
Abstract
Emerging resistance to all available antibiotics highlights the need to develop new antibiotics with novel mechanisms of action. Most of the currently used antibiotics target Gram-positive bacteria while Gram-negative bacteria easily bypass the action of most drug molecules because of their unique outer membrane. This additional layer acts as a potent barrier restricting the entry of compounds into the cell. In this scenario, several approaches have been elucidated to increase the accumulation of compounds into Gram-negative bacteria. This review includes a brief description of the physicochemical properties that can aid compounds to enter and accumulate in Gram-negative bacteria and covers different strategies to target or bypass the outer membrane-mediated barrier in Gram-negative bacterial pathogens.
Similar content being viewed by others
Introduction
The discovery and clinical utilization of antimicrobials was a very significant turning point in the history of medicine, which significantly reduced morbidity and mortality worldwide. However, this advancement was soon followed by the development of resistance to these antimicrobials, thus raising the prospect of returning to the “pre-antibiotic era” with high lethality associated even with simple infections. This resistance amongst pathogenic microbes is a complex and dynamic process resulting from certain direct or indirect factors including overuse and misuse of antibiotics, poor sanitation, antibiotic disposal, and others. Although antimicrobial resistance (AMR) is a global problem, densely populated areas such as Asia-Pacific regions are at higher risk of spread owing to socioeconomic factors. This increasing threat of multidrug-resistant (MDR) pathogens is widely acknowledged by global and national organizations, including the World Health Organization (WHO) and the Centers for Disease Control and Prevention, USA1.
In order to spur antibiotic discovery, WHO has classified carbapenem-resistant Enterobacterales, carbapenem- and colistin-resistant Acinetobacter baumannii, and carbapenem-resistant Pseudomonas aeruginosa as pathogens of global critical threat in its pathogen priority list. Additionally, the emergence of extensively drug-resistant (XDR) and pan-drug-resistant (PDR) strains of Klebsiella pneumoniae worsens the situation1. Ameta-analysis of 175 studies conducted in different countries of Southeast Asia revealed 73.0%, 29.8%, and 2.8% cases of carbapenem-resistant A. baumannii (CRAB), carbapenem-resistant P. aeruginosa (CRPA) and carbapenem-resistant Enterobacterales (CRE), respectively1. A survey conducted in 2019 involving 204 countries and territories reported bacterial AMR to be associated with 1.27 million deaths, with the highest death rate recorded in western sub-Saharan Africa (27.3 deaths per 100,000 individuals) and the lowest in Australasia (6.5 deaths per 100,000 individuals). The research highlights the fatality associated with quickly emerging drug-resistant strains of Escherichia coli, followed by Staphylococcus aureus, K. pneumoniae, Streptococcus pneumoniae, A. baumannii, and P. aeruginosa, which were directly responsible for a total of 929,000 deaths2. According to a report published by the UK government, AMR will be associated with around 300 million premature deaths, with an associated global economic loss of $100 trillion by 20503. The increasing numbers of MDR bacterial strains, especially Gram-negative bacterial (GNB) pathogens, at an alarming rate highlights the urgent need to develop new antibacterial agents with novel modes of action targeting these pathogens in order to prevent future catastrophic pandemic outbreaks4.
Gram-negatives are far more difficult to treat as compared to Gram-positive bacterial (GPB) pathogens because of their uniquely designed lipopolysaccharide (LPS) containing additional outer membrane (OM), their efflux pumps and other resistance mechanisms. Using one or the other mechanism, these bacteria easily bypass the action of most of the commonly used antibiotics and thus, are responsible for a variety of serious community and healthcare-associated infections5,6. In this context, the Infectious Diseases Society of America issued a report entitled “Bad Bugs, No Drugs: As Antibiotic Discovery Stagnates, A Public Health Crisis Brews” already in 2004 to encourage and promote pharmaceutical investment in antibiotic research and development4. This is however still lagging, with most big pharma companies having left the development of antibiotics and the situation, therefore worsens critically4,7.
