dataset cross-reference
See Supplementary Table 1 to cross-reference data files available through the Open Connectome Project (OCP) and FigShare. Files comprising the Chessboard and Elute-Test Datasets (Data Citation 1) are listed alongside the animal and ribbon they derive from, the antibody stains they include, and the accession addresses for the data at both OCP and FigShare.
Note that the OCP web addresses as written will display the full extent of the first slice (z-section) of a multi-slice volume image. To view a smaller region in x or y, adjust the values in the second-to-last and third-to last terms in the HDF5 web address. To view a different slice, adjust the final term in the HDF5 address. (For additional instructions regarding the OCP web interface, see Usage Notes: OCP Data Access.)
Technical Validation
Antibody specificity
All antibodies used in this study have been validated by their originators as reliable and specific to the antigens of interest, and tested within our laboratory for reliable and repeatable staining of ATomo ribbons of LR-white embedded tissue sections. We make sure all antibodies stain in patterns that correspond to their expected synaptic localization. That is, for example, each GABAergic antibody was screened to confirm that it co-stained a significant number of putative GABAergic synapses defined by multiple other GABAergic antibodies, and that it did not excessively stain within cell bodies or blood vessels, where synapses would not be expected.
We have quantified these expected staining patterns by performing a pairwise 2-dimensional cross-correlation analysis to quantify spatial correlations (colocalization) between the principal antibodies used in these experiments. Pairwise cross-correlation plots were generated by shifting pairs of registered antibody channels against each other in x and y and calculating 2d image correlations for each spatial shift.
These pairwise cross-correlations demonstrated sharp central peaks for colocalized antibodies, and broader noisier values for poorly colocalized antibodies. Examining the cross-correlations at small 2d shifts between images reveals that pairs of antibodies which are expected to colocalize within either pre- or postsynaptic compartments (for example, Synapsin1 and vGluT1 or PSD95 and GluR2, respectively) have sharp peaks of correlation, while pairs of antibodies which represent associated pre- and postsynaptic compartments (for example, Synapsin1 and PSD95) have broader, more diffuse cross-correlation peaks (Figure 3).

(a–c) Multi-channel composite images of related synaptic protein stains in a subregion of a single serial section of the Ex10-R55 ribbon. Scale bar 5 um. (d–g) 2-dimensional cross-correlation analysis of the colocalization of antibody stains specific to the pre- and postsynaptic compartments of glutamatergic and GABAergic synapses. Each plot contains an array of panels representing correlation between different stains of a subregion of the Ex10-R55 ribbon as a function of spatial shifts in x and y (shifts up to 10 px shown), averaged over stacks of 10 serial sections. (a,d) Colocalization of stains for presynaptic glutamatergic proteins (Synapsin1, vGluT1 and vGluT2) and comparison with postsynaptic PSD95 and nuclear DAPI stains. (b,e) Colocalization of stains for postsynaptic glutamatergic proteins (PSD95, GluR1, GluR2, NR2A and NR2B) and comparison to nuclear DAPI stain. (c,f) Colocalization of stains for presynaptic (Synapsin1, vGAT, GAD, and PV25) and postsynaptic (GABAARa1 and Gephyrin) GABAergic proteins and comparison with nuclear DAPI stain. (g) Colocalization of glutamatergic and GABAergic vesicular neurotransmitter transporters (vGluT1, vGluT2, vGAT) relative to presynaptic marker Synapsin1, postsynaptic markers PSD95 (glutamatergic) and gephyrin (GABAergic), and DAPI-stained cell nuclei.
To quantify these correlations, we measured the absolute peak of the cross-correlation, and calculated a half-maximum value based on the average background correlation. Using Matlab, we also calculated a 2d contour of the cross-correlation peak at the half-maximum value, and measured the area of this contour and calculated the diameter of a circular contour with the same area. These values are displayed in Table 9.
Antibody consistency
In previously published work, we demonstrated that multiple rounds of elution have minimal effect on tissue antigenicity as quantified by the consistency of staining with an antibody against Synapsin1 across multiple rounds of imaging9. In the present datasets, we also observed comparable staining quality irrespective of the multiple rounds of stripping and staining.
To further test the consistency of our staining procedure, we have performed three rounds of repeated staining with fifteen key antibodies on ribbons equivalent to those used to produce the Chessboard Dataset. We used 2d image correlation to assess the similarity of staining between the first and third rounds of imaging for each antibody (Figure 4a,c1,e1,g1). To compare the observed level of correlation to a proxy for ‘random’ colocalization, we rotated the images of the third imaging session 180° and repeated the analysis (Figure 4b,c2,e2,g2).

(a) False-color composite image comparing the first stain of a subregion of ribbon Ex1-R02A with an antibody against Synapsin1 protein (green) with the third sequential stain of the same region (red). Areas of overlap (yellow) represent consistent patterns of staining despite intervening rounds of stripping and restaining. (b) Here the image of the third round of staining has been rotated 180° to illustrate that the chance occurrence of overlapping staining is quite low, despite the high stain density in the two images. (c1) Synapsin1 immunofluorescence intensity at individual pixels compared between rounds 1 and 3. The R value represents Pearson’s correlation coefficient between the two images. An approximate threshold between foreground and background pixels (set at 1,000 a.u.) in each image is illustrated by horizontal and vertical red dashed lines. (c2) The same analysis with the image of round 3 rotated 180° to serve as a control for random colocalizations. (d,e) The same analyses for PSD95 and Gephyrin staining. (f) Comparison of correlation coeffients (R) for images of the first and third round of staining for 15 different antibodies. (g) For the same set of antibody stains, comparison of the ‘percent consistency’: the percent of pixels that were bright (above threshold) in either imaging session which were also bright in the other imaging session. See Table 10 for a full list of these values.