The outer membrane of Gram-negative bacterial pathogens as major resistance determinant
Structural features of outer membrane
The OM of Gram-negative bacteria has a very complex architecture with phospholipids, lipopolysaccharides, lipoproteins and β-barrel porins, combining together to act as an asymmetrical lipid bilayer barrier that prevents the entry of foreign molecules including host defense molecules and antibiotics8. The inner leaflet of the OM is made up of phospholipids while the outer leaflet consists of a well-organized, densely packed LPS whose phosphates are stabilized by Mg2+/Ca2+ ions. LPS consists of the glucosamine-based glycolipid Lipid A and the tight packing of saturated fatty acid chains maintains a low level of membrane fluidity, thus limiting the permeation of hydrophobic compounds across the OM9. Additional core and O-antigen oligosaccharides present in LPS of wild-type bacterial strains are required for microbial virulence as they protect bacteria from antibiotics and complement-mediated lysis10. Lipoproteins localized on the periplasmic face of OM are involved in various cellular processes such as the transportation of molecules, maintenance of envelope integrity, transduction of signals and pathogenesis11. The OM contains certain β-barrel protein channels called porins that act as size exclusion channels, allowing diffusion of small hydrophilic molecules such as nutrients from the extracellular environment into the periplasm but preventing entry of large and/or hydrophobic molecules, including several classes of antibiotics9.
Efflux pumps as major resistance determinant
Unfortunately, even if lipophilic or amphiphilic antibiotics succeed in crossing the OM, there are further lines of defense, e.g., effluxpumps (EPs) that transport these molecules back to the outer environment. These efflux pumps are abundantly used by MDR bacteria to prevent the accumulation of antibiotics in the intracellular environment, thus further decreasing the susceptibility to antibiotics. Naturally, bacteria utilize these pumps to excrete bacterial metabolic products and to recognize harmful exogenous compounds that have penetrated through the cell wall12. Efflux pumps were first identified as a resistance mechanism in 1970s13 and since then, they are recognized as one of the major factors responsible for, both, intrinsic and acquired resistance in MDR Gram-negative bacterial pathogens. They generally fall under six families based on their mode of activation/regulation and function: 1. ATP-binding cassette (ABC) family, 2. Resistance-nodulation-cell division (RND) family, 3. Major facilitator superfamily, 4. Small multi-drug resistance family, 5. Multi-drug and toxic compound extrusion family and 6. Related proteobacterial antimicrobial compound efflux14. All those families of efflux pumps are present in both Gram-positive and Gram-negative bacteria, except for the RND superfamily that is specifically present in Gram-negative bacterial pathogens only and extrude xenobiotics such as antibiotics, dyes, heavy metals, detergents, and many other substrates12. Altogether, these unique features of the OM of Gram-negatives combined with the presence of efflux pumps provide a strong defense against commonly used antibiotics9.
Bacterial enzymes targeting antibacterial agents
Apart from these features of the OM and efflux pumps as mechanisms of limited permeability and resistance respectively, Gram-negative bacteria further produce antibiotic modifying enzymes to render them ineffective, such as β-lactamasese.g., serine β-lactamases, metallo-β-lactamases (MBLs) (including Verona integron-encoded metallo-β-lactamases (VIM encoded MBL) and New Delhi metallo-β-lactamases (NDM)), cefotaximase, K. pneumoniae carbapenemase (KPC), imipenemase (IMP)and oxacillinase (OXA) that breaks the β-lactam ring. To worsen the situation, bacterial strains producing extended-spectrum β-lactamases (ESBLs) have evolved to target even third-generation cephalosporins, thus further reducing the treatment options available1. Besides the β-lactams, also other classes of antibiotics are not left untouched by bacterial enzymatic action. Bacterial ribosome-targeting antibiotics such as aminoglycosides, macrolides, lincosamides and streptogramins are inactivated by bacterial rRNA methyltransferases that modify the drug target site by methylation. The efficacy of peptidoglycan targeting glycopeptides is reduced by peptidoglycan-modifying enzymes such as dehydrogenase, serine racemase, ligase and others. Further worsening the situation, even the last resort antibiotic, colistin is made ineffective by bacterial phosphoethanolamine transferases that modify its target lipid A15. Apart from these, thousands of other bacterial enzymes targeting major classes of antibiotics include phosphotransferases, glycosyltransferases and esterases (targeting macrolides), nucleotidyltransferases (targeting lincosamides and aminoglycosides), acetyltransferases (targeting Streptogramin A, phenicols, aminoglycosides and fluoroquinolones), hydrolases (targeting phenicols), phosphotransferases (targeting aminoglycosides, phosphomycin, rifamycins) and monoxygenases (targeting tetracyclins, rifamycins)16. A detailedoverviewofthe molecular mechanismofantibioticresistanceamong Gram-negatives has recentlybeenreviewed17.
Membranevesicles, the un-intentionalresistancemechanism
Additionally, both Gram-positive and Gram-negative bacteria possess an additional mechanism of protection through the formation of membrane vesicles (MVs) which are spherical nanoparticles formed of a lipid bilayer. In Gram-negatives, these vesicles are formed by blebbing of OM as a result of cell envelope disruption. While pinching off from the membrane, these vesicles encapsulate some of the periplasmic components of the cell including misfolded proteins, toxic intermediates or foreign substances including certain antibiotics and thus, helping bacteria in detoxification and increasing chances of survival. Sometimes, these vesicles also carry certain antibiotic-inactivating enzymes. However, the molecular mechanism of how these vesicles target antibiotics remains elusive18.
Along with OM, inner membrane in Gram-negative bacteria also acts as a permeability barrier for polar and highly charged compounds compared to neutral lipophilic solutes. This phospholipid bilayer also carries a number of solute-specific energy-dependent transporters which restricts the entry of compounds into the cytoplasm. The entry of compounds via inner membrane has been reviewed and is out of scope of this review19,20.
Possible ways of entry of antibiotics in Gram-negative bacteria
Even though a large number of potential intracellular targets have been studied in Gram-negative bacterial pathogens such as peptide deformylase (PDF), enoyl-acyl carrier protein reductase (FabI), 3-ketoacyl-acyl carrier protein III (FabH), methionyl, tRNA synthetase (MetRS) and phenylalanyl-tRNA synthetase (PheRS), identifying inhibitors that accumulate into Gram-negative bacteria has been a major challenge, which is best exemplified by >70 high throughput screening (HTS) campaigns run by GlaxoSmithKline with poor success. This study therefore highlights the limited activity of intracellular-enzyme targeting compounds against Gram-negative bacteria, mainly owing to the extremely limited permeability of OM21.
The major routes of entry of compounds into Gram-negative bacteria pathogens are discussed below:
Self-promoted uptake
The hydrophobic, fatty acid chain bearing lipid A, a core oligosaccharide and the O-antigen together constitute LPS. The phosphorylated glucosamine backbone of lipid A and the core region carry several anionic groups that strongly attract divalent cations to recompense electrostatic repulsion between neighboring LPS molecules. The mechanism of action of antibacterial agents via self-promoted uptake has already been reviewed6,22. Hydrophobic antibacterial agents like macrolides (erythromycin, clarithromycin), rifamycins, novobiocin and fusidic acid make use of the hydrophobic nature of LPS lipid A to enter the bacterial cells. On the other hand, membrane permeabilizers, such as Tris/EDTA, polymyxin B, polymyxin B nonapeptide (PMBN), aminoglycosides (gentamicin, kanamycin) and cationic peptides compete with divalent cations to bind with LPS, resulting in release of LPS into the medium. This reduced LPS in the OM is compensated by glycerophospholipids, which results in phospholipid bilayer patches. These patches are several times more permeable to lipophilic antibiotics. In this way, membrane permeabilizers increase susceptibility of Gram-negative bacteria towards hydrophobic antibiotics6.
Entry via porins
The most common way of entry into Gram-negative bacteria is via hydrophilic β-barrel protein pores. Via these so-called porins, small hydrophilic ions and molecules with a molecular mass below roughly 600 Da, such as amino acids and sugars can diffuse to the bacterial periplasm. The central hydrophilic portion of these proteins makes them compound-selective for hydrophilic molecules. Interestingly, antibiotics like β-lactams, tetracyclines, chloramphenicol and fluoroquinolones find their way into the cell via these porins. However, entry through these porins is affected by physicochemical parameters, e.g., nature and position of specific charges. This dependency was illustrated by Bezrukov’s group, where a sharp increase in the selectivity of the porin OmpF for cations relative to anions was observed in solutions of low ionic strength6,23. Interestingly, ampicillin and amoxicillin (both β-lactams) have pH-dependent actions on OmpF porins as they transiently block the open channels of the porins and show maximum blockage at a pH corresponding to their isoelectric point. The interaction between the specific charges of the zwitterionic molecules and the OmpF porins aids the antibiotic drug translocation across the membrane. Quinolone derivatives are bactericidal antibiotics that primarily translocate over the OM via porins (in particular OmpF). Less hydrophobic quinolones like norfloxacin and ciprofloxacin are proven to cross the membrane via other porins24,25.
Despite being an excellent way of entry of drugs, studies have revealed the emergence of a large number of resistant strains of E. coli, P. aeruginosa, N. gonorrhoeae, Enterobacteraerogenes and K. pneumoniae in which either there is loss/severe reduction of porins or replacement of one or two major porins by another type; or altered function due to specific mutations6. A wide spread of porin-based drug resistance was reported by Pagès et al. where 44% among 45 β-lactam resistant clinical isolates of E. aerogenes obtained from French hospitals showed a lack of porins26.