Since the large proportion of dark background pixels could be contributing significantly to these image correlations, we also computed a ‘percent consistent’ metric that measured the proportion of bright pixels that were bright in both images. We set an approximate threshold between foreground and background at 1,000 a.u., and quantified the proportion of pixels above this threshold in either imaging session that were also above threshold in the other imaging session. As with the correlation metric, we compared this ‘percent consistent’ metric between the observed colocalization and the ‘random’ colocalization achieved by rotating one image 180° (Figure 4g). This pixelwise metric is only a rough quantification of antibody consistency, but it provides a simple supplement to the correlation values described above.
We find that the antibodies against Synapsin1 and PSD95 produce highly consistent staining between sessions, while the rest (with the exception of NR2A) ranged from relatively good to relatively poor consistency, when compared with the extremely low values expected by random colocalization (Figure 4f,g, Table 10). This analysis corresponds well with previous unpublished observations by our lab, and suggests that each antibody channel represents a mixture of robust signal and stochastic variability, with a signal-to-noise ratio that varies between antibodies.
An important result of this analysis was that the NR2A antibody appeared to be completely unreliable, performing much more poorly than expected compared to random colocalization. Previous observations by our lab (data unpublished) and the correlation analysis presented in Figure 3 demonstrate a reasonable level of colocalization of this antibody with PSD95 and other glutamatergic post-synaptic markers. However, given the results of this analysis, we must recommend extreme caution in any interpretation of this antibody channel within the present dataset.
The stochasticity observed in some antibodies will be an important consideration in any analysis of the Chessboard Dataset. We do not yet have an adequate biophysical model to explain the source of this variability, which is why we have made this Elution-Test dataset available through Figshare for download and further analysis (Data Citation 1, Files 1–7 Supplementary Table 1; see Data Records). Fortunately, the component of stochasticity we observe in some stains is not as concerning as it might be if one hoped to use these antibodies on their own for synapse identification. ATomo presents the opportunity to use over a dozen antibodies in parallel to define synapses based on the convergence of multiple lines of immunohistochemical evidence. Therefore even a stain with low signal-to-noise can be useful in characterizing synapse diversity in combination with other, more reliable stains. For further discussion of recommended approaches to proteomic analysis of synapses using the Chessboard Dataset, please see Usage Notes, below.
Usage Notes
OCP data access
The HDF5 web interface enables anyone with an internet connection to access arbitrary data cutouts through their web browser or to download data cutouts using python or Matlab scripts. Several usage examples are listed below, followed by an explanation of the meanings of the HDF5 arguments used.
Data access examples
Single-Channel 2d Web-View (8 bit grayscale)
Template: http://openconnecto.me/ocp/ca/ [data token]/[orthogonal-view]/[channel token]/[resolution]/[x-range]/[y-range]/[z-range]/
Example: http://openconnecto.me/ocp/ca/Ex6R15C2/xy/DAPI-1/0/700,1700/500,1500/15/
Result: Produces cutout of DAPI-1 channel from data volume Ex6R15C2 at full resolution (‘0’) for x-range 0–1400, y-range 0–700, and z-slice 15.
Multi-Channel 2d Web-View (CYMRGB false color)
Template: http://openconnecto.me/ocp/ca/[data token]/mcfc/[orthogonal-view]/[channel tokens]/[resolution]/[x-range]/[y-range]/[z-range]/
Example: http://openconnecto.me/ocp/ca/Ex6R15C2/mcfc/xy/0,0,0,Synapsin1-2,PSD95-1,DAPI-1/0/700,1700/500,1500/20/
Result: Produces image of Synapsin1-2 (red), PSD95-1 (green), and DAPI-1 (blue) channels from data volume Ex6R15C2 at resolution 0 for x-range 0–1400, y-range 0–1400, and z-slice 20.
Note: Channels are displayed as Cyan, Yellow, Magenta, Red, Green, Blue in order listed. Use placeholder channel token ‘0’ as above to skip colors.
Multi-Channel 2d Matlab Download (CYMRGB false-color png file)
Script: https://gist.github.com/ncweiler/8075665
Result: This script downloads a specified 2d data cutout as a false-color png file. It also creates a data object in Matlab containing an RGB image of the cutout and displays the data as a false-color image. Channel colors are determined as in CYMRGB multi-channel false color web view above.
Multi-channel Volume Matlab Download (hdf5 data file)
Script: https://gist.github.com/ncweiler/7953445
Result: This script downloads a specified multichannel 3d data cutout as an hdf5 file. It creates a data object in Matlab containing one image stack for each channel. A user can then display specified slices of each channel stack.
HDF5 arguments
As in the above examples, the following arguments will be needed to access data cutouts through the HDF5 interface:
Data token
To create a data cutout from a particular data volume, you must use the associated data volume token. For example, the token ‘Ex10R55’ would be used to access the volume reconstruction of that ribbon.
Channel Token(s)
Desired protein channels may be specified either by name or by numeric channel token (1–29, though NB that not all data volumes have 29 channels, and tokens may refer to different channels in different data volumes (See Tables 4 and 7 (available online only)).
Resolution
Data cutouts may be viewed in original or down-sampled resolution. ‘0’ specifies original resolution, and ‘1’ specifies half resolution.
Orthogonal view
The plane of view for 2d cutouts may be ‘xy’, ‘xz’, or ‘yz’.
X-, Y-, and Z-Range
These arguments specify the 2d or 3d extent of the data cutout. Use of the range arguments will depend on whether the cutout is a 2d plane or 3d volume. If accessing a 3d volume, a user must specify min and max values for all three axes, which set the height, width, and depth of the 3d cutout. In contrast, if accessing an xy plane, a user would specify min and max values for the x and y axes to determine the width and height of the 2d cutout, but would specify a single value for z to indicate the desired z-axis cross-section to be accessed.