Facilitated diffusion
Facilitated diffusion is a process of transportation of molecules across the OM utilizing substrate-specific carrier proteins against concentration gradients. The process is independent of metabolic energy but is facilitated by factors like pH and drug concentration. Two such carrier proteins studied in E. coli are TonA and TonB. These proteins are naturally used by E. coli for the accumulation of ferrichrome (a naturally occurring iron chelator). The antibiotic albomycin, a structural analog of ferrichrome, follows the same path to enter bacterial cell via binding to TonA and TonB27.
Entry via transport proteins [specific and non-specific proteins]
The OM of Gram-negative bacteria comprises a large number of proteins such as murein lipoprotein (Lpp) (provide OM-peptidoglycan interactions and to maintain OM integrity), OmpA (to maintain the OM integrity) and general diffusion porins. Apart from these proteins, some other specialized protein channels and receptors are found on OM, which are responsible for the uptake of specific substrates. For example, LamB and BtuB help in transportation of maltodextrins and vitamin B12. Proteins such as Omp85 are involved in OM and surface appendage biogenesis. A variety of translocators are employed for the assembly of adhesins, pili and flagella. Additionally, several translocons present in type II secretion systems are involved in the release of toxins and different types of enzymes and proteins are also involved in the assembly of LPS. These transport proteins can be attractive targets for transporting antibiotics. However, most of these targets are not extensively studied as antibiotic entry routes6. One exception is siderophore transporters where the first drug, Fetroja (cefiderocol), exploiting this mechanism, combined with facilitated diffusion, for uptake has reached approval. The compound binds to the ferric ion and is transported to the periplasmic space via siderophores28.
Entry due to Donnan potential
Donnan potential was initially identified as a surface potential in Gram-negative bacteria that are involved in cell mobility and adaptation to different environmental stimuli including charge, ionic strength and pH. Donnan potential is basically the imbalance of charge caused by freely entering, small, permeable ions in presence of large impermeable charged molecules that are trapped in the periplasmic space of Gram-negative bacteria. Generally, membrane potential is negative towards the periplasmic side (40–80 mV), which drives small positive molecules across the membrane. Thus, small positively charged antibacterial compounds can utilize this mode of entry into Gram-negative bacteria14.
Treatment options against Gram-negative bacterial pathogens
Several classes of antibiotics have been developed to target multi-drug resistant Gram-negative bacterial pathogens. For example, fosfomycin was earlier used as an effective treatment option in urinary tract infections (UTIs)29,30. Similarly, tigecycline (targeting CRE and CRAB)31,32, fluoroquinolones (broad spectrum antibiotic effective even against drug-resistant strains), aminoglycosides (to treat Gram-negative bacteria associated nosocomial pneumonia) and piperacillin/tazobactam (against multiple Gram-negative pathogens)33 are other effectively used antibiotics. The membrane-disrupting drug colistin is employed only as an antibiotic of last resort against serious, MDR- Gram-negative bacterial nosocomial infections. However, colistin is highly nephrotoxic and the emergence of colistin-resistant strains limits its clinical use1. Tables 1, 2 list various antibiotics discovered or under development targeting Gram-negative bacterial pathogens.
More recently, new antibacterial molecules targeting Gram-negative bacterial pathogens have entered the clinical pipeline. ARX-1796, a diazabicyclooctane (DBO) β-lactamase inhibitor (DBO-BLI) is under development by Arixa Pharmaceuticals for the treatment of Gram-negative Enterobacterales infections. Cefepime/Zidebactam is another cephalosporin with a DBO-BLI/PBP2 binder combination approved for treatment of Gram-negative pathogens including A. baumannii, P. aeruginosa and Enterobacterales34.
Apart from these, antimicrobials targeting novel bacterial targets such as bacterial riboswitches (e.g., roseoflovin), quorum sensing (e.g., Gram-negative-specific N-acylhomoserine lactones, AHLs), type III secretion systems (e.g., salicylideneacylhydrazides), biofilm (e.g., glycomimetic compounds)35, ClpP, ClpX, MgrA, and ToxT modulators (e.g., β-lactone U1, phenyl esters AV170, and AV286) are also under study36.
Although numerous classes of antibiotics are available to combat Gram-negative pathogens, rapidly emerging MDR strains limit our treatment options. In this scenario, clinicians are forced to use last-resort drugs such as colistin, etc., consequently increasing the pressure on even last-resort antibiotics37.