Token/argument reference
A list of available public data tokens is available here: http://openconnecto.me/ocp/ocpca/public_tokens/. Available channel tokens, resolutions, and x-, y-, and z-ranges for each data volume may be viewed using the following url template: http://openconnecto.me/ocp/ocpca/[data token]/info/ (EG: http://openconnecto.me/ocp/ocpca/Ex10R55/info/). Alternatively, see Tables 3, 4, 6 and 7 (available online only).
Choice of antibodies to screen for plasticity among diverse synapse subpopulations
ATomo’s ability to sequentially stain multiple different synaptic proteins and image their localization at high resolution presents the opportunity to define synapses by the spatial colocalization of multiple synaptic molecular constituents, whereas measurements of single antibodies could be noisy or misleading. See Figure 5 for a summary of the subsynaptic localizations of the antigens stained against in these experiments.

This figure presents the synaptic organization of the molecules used in the above experiments in cartoon form for glutamatergic (a) and GABAergic (b) synapses. Presynaptic axonal boutons are at the top for both synapses classes, and postsynaptic targets (here a dendritic spine and the surface of a cell body, as is typical of glutamatergic and GABAergic synapses, respectively). Molecules or structures not explicitly stained for in the above experiments are labeled with parentheses. Please see Usage Notes: Choice of Antibodies to Screen for Plasticity among Diverse Synapse Subpopulations, above, for further descriptions and references relevant to this figure.
Markers of glutamatergic synapses
Synapsin1 is a vesicle-associated actin-binding protein. Staining for Synapsin1 is found at the majority of glutamatergic and GABAergic synapses in cortex, although we have observed it to be brighter at glutamatergic synapses10. This highly robust stain (see Figure 4) is a useful proxy for identification of loci of putative synapses, which can then be subject to additional classification and molecular characterization10,40. Expression of distinct isoforms of the vesicular glutamate transporter (vGluTs) has commonly been used to distinguish between axonal boutons of vGluT1-expressing intracortical synapses and vGluT2-expressing thalamocortical synapses. Some presynaptic terminals stain for both vGluT1 and vGluT2, which are likely to originate from a subpopulation of thalamocortical neurons expressing vGluT1 in addition to the typical thalamically expressed vGluT2 (refs 41,42), though others have reported vGluT2 expression in normally vGluT1-expressing intracortical synapses43.
PSD95 is postsynaptic scaffolding molecule specific to glutamatergic synapses44–46. It is located close to the post-synaptic plasma membrane, and can thus also be used to define the post-synaptic compartment of the synapse adjacent to a Synapsin1-stained presynaptic compartment. Antibodies against glutamate receptor subunits including GluR1, GluR2, NR2A, and NR2B also stain the postsynaptic membrane of glutamatergic synapses47–49, and are therefore expected to colocalize with PSD95 (Figure 3). Different components of cortical microcircuits are also thought to exhibit characteristic differences in NR2 subunit expression48,50. However, a considerable literature describes additional presynaptic and extrasynaptic expression of these molecules51–55, suggesting that these stains should primarily be used in combination with other synaptic markers such as PSD95 and Synapsin1 to characterize receptor expression in different synaptic compartments.
Markers for GABAergic synapses
Antibodies against glutamate decarboxylase (GAD), an enzyme responsible for synthesizing the neurotransmitter GABA, and vGAT, a vesicular GABA transporter, both stain presynaptic terminals of inhibitory synapses and can be used to differentiate these from excitatory synapses56–58. vGAT protein is thought to be translated locally at GABAergic and glycinergic boutons, making this stain highly specific for inhibitory synaptic (as opposed to extrasynaptic) loci58.
GABAergic synapses express a distinct postsynaptic scaffolding protein, gephyrin, which can be used to identify these inhibitory synapses from PSD95-expressing glutamatergic synapses59–61. As with glutamatergic synapses, stains for postsynaptic GABA receptor subunits can be used to further identify and characterize these postsynaptic compartments. A multitude of GABA receptor subunits are expressed in the brain61. One common subunit, GABA(A)Ra1, has been stained for in these experiments. This subunit is present in one of the largest subclasses of GABA(A) receptors and so can be used in combination with gephyrin staining to identify and subclassify the postsynaptic elements of GABAergic synapses62.
Parvalbumin (PV) is a calcium binding protein expressed by a large subclass of GABAergic neurons, including fast-spiking basket cells and chandelier cells. This stain can therefore be used to identify presynaptic GABAergic terminals belonging to these cell classes26,63. PV+ basket cell and chandelier cell synapses can be readily distinguished from each other, as basket cells make en passant and terminaux synapses primarily onto postsynaptic cell bodies, whereas chandelier cells make distinctive long ‘cartridge’ synapses onto the axon initial segments of postsynaptic neurons64,65.
Markers for plasticity at glutamatergic synapses
The size of both pre- and post-synaptic compartments has been correlated with synaptic strength, and so differences in the size and intensity of Synapsin1 and PSD95 staining could be used as measure of synaptic potentiation or deprivation as a result of experience66–68. Whisker deprivation in adult mice has recently been shown to increase the total density of Synapsin1 staining and postsynaptic spines in spared cortex, but only after complete unilateral whisker trimming69. Chessboard deprivation, which we chose to promote Hebbian circuit-level plasticity rather than the wholesale homeostatic plasticity expected from a complete removal of whisker input19,21,70,71, may not have such drastic effects. Indeed, several long-term in vivo imaging studies have shown significant dendritic spine turnover, but not wholesale changes in total spine number in particular populations of L5 pyramidal neurons16–18,72, implying that chessboard map plasticity may have more to do with altering specific circuit connectivity than massively altering connectivity per se.