Physicochemical parameters of compounds to target outer membrane for accumulation in Gram-negative pathogens
An important step of designing a drug against Gram-negative bacteria is focusing on the increased accumulation of the compound into the bacterial cell. This can be done by tuning the physicochemical parameters of the compound. Several studies have been conducted to study the correlation between the physicochemical properties of compounds and their accumulation in Gram-negatives. Studies involving entry of compounds via porins revealed increased penetration of zwitterionic molecules and reduced penetration of analogs with dianionic charges and bulky/protruding side chains. Thus, highlighting the importance of charge and size of a compound to allow its passage through the constriction zone of porins. Adding on to the physicochemical properties, intracellular accumulation of tetracyclines and fluoroquinolones in E. coli was found to display moderate inverse correlation with hydrophobicity38.
The initial correlation between the physicochemical properties of a drug and its oral bioavailability was proposed by Lipinski et al. They set five general rules as drugability guidelines for New Molecular Entities (NMEs) based on HTS results. According to these rules, bioavailability of a drug molecule depends on key physico-chemical properties like molecular weight, lipophilicity and hydrogen bond counts39. The molecule should possess not more than five hydrogen bond donors, hydrogen bond acceptors (≤10), molecular mass (<500 Da) and calculated octanol-water partition coefficient (clogP ≤ 5). Although the rule is applicable to several drugs, many antibiotics, anti-fungal, vitamins and cardiac glycosides fall outside the “rule of 5”. O’Shea et al. analyzed a set of 147 active antibacterials and reported that anti-bacterial compounds share a different property space as compared to other therapeutic drug classes, i.e., comparatively high molecular weight and low lipophilicity. Narrowing down the antibacterial properties, they pointed out that compounds targeting Gram-positive and Gram-negative bacteria have different physicochemical properties. For instance, the average molecular weight of compounds active against Gram-positive bacteria was 813 Da, while for Gram-negative bacteria, it was 414 Da. Additionally, drugs targeting Gram-negatives were found to have higher polarities (clogP = −0.1) as compared to those targeting Gram-positive bacteria (clogP = 2.1). Higher polarity of compounds targeting Gram-negative bacteria corresponds to the fact that the compounds are seeking their way through the hydrophilic porin channels. Therefore, increased polarity and reduced size aid the entry of a compound into Gram-negative bacteria40.
Optimizing the physicochemical parameters of a compound is one of the most challenging ways of initiating the entry of compounds into Gram-negative bacteria. Although low molecular weight and low lipophilicity were identified as two important parameters to potentiate drug entry into Gram-negative bacteria OM via porins, these parameters were not sufficient to develop a broad-spectrum antibacterial drug. Moreover, differences in the LPS composition and OM proteins amongst various bacterial pathogens call for a more exhaustive study11.
In this scenario, Hergenrother and colleagues investigated the accumulation of over 180 diverse compounds, including ~70 Gram-positive-only antibiotics in E. coli MG1655 to identify the common physicochemical properties of compounds to get accumulated within cells. Based on their observation, they proposed general rules of compound accumulation for E. coli, i.e., the presence of an ionizable Nitrogen, low Three-dimensionality, and Rigidity, further codified as “eNTRy rules”41. Accumulation data highlighted the contribution of charge in accumulating compounds in E. coli. Compounds with a positive charge were more likely accumulated as compared to other compounds. Further investigation identified the important contribution of primary amines to compound accumulation, as compared to secondary, tertiary, or quaternary amines, since most of the accumulating compounds were found to contain primary amines. Therefore, structure-activity relationship (SAR) studies were conducted to validate the role of primary amines by displacing them with other functional groups in different classes of accumulating compounds. However, the primary amine was not the only factor contributing to compound accumulation as from the total of 68 compounds containing primary amines tested, 32 primary amines did not show accumulation. Therefore, Hergenrother and group investigated other factors essential for drug accumulation using a set of 68 primary amines which were studied for a total of 297 molecular descriptors by computational analysis. Additionally, high rigidity (as described by the number of rotatable bonds) and low three-dimensionality (as measured by a computed globularity score) of compounds seem to contribute to accumulation in E. coli. The probable reason for increased drug accumulation was the lower susceptibility of rigid and planar compounds to the action of efflux pumps41. Based on the observed results, guidelines for compound accumulation in E. coli were postulated. The major advantage of these chemical traits was that these parameters can be implemented by medicinal chemists into new compounds. Additionally, the compound’s amphiphilic moment and amine steric hindrance were also found to influence drug accumulation. In some cases, guanidiniums and N-alkylpyridiniums were also found to facilitate compound accumulation in E. coli41. Gram-positive specific antibacterial compounds optimized to broaden their anti-microbial spectrum by applying eNTRY rules are described in Table 3.
A novel fluoroquinolone, delafloxacin, was designed for the treatment of acute bacterial skin and skin structure infections caused by both Gram-positive and Gram-negative bacterial pathogens. Three key structural features of delafloxacin include the lack of a strong basic group at position C7 (making the compound anionic at physiological pH), the presence of a chlorine atom in position C8 (stabilizing the heterocycle) and an aromatic ring at N1 (increases the molecular surface). The presence of the carboxylic acid in delafloxacin leads to a higher accumulation of compound at acidic pH, which is physiologically relevant in acute bacterial skin and skin structure infections. Delafloxacin has further shown good potency in treating respiratory, urinary, and sexually transmitted infections42.
OM disruption as a strategy to increase permeabilization
Pentamidine is a clinical antiprotozoal drug that is used in the treatment of Pneumocystis pneumonia43,44, trypanosomiasis, and leishmaniasis45. It is known to have a moderate anti-bacterial activity against Gram-positive bacteria46,47. In addition, it potentiates novobiocin, erythromycin and rifampicin against Gram-negative bacteria, including wild-type and polymyxin-resistant strains of A. baumannii48,49. Based on the structure of pentamidine, Wesseling and group synthesized similar bis-amidine analogs50. SAR studies highlighted the effect of chain length, rigidity, and hydrophobicity of the linker units in bis-amidines in potentiating Gram-positive antibiotics. Long, hydrophobic linkers were generally found to be associated with non-specific membrane disruption, along with strong hemolytic activity. In contrast, linker motifs with a single aromatic ring displayed enhanced synergistic activity compared to pentamidine, without being hemolytic. Furthermore, the relative positioning of the benzamidine groups on the aromatic linker and ortho-, meta- and para-substitution pattern of the amidine moieties themselves, played an important role in potentiating the activity of erythromycin, rifampicin and novobiocin against a number of E. coli strains including polymyxin-resistant and carbapenem-resistant variants with MICs being reduced by factor 33- to ≥60. Additionally, bis-amidines displayed a strong synergistic activity with antibiotics targeting Gram-positive bacteria against other Gram-negative bacterial pathogens including A. baumannii, K. pneumoniae and P. aeruginosa. Mechanistic studies revealed that these compounds act by disrupting the OM of Gram-negatives, thus increasing the antibiotic’s permeability9,50.
Zhou et al. screened a library of 170 FDA-approved antibiotics in combination with pentamidine. Pentamidine was found to increase the susceptibility of E. coli K12 for most fluoroquinolones, tetracyclines, amphenicols, pleuromutilins, ansamycins, macrolides, oxazolidinones and a small fraction of β-lactams and nitrofurans. However, aminoglycosides, aminocoumarin, sulfonamides and lincosamide classes of drugs were poorly boosted. Further, pentamidine was found to potentiate minocycline, linezolid, valnemulin and nadifloxacin against all the standard laboratory strains of A. baumannii, P. aeruginosa, and K. pneumoniae. Moreover, tetracyclines and oxazolidinones displayed a strong synergistic effect while aminoglycosides displayed a strong antagonistic effect with pentamidine. Molecular descriptor-based approaches to understand the physicochemical properties of synergistic antibiotics revealed six descriptors describing four molecular characteristics, namely hydrophobicity, partial charge, rigidity and surface rugosity51. These descriptors are in continuation with the eNTRy rules described above. For example, the potent Gram-positive antibiotic linezolid possesses high hydrophobicity and rigidity. This was further justified by a stable MIC of the drug even with the addition of exogenous phosphatidylethanolamine (PE)51.
The safety of primary amine-containing compounds was questioned due to the increased risk of hERG (human ether-a-go-go-related gene) inhibition and a very limited number of commercially available antibiotics possess a primary amine group in their structure. However, the lack of primary amines in most of the commercially available antibiotics was justified by difficulties in the synthesis and purification of primary amine-containing compounds. Richter and colleagues studied and reported the safety pharmacology profiles of several primary amine-containing compounds including commercially available antibiotics β-lactams (ampicillin, amoxicillin, ceftolozane, cefadroxil, cephalexin etc.), aminoglycosides (kanamycin, amikacin, sisomicin, netilmicin etc.), vancomycin, daptomycin, colistin and many antibiotics at their discovery phase (nemonoxacin, plazomicin, surotomycin, cefilavancin, cefamulin, LTX-109, ACHN-975, epetraborole, and murepavadin)9,43.