Differential expression of glutamate receptor subunits is also indicative of synaptic state, and can be used to characterize synaptic strength and explore the mechanisms of experience dependent synaptic change53,73–75. The relative brightness of immunostaining for the different receptor subunits at individual synapses could be used to define postsynaptic receptor ratios, which are thought to be highly important for regulating Hebbian, homeostatic and meta-plasticity. In particular, the ratio of NMDARs and AMPARs can been used as a proxy for synaptic potentiation76,77. In addition, changes in AMPA receptor subunits GluR1 and GluR2 are associated with Hebbian and homeostatic synaptic plasticity67,78–81, while changes in expression of NMDA receptor subunits NR2A and NR2B are associated with developmental and adult metaplasticity49,52,53,82–88. GluR1, in particular, has been implicated as a key player in map plasticity driven by chessboard deprivation89.
We should also note that although we have frequently observed good spatial colocalization between all of these glutamate receptor subunit stains and PSD95-stained postsynaptic compartments (unpublished observations, but also see Figure 3), our analysis of antibody consistency across imaging sessions (Figure 4) reveals that the NR2A antibody we have used exhibits a very low signal to noise ratio. As a result, we recommend being highly cautious in interpreting this particular antibody channel.
Differential expression of the vGluT1 and vGluT2 isoforms has also associated with differences in the kinetics of presynaptic vesicle release as a result of their different rates of vesicle loading90, and expression of these two isoforms may be regulated to produce homeostatic plasticity43.
Markers for plasticity at GABAergic synapses
Alterations in GABA(A)R concentration at GABAergic synapses have been linked to forms of inhibitory plasticity, which could be detected by changes in staining intensity of these molecules at synaptic subsets91–97. Shifts in inhibition of spared versus deprived whisker responses are thought to be involved in certain types of whisker map plasticity98,99, and chessboard deprivation, in particular, is thought to keep open cortical critical periods through an effect on PV+ intracortical inhibition71.
Other synaptic markers
Arc is an immediate early gene product whose synaptic expression is modified by neural activity. It is thought to have a role in synaptic depression and potentially other forms of synaptic plasticity such as synaptic homeostasis100–103. However, the antibody we used here proved extremely noisy in the initial ribbons we stained for it, and we discontinued its use in later ribbons. Any analysis of this channel should be approached with caution.
Intrinsic transgenic YFP expression, in some cases amplified by an antibody against GFP, labels a subpopulation of layer 5 pyramidal neurons in the thy1-yfp line-H mice used in these experiments22,23,104. In vivo two-photon microscopy studies have observed increased dendritic spine turnover in the distal dendritic tufts of these neurons following chessboard deprivation similar to that performed in this experiment16–18. Analysis of this ATomo dataset may permit quantification of additional spine plasticity in deeper layers of the cortex. Synaptopodin is a protein associated with the spine apparatus, a structure observed at a subset of large dendritic spines and thought to be involved in synaptic plasticity105,106.
Analysis recommendations & caveats
Given the-well known variability of immunohistochemical techniques in general107,108, and our own analysis of the antibodies used in this study (see Technical Validation, Figure 4, Table 10) we suggest that the most robust analyses of this data will be focused on comparisons of stains between cortical layers and columns within individual ribbons. Because these different regions of interest (ROIs) within each ribbon were necessarily stained under exactly the same conditions, such internal comparisons will be relatively immune to variability between stains, whether inherent to the antibody staining itself or as a result of slight differences in the staining procedure between different ribbons.
As a simple example of this sort of analysis, Figure 6 illustrates a quantification of differences in vGluT2 staining between L4 and L5a across ribbons. The higher density of vGluT2+ synapses in L4 is well documented8,10,109–111, and so we present this analysis as a simple example of how synaptic molecular differences between anatomical regions may be detected using the Chessboard Dataset.

(a) Maximum intensity projection of the C2-L4 ROI of ribbon Ex10-R55 stained for vGluT2 (green) and DAPI (blue). (b) The same for the C2-L5a ROI. (c) Maximum intensity projection of the C2-L4 ROI of ribbon Ex10-R55 stained for Synapsin1 (red). Neuropil volume in each ROI is estimated by using a geometric expansion of the Synapsin1 channel’s negative space (blue), as this antibody is strongly excluded from cell bodies. The adjusted volume is used to calculate neuropil puncta densities for comparison between ROIs. (d) Neuropil-normalized vGluT2 puncta densities across all Chessboard Dataset data volumes for L4 ROIs (left) and L5a ROIs (right), with spared and deprived ROIs within each layer averaged together. vGluT2 density in L5a was significantly less than in L4: median difference: 46.24%; P=0.0313 (Wilcoxon signed-rank nonparametric test for difference of group medians); n=7 ribbons. (e) vGluT2 punctum intensity distributions for ribbon Ex10-R55 ROI C2-L4 (median 25,923 a.u.; n=24,189 puncta) and C2-L5a (median 24,454 a.u.; n=14,888 puncta). Shown are median values (red horizontal line), comparison interval (notch in box plot), interquartile range (vertical box extent), and maximum and minimum values (whiskers). Population medians are significantly different: P=4.85*10−21 (Wilcoxon signed-rank nonparametric test for difference of group medians). (f) Median punctum intensity across all ribbons for L4 ROIs (left) and L5a ROIs (right), with spared and deprived ROIS within each layer averaged together. Median vGluT2 punctum total intensity in L5a was significantly less than in L4: median difference: 21.56%; P=0.0156 (Wilcoxon signed-rank nonparametric test for difference of group medians); n=7 ribbons.