Limitations of eNTRy rules and other identified physicochemical identifiers
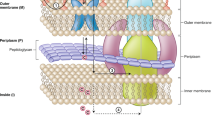
Although the presence of a primary amine was in several examples found to be an important factor in promoting drug accumulation in Gram-negative bacteria, other structural features such as flexibility and shape have an important contribution in the uptake of compounds via self-promoted cell uptake. Apart from these parameters, charges present on the compounds play an important role in transporting molecules via porins for small, hydrophilic molecules and OM disruption for lipophilic and positively charged molecules. Figure 1 illustrates the mode of entry of a wide range of antimicrobials through the outer membrane components in Gram-negative bacteria e.g., porins, transport proteins and OM proteins. However, the limited accumulation and efflux of antimicrobials in Gram negative bacteria tags Gram-negative bacteria-a difficult-to-treat-microbe. In this scenario, Fig. 2 describes the rules governing the physicochemical parameters of antibacterial agents to improve their accumulation in Gram-negative bacteria.

The figure represents the complex architecture of the cell envelope of Gram-negative bacteria which acts as a barrier for antimicrobial agents to enter the cell because of their specific physio-chemical composition. The figure illustrates the routes of entry followed by a wide range of antimicrobials to enter the outer membrane. The ways include membrane disruption by competing with divalent cations of membrane (as followed by agents such as PMBN, dendrimers, anti-microbial peptides), hydrophilic β-barrel proteins called porins (as followed by fluoroquinolones, β-lactams, tetracyclines, chloramphenicol), targeting outer membrane proteins (e.g.,darobactin, murepavadin, MRL-494). Further, the antibacterials such as macrolides utilizes membrane diffusion mechanism to enter the bacterial outer membrane. Adding on to the entry routes, antibacterials such as albomycin make use of transport proteins to cross outer membrane of bacterial cell. However, the accumulation of antimicrobials inside bacterial cells is compensated by the presence of outer membrane efflux pumps that export the entered antimicrobial agents and other foreign substances out of the cell. Figure created with BioRender.com.

Of these rules, traditional drug space of orally bioavailable compounds (Lipinski rules) suggests that a druggable molecule should possess an av. mol. wt. of <500 Da, with <5 hydrogen bond donors, ≤10 hydrogen bond acceptors and calculated octanol-water partition coefficient (clogP ≤ 5). These physicochemical properties were further narrowed down by O’Shea and Moser’s, according to which, a Gram-negative targeting compound must possess an average mol. wt. of 414 Da with clogP ~ 0.1. Later, according to the eNTRy rules proposed by Hergenrother and colleagues, compounds with ionizable nitrogen, globularity ≤0.25 and rotational bond ≤5 are more likely to accumulate in Gram negatives. Glob globularity, RB number of rotatable bonds. Figure created with BioRender.com.
An example where one of the modifications made was the addition of a primary amine that does not broaden the antibacterial spectrum was the conversion of erythromycin to (9S)-erythromycylamine52,53. In contrast, azithromycin, a macrolide with RB = 7; Glob = 0.24 was found to enter Gram-negative bacteria via the self-promoted pathway54. The structures of erythromycin and pleuromutilin along with their modified analogs are depicted in Fig. 3.

A Erythromycin, a macrolide class antibiotic, was modified to 9(S)-erythromycyclamine to improve antibacterial spectrum by introducing primary amine to the structure. In contrast to the eNTRy rules, introduction of primary amine does not make any difference in the anti-bacterial spectrum of the compound. B Similarly, modification of Pleuromutilin to Pleuromutilin-amine by introduction of primary amine was not effective in improving its antibacterial activity. C Azithromycin, a macrolide class antibiotic, having RB=7 is studied to enter Gram-negative bacteria by self-promoted uptake, which is again in contrast to the eNTRy rules where RB of the compound to accumulate in Gram-negatives should ≤5.
Hirsch and colleagues evaluated the applicability of eNTRy rules for repositioning an antimalarial chemical class targeting Plasmodium falciparum (Pf) IspE, which lacked activity against bacterial pathogens even in the presence of PMBN. They designed and synthesized a library of amino acid-modified compounds together with the respective Boc-protected analogs. Of these analogs, only glycine-containing compounds were found to show activity against E. coli while suffering efflux issues55. Taken together, there are several physicochemical factors that affect the entry and accumulation of compounds in Gram-negative bacteria. These parameters alone or in combination need to be analyzed for developing broad-spectrum antibiotics against MDR Gram-negative bacterial pathogens.