To compare the density and intensity of staining between ROIs, we quantified individual puncta of antibody label using a 3d-Gaussian Segmentation method available in the 3d ImageJ Suite developed by Thomas Boudier and colleagues112. In a first pass, putative staining puncta were defined based on local maxima of staining intensity with a 3-pixel filter radius. A watershed algorithm was then used to define boundaries between these putative puncta. Within the watershed boundaries, each punctum was modeled using a 3d Gaussian filter, and segmented at a width of two standard deviations to encompass 90% of its intensity distribution. An advantage of this approach over simpler intensity threshold-based segmentation methods is that punctum size is determined by the standard deviation of the gaussian curve, and is therefore more independent of the absolute intensity of the punctum.
Within anatomically defined regions of each individual ribbon volume (Figure 2j), individual punctum fluorescence intensity and overall punctum density were calculated for each antibody stain. To avoid effects produced by differences in cell body distribution between ROIs, density measurements were calculated using estimated neuropil volume, calculated based on a dilation of the Synapsin1 channel, which is almost completely excluded from cell bodies (Figure 6c). Within individual ribbons, median punctum fluorescence intensity was compared between L4 and L5a regions using using a nonparametric Wilcoxon signed-rank test (Figure 6e). Differences in punctum density and median punctum intensity between L4 and L5a were compared across ribbons, and tested for significance using the Wilcoxon signed-rank test (Figure 6d,f).
This analysis demonstrates clearly the capacity for detecting synaptic molecular differences at the scale of the concentration of vGluT2+ thalamic inputs to L4 of barrel cortex (Figure 6). However, the same approach does not produce convincing evidence of similar wholesale differences in antibody staining between spared and deprived columns. Chessboard whisker trimming has been shown to robustly induce functional map plasticity and changes in dendritic spine density in subsets of neurons in the adult mouse16–21,89, but to our knowledge, no previous work has reported shifts in overall levels of synaptic proteins following chessboard trimming in the adult that approach the scale of the difference in vGluT2 expression between layers.
Indeed, the synaptic molecular changes underlying chessboard map plasticity are likely to be relatively subtle. Diverse synaptic components of the barrel cortex circuit are affected differently during map plasticity, and in many cases seem to exhibit bidirectional compensatory shifts, perhaps as a homeostatic mechanism to maintain stable overall synaptic drive13,14,70,113–118. As a result, analysis of wholesale differences in expression of individual synaptic proteins is likely to miss these more subtle changes occurring at specific circuit elements.
We anticipate that an important first step in analyzing these data will be to classify synaptic loci and define synaptic subtypes, which can then be analyzed for molecular changes between spared and deprived conditions. This is particularly important given the fact that many of the synaptic proteins being measured here are also expressed at non-synaptic sites8,59,61,62,119, which ought to be excluded from analysis of synaptic molecular changes.
We anticipate that analysis of the spatial distributions and plasticity-induced differences among these synaptic molecules will help to elucidate synaptic subsets affected by the altered sensory experience of chessboard whisker deprivation, and quantify changes in the molecular composition of these synapses in different layers of spared and deprived cortical columns. We believe some particularly promising areas of analysis include comparisons of punctum intensity ratios across synapse classes, analysis of the spatial relationships between puncta of different channels within individual synapses, and characterization of the incidence of distinct synapse classes and synapses with distinct molecular compositions onto different postsynaptic cell types.
Thus far the machine learning40 and other statistical approaches we have developed to define these synaptic subtypes and their molecular diversity have not proven sufficiently robust or efficient for analysis of datasets of this complexity and scale, and at any rate are well beyond the scope of the present data descriptor. We hope that others, given access to these rich data, will devise novel, creative approaches to characterizing synapses and their molecular diversity beyond what we have imagined.
Additional information
References
References
Takamori, S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006).
McPherson, P. S. Proteomic analysis of clathrin-coated vesicles. Proteomics 10, 4025–4039 (2010).
Sugiyama, Y., Kawabata, I., Sobue, K. & Okabe, S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nat. Methods 2, 677–684 (2005).
Collins, M. O. et al. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97(Suppl 1), 16–23 (2006).
Dosemeci, A. et al. Composition of the synaptic PSD-95 complex. Mol. Cell. Proteomics 6, 1749–1760 (2007).
Zipursky, S. L. & Sanes, J. R. Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell 143, 343–353 (2010).
Dalva, M. B., McClelland, A. C. & Kayser, M. S. Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220 (2007).
O’Rourke, N. A., Weiler, N. C., Micheva, K. D. & Smith, S. J. Deep molecular diversity of mammalian synapses: why it matters and how to measure it. Nat. Rev. Neurosci. 13, 365–379 (2012).
Micheva, K. D. & Smith, S. J. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron 55, 25–36 (2007).
Micheva, K. D., Busse, B., Weiler, N. C., O’Rourke, N. A. & Smith, S. J. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron 68, 639–653 (2010).
Diamond, M. E., von Heimendahl, M. & Knutsen, P. M. ‘ Where ’ and ‘ what ’ in the whisker sensorimotor system. Nat. Rev. Neurosci. 9, 601–612 (2008).
Feldmeyer, D. et al. Barrel cortex function. Prog. Neurobiol. 103, 3–27 (2013).
Feldman, D. E. & Brecht, M. Map plasticity in somatosensory cortex. Science 310, 810–815 (2005).
Fox, K. Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience 111, 799–814 (2002).
Fox, K . The Barrel Cortex (Cambridge University Press, 2008).
Trachtenberg, J. T. et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794 (2002).
Holtmaat, A., Wilbrecht, L., Knott, G. W., Welker, E. & Svoboda, K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 441, 979–983 (2006).
Wilbrecht, L., Holtmaat, A. & Wright, N. Structural plasticity underlies experience-dependent functional plasticity of cortical circuits. J. Neurosci. 30, 4927–4932 (2010).
Wallace, H. & Fox, K. The effect of vibrissa deprivation pattern on the form of plasticity induced in rat barrel cortex. Somatosens. Mot. Res. 16, 122–138 (1999).
Hardingham, N., Wright, N., Dachtler, J. & Fox, K. Sensory deprivation unmasks a PKA-dependent synaptic plasticity mechanism that operates in parallel with CaMKII. Neuron 60, 861–874 (2008).
Wallace, H., Glazewski, S., Liming, K. & Fox, K. The role of cortical activity in experience-dependent potentiation and depression of sensory responses in rat barrel cortex. J. Neurosci. 21, 3881–3894 (2001).
Feng, G. et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 1–11 (2000).
Yu, J. et al. Local-circuit phenotypes of layer 5 neurons in motor-frontal cortex of YFP-H mice. Front. Neural Circuits 2, 6 (2008).
Hestrin, S. & Galarreta, M. Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 28, 304–309 (2005).
Markram, H. et al. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807 (2004).
Ascoli, G. A. et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568 (2008).
Garner, J. P., Weisker, S. M., Dufour, B. & Mench, J. A. Barbering (fur and whisker trimming) by laboratory mice as a model of human trichotillomania and obsessive-compulsive spectrum disorders. Comp. Med. 54, 216–224 (2004).
Grinvald, A., Lieke, E., Frostig, R., Gilbert, C. & Wiesel, T. Functional architecture of cortex revealed by optical imaging of intrinsic signals. Nature 324, 361–364 (1986).
Grinvald, A. et al. in Mod. Tech. Neurosci. Res. (eds Windhorst, U. & Johansson, H.) 893–969 (Springer, 1999).
Kalatsky, V. A. & Stryker, M. P. New paradigm for optical imaging: temporally encoded maps of intrinsic signal. Neuron 38, 529–545 (2003).
Micheva, K. D., O’Rourke, N. A., Busse, B. & Smith, S. J. Array tomography: rodent brain fixation and embedding. Cold Spring Harb. Protoc. 2010, 1264–1266 (2010).
Micheva, K. D., O’Rourke, N. A., Busse, B. & Smith, S. J. Array tomography: production of arrays. Cold Spring Harb. Protoc. 2010, 1214–1218 (2010).
Micheva, K. D., O’Rourke, N. A., Busse, B. & Smith, S. J. Array tomography: production of arrays. Cold Spring Harb. Protoc. 2010, 1280–1281 (2010).
Micheva, K. D., O’Rourke, N. A., Busse, B. & Smith, S. J. Array tomography: immunostaining and antibody elution. Cold Spring Harb. Protoc. 2010, 1270–1273 (2010).
Micheva, K. D., O’Rourke, N. A., Busse, B. & Smith, S. J. Array tomography: high-resolution three-dimensional immunofluorescence. Cold Spring Harb. Protoc. 2010, 1274–1279 (2010).
Wang, G. X. & Smith, S. J. Sub-diffraction limit localization of proteins in volumetric space using Bayesian restoration of fluorescence images from ultrathin specimens. PLoS Comput. Biol. 8, e1002671 (2012).
Lowe, D. G. Object recognition from local scale-invariant features. in Proc. Seventh IEEE Int. Conf. Comput. Vis. 2, 1150–1157 (1999).
Fischler, M. & Bolles, R. Random sample consensus: A paradigm for model fitting with applications to image analysis and automated cartography. Commun. ACM 24, 381–395 (1981).
Saalfeld, S., Fetter, R., Cardona, A. & Tomancak, P. Elastic volume reconstruction from series of ultra-thin microscopy sections. Nat. Methods 9, 717–720 (2012).
Busse, B. & Smith, S. Automated analysis of a diverse synapse population. PLoS Comput. Biol. 9, e1002976 (2013).
Barroso-Chinea, P. et al. Expression of the mRNAs encoding for the vesicular glutamate transporters 1 and 2 in the rat thalamus. J. Comp. Neurol. 501, 703–715 (2007).
Boulland, J.-L. et al. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J. Comp. Neurol. 480, 264–280 (2004).
De Gois, S. et al. Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J. Neurosci. 25, 7121–7133 (2005).
Hunt, C. A., Schenker, L. J. & Kennedy, M. B. PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J. Neurosci. 16, 1380–1388 (1996).
Chen, X. et al. Organization of the core structure of the postsynaptic density. Proc. Natl Acad. Sci. USA 105, 4453–4458 (2008).
Chen, X. et al. PSD-95 is required to sustain the molecular organization of the postsynaptic density. J. Neurosci. 31, 6329–6338 (2011).
Granger, A. J., Gray, J. A., Lu, W. & Nicoll, R. A. Genetic analysis of neuronal ionotropic glutamate receptor subunits. J. Physiol. 589, 4095–4101 (2011).
Cull-Candy, S. G. & Leszkiewicz, D. N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, re16 (2004).
Yashiro, K. & Philpot, B. D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094 (2008).
Kumar, S. S. & Huguenard, J. R. Pathway-specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J. Neurosci. 23, 10074–10083 (2003).
Pinheiro, P. S. & Mulle, C. Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat. Rev. Neurosci. 9, 423–436 (2008).
Sanz-Clemente, A., Nicoll, R. A. & Roche, K. W. Diversity in NMDA receptor composition: many regulators, many consequences. Neurosci 19, 62–75 (2013).
Paoletti, P., Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400 (2013).
Santos, S. D., Carvalho, A. L., Caldeira, M. V. & Duarte, C. B. Regulation of AMPA receptors and synaptic plasticity. Neuroscience 158, 105–125 (2009).
Lau, C. G. & Zukin, R. S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426 (2007).
McLaughlin, B., Wood, J. & Saito, K. The fine structural localization of glutamate decarboxylase in synaptic terminals of rodent cerebellum. Brain Res. 76, 377–391 (1974).
Buddhala, C., Hsu, C.-C. & Wu, J.-Y. A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem. Int. 55, 9–12 (2009).
Chaudhry, F. A. et al. The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J. Neurosci. 18, 9733–9750 (1998).
Harris, K. M. & Weinberg, R. J. Ultrastructure of synapses in the mammalian brain. Cold Spring Harb. Perspect. Biol. 4, a005587 (2012).
Sassoè-Pognetto, M. et al. Understanding the molecular diversity of GABAergic synapses. Front. Cell. Neurosci 5, 4 (2011).
Luscher, B., Fuchs, T. & Kilpatrick, C. L. GABA(A) receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409 (2011).
Kasugai, Y. et al. Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur. J. Neurosci. 32, 1868–1888 (2010).
Rudy, B., Fishell, G., Lee, S. & Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61 (2011).
Fish, K. N., Sweet, R. A., Deo, A. J. & Lewis, D. A. An automated segmentation methodology for quantifying immunoreactive puncta number and fluorescence intensity in tissue sections. Brain Res. 1240, 62–72 (2008).
Sweet, R. A., Fish, K. N. & Lewis, D. A. Mapping synaptic pathology within cerebral cortical circuits in subjects with schizophrenia. Front. Hum. Neurosci 4, 44 (2010).
Gray, N. W., Weimer, R. M., Bureau, I. & Svoboda, K. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 4, e370 (2006).
Kessels, H. W. & Malinow, R. Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350 (2009).
Cheetham, C. E. J., Barnes, S. J., Albieri, G., Knott, G. W. & Finnerty, G. T. Pansynaptic enlargement at adult cortical connections strengthened by experience. Cereb. Cortex 24, 521–531 (2014).
Chau, L. S., Akhtar, O., Mohan, V., Kondilis, A. & Galvez, R. Rapid adult experience-dependent anatomical plasticity in layer IV of primary somatosensory cortex. Brain Res. 1543, 93–100 (2014).
Feldman, D. E. Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55 (2009).
Nowicka, D., Soulsby, S., Skangiel-Kramska, J. & Glazewski, S. Parvalbumin-containing neurons, perineuronal nets and experience-dependent plasticity in murine barrel cortex. Eur. J. Neurosci. 30, 2053–2063 (2009).
Holtmaat, A. & Svoboda, K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658 (2009).
Anggono, V. & Huganir, R. L. R. Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 22, 461–469 (2012).
Clem, R. L. & Barth, A. L. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 49, 663–670 (2006).
Kopp, C., Longordo, F. & Lüthi, A. Experience-dependent changes in NMDA receptor composition at mature central synapses. Neuropharmacology 53, 1–9 (2007).
Crair, M. & Malenka, R. A critical period for long-term potentiation at thalamocortical synapses. Nature 375, 325–328 (1995).
Carroll, R. C., Beattie, E. C., von Zastrow, M. & Malenka, R. C. Role of AMPA receptor endocytosis in synaptic plasticity. Nat. Rev. Neurosci. 2, 315–324 (2001).
Sheng, M. & Kim, M. J. Postsynaptic signaling and plasticity mechanisms. Science 298, 776–780 (2002).
Malinow, R. & Malenka, R. C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126 (2002).
Song, I. & Huganir, R. L. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588 (2002).
Isaac, J. T. R., Ashby, M. C. & McBain, C. J. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871 (2007).
Evans, R. C. et al. The effects of NMDA subunit composition on calcium influx and spike timing-dependent plasticity in striatal medium spiny neurons. PLoS Comput. Biol. 8, e1002493 (2012).
Hunt, D. L. & Castillo, P. E. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr. Opin. Neurobiol. 22, 496–508 (2012).
Yashiro, K., Corlew, R. & Philpot, B. D. Visual deprivation modifies both presynaptic glutamate release and the composition of perisynaptic/extrasynaptic NMDA receptors in adult visual cortex. J. Neurosci. 25, 11684–11692 (2005).
Lee, M.-C., Yasuda, R. & Ehlers, M. D. Metaplasticity at single glutamatergic synapses. Neuron 66, 859–870 (2010).
Philpot, B. D., Cho, K. K. A & Bear, M. F. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron 53, 495–502 (2007).
Philpot, B. D., Sekhar, A. K., Shouval, H. Z. & Bear, M. F. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron 29, 157–169 (2001).
Bellone, C. & Nicoll, R. A. Rapid bidirectional switching of synaptic NMDA receptors. Neuron 55, 779–785 (2007).
Wright, N. et al. Laminar analysis of the role of GluR1 in experience-dependent and synaptic depression in barrel cortex. Nat. Neurosci. 11, 1140–1142 (2008).
Fremeau, R. T. Jr et al. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304, 1815–1819 (2004).
Fritschy, J.-M. Epilepsy, E/I Balance and GABA(A) receptor plasticity. Front. Mol. Neurosci 1, 5 (2008).
Mody, I. Aspects of the homeostaic plasticity of GABAA receptor-mediated inhibition. J. Physiol. 562, 37–46 (2005).
Nusser, Z., Cull-Candy, S. & Farrant, M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 19, 697–709 (1997).
Nusser, Z., Roberts, J. D., Baude, A, Richards, J. G. & Somogyi, P. Relative densities of synaptic and extrasynaptic GABAA receptors on cerebellar granule cells as determined by a quantitative immunogold method. J. Neurosci. 15, 2948–2960 (1995).
Nusser, Z., Hajos, N., Somogyi, P. & Mody, I. Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature 395, 172–177 (1998).
Li, P., Rudolph, U. & Huntsman, M. M. Long-term sensory deprivation selectively rearranges functional inhibitory circuits in mouse barrel cortex. Proc. Natl Acad. Sci. USA 106, 12156–12161 (2009).
Sun, Q.-Q. Experience-dependent intrinsic plasticity in interneurons of barrel cortex layer IV. J. Neurophysiol. 102, 2955–2973 (2009).
Foeller, E., Celikel, T. & Feldman, D. E. Inhibitory sharpening of receptive fields contributes to whisker map plasticity in rat somatosensory cortex. J. Neurophysiol. 94, 4387–4400 (2005).
Gambino, F. & Holtmaat, A. Synapses let loose for a change: inhibitory synapse pruning throughout experience-dependent cortical plasticity. Neuron 74, 214–217 (2012).
Bramham, C. R. et al. The Arc of synaptic memory. Exp. Brain Res. 200, 125–140 (2010).
Shepherd, J. D. & Bear, M. F. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 14, 279–284 (2011).
Korb, E. & Finkbeiner, S. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci. 34, 591–598 (2011).
Béïque, J.-C., Na, Y., Kuhl, D., Worley, P. F. & Huganir, R. L. Arc-dependent synapse-specific homeostatic plasticity. Proc. Natl Acad. Sci. USA 108, 816–821 (2011).
Porrero, C., Rubio-Garrido, P., Avendaño, C. & Clascá, F. Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Res. 1345, 59–72 (2010).
Zhang, X.-L. et al. Essential role for synaptopodin in dendritic spine plasticity of the developing hippocampus. J. Neurosci. 33, 12510–12518 (2013).
Segal, M., Vlachos, A. & Korkotian, E. The spine apparatus, synaptopodin, and dendritic spine plasticity. Neurosci. 16, 125–131 (2010).
Rhodes, K. J. & Trimmer, J. S. Antibodies as valuable neuroscience research tools versus reagents of mass distraction. J. Neurosci. 26, 8017–8020 (2006).
Fritschy, J.-M. Is my antibody-staining specific? How to deal with pitfalls of immunohistochemistry. Eur. J. Neurosci. 28, 2365–2370 (2008).
Micheva, K. D. & Beaulieu, C. An anatomical substrate for experience-dependent plasticity of the rat barrel field cortex. Proc. Natl Acad. Sci. USA 92, 1–5 (1995).
Fremeau, R. T. et al. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31, 247–260 (2001).
Graziano, A., Liu, X.-B., Murray, K. D. & Jones, E. G. Two sets of glutamatergic afferents to the somatosensory thalamus and two thalamocortical projections in the mouse. J. Comp. Neurol. 507, 1258–1276 (2008).
Ollion, J., Cochennec, J., Loll, F., Escudé, C. & Boudier, T. TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841 (2013).
Margolis, D. J., Lütcke, H. & Helmchen, F. Microcircuit dynamics of map plasticity in barrel cortex. Curr. Opin. Neurobiol. 24, 76–81 (2014).
Margolis, D. J. et al. Reorganization of cortical population activity imaged throughout long-term sensory deprivation. Nat. Neurosci. 15, 1539–1546 (2012).
Jacob, V., Petreanu, L., Wright, N., Svoboda, K. & Fox, K. Regular spiking and intrinsic bursting pyramidal cells show orthogonal forms of experience-dependent plasticity in layer V of barrel cortex. Neuron 73, 391–404 (2012).
Oberlaender, M., Ramirez, A. & Bruno, R. M. Sensory experience restructures thalamocortical axons during adulthood. Neuron 74, 648–655 (2012).
Kätzel, D. & Miesenböck, G. Experience-dependent rewiring of specific inhibitory connections in adult neocortex. PLoS Biol. 12, e1001798 (2014).
Fox, K. & Wong, R. O. L. A comparison of experience-dependent plasticity in the visual and somatosensory systems. Neuron 48, 465–477 (2005).
Köhr, G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 326, 439–446 (2006).
Data Citations
Weiler, N. C., Collman, F., Vogelstein, J. T., Burns, R., & Smith, S. J. Figshare (2014) http://dx.doi.org/10.6084/m9.figshare.1136081
Weiler, N. C. Figshare (2014) http://dx.doi.org/10.6084/m9.figshare.1183512
Acknowledgements
Our thanks to Kristina Micheva for her expertise in ATomo staining and imaging, to Nafisa Ghori for her work producing hundreds of ATomo ribbons for this project, to Gordon Wang and Nancy O’Rourke for their helpful commentary on this manuscript, and to Priya Manavalan for her assistance making these data available at OCP. We would also like to thank our funding sources for their generous support, including NIH grants 1R21MH099797, 1R01NS075252 and 1R01NS077601 and CRCNS Data Sharing: The EM Open Connectome Project. The authors declare that they have had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Contributions
N.C.W. and S.J.S. designed the experiment. N.C.W. and F.C. developed ISI-tissue punch dissection technique and wrote volume reconstruction software. N.C.W. collected the data. N.C.W. and J.T.V. designed technical validation. J.T.V. and R.B. processed image data for hosting through OCP. N.C.W. wrote the paper with input from all authors.
Corresponding author
Ethics declarations
Competing interests
S.J.S. has a founder’s interest in a company (Aratome) that is seeking to commercialize array tomography. The remaining authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Weiler, N., Collman, F., Vogelstein, J. et al. Synaptic molecular imaging in spared and deprived columns of mouse barrel cortex with array tomography. Sci Data 1, 140046 (2014). https://doi.org/10.1038/sdata.2014.46
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2014.46
This article is cited by
-
An open-source, end-to-end workflow for multidimensional photoemission spectroscopy
Scientific Data (2020)
-
Ctrl alt share
Scientific Data (2015)