Other strategies targeting the outer membrane of Gram-negative bacteria
In addressing the problem of OM permeability in Gram-negative bacteria, a number of new and innovative approaches are currently being investigated. The strategies include interfering with LPS biosynthesis, targeting OM proteins such as the β-barrel assembly machine (BAM) complex, entry of compounds via siderophores via a Trojan horse strategy, co-administration of drugs with membrane disrupting molecules or efflux pump blockers9. Details of these strategies are discussed in the following section.
Targeting outer membrane proteins
OM proteins are attractive and easy-to-approach targets for antibacterial drugs56. Drugs targeting these proteins indirectly lead to OM disruption. The acetyltransferases LpxA and LpxD are involved in the first and third steps respectively of LPS biosynthesis in Gram-negative bacteria. Both these enzymes are essential for bacterial growth56. New England Biolabs screened a library of ~1.9 billion random 12-amino acid peptides, fused to M13 phages. Peptide RJPXD33 (TNLYMLPKWDIP) was found to exhibit moderate affinity with LpxD (Kd = 6 µM). Additionally, the compound was also found to bind and inhibit LpxA (Kd = 22 µM). RJPXD33 contains a conserved fatty acid binding groove similar to LpxA and LpxD, thus highlighting its dual-target potency. Although no whole-cell activity was reported for RJPXD33, the property of the compound to target essential proteins opens new drug targets for drug discovery56,57.
Similarly, other enzymes involved in lipid biosynthesis, Kdo biosynthesis, and LPS transport were studied to identify new potential targets for drug discovery56. A list of compounds under study aiming LPS disruption or targeting the outer membrane proteins of Gram-negative bacteria is mentioned in Table 4.
García-Quintanilla and coworkers have studied the efficacy of the LpxC inhibitor PF-5081090, which at 32 µg/mL effectively potentiates rifampin, vancomycin, azithromycin, imipenem and amikacin against MDR A. baumannii strainsleading to significant increase in cell permeability58.
Vinuesa et al. studied the synergistic effect of LpxC inhibitors with iron chelators, 2,2′-bipyridyl (BIP) and deferiprone, and gallium nitrate (Ga(NO3)3) against A. baumannii including MDR strains. LpxC inhibitor LpxC-2 displayed synergy with BIP against 30% of strains tested (FICI values: 0.38–1.02), and with Ga(NO3)3 against 50% of tested strains (FICI values: 0.27–1.0). Another LpxC inhibitor, LpxC-4 displayed potent synergy with Ga(NO3)3 against all the tested strains with FICI values: 0.08– ≤ 0.5059.
Imai and group studied the antibacterial property of 67 strains of nematode symbiontsPhotorhabdus and Xenorhabdusagainst E. coli. A small zone of E. coli inhibition was seen for P. khanii HGB1456. Bioassay-guided isolation of bacterial extracts led to the identification of a modified heptapeptide, darobactin (W1-N2-W3-S4-K5-S6-F7 with two cycles formed between W1-W3 and W3-K5). Darobactin significantly inhibits MDR E. coli, K. pneumoniae, and some strains of P. aeruginosa, including polymyxin-resistant, ESBL (extended-spectrum β-lactamase), and carbapenem-resistant clinical isolates at 2–4 μg/ml. The compound was found to be bactericidal against E. coli with a minimum bactericidal concentration (MBC) of 8 μg/ml. Additionally, the compound was not cytotoxic against HepG2, FaDu and HEK293 cell lines with IC50 > 128 μg/mL. Sequencing of darobactin-resistant mutants highlighted 2–3 mutations in the OM essential protein BamA, the core protein of the BAM machinery. By targeting BamA, darobactin disrupts the complete β-barrel assembly machinery (BAM) of GNB, thereby disrupting the folding and assembly of OM proteins60. Müller and group designed and biosynthesized new darobactin analogs based on cryo-EM structure of BamABCDE (BAM)-D9 complex. Analogs were generated by the core peptide modification strategy, which includes addition of multiple halogenated tryptophans. Of these derived analogs, darobactin 22 (D22) was found effective against clinical strains of carbapenem-resistant A. baumannii and P. aeruginosa with up to 32- and 4-fold reduction in MIC values. In fact, the compound was equipotent to colistin61.
Efflux pumps inhibitors (EPIs)
EPs play a predominant role in the development of intrinsic resistance among microbes rendering them attractive targets in the field of antibacterial therapy. Most antibiotics that penetrate the OM or reach the cytosol of Gram-negatives cells are ejected by these efflux pumps. They prevent the accumulation of compounds within the periplasmic space and the bacterial cytosol, thus protecting the bacteria from the action of an anti-bacterial agent12. Several studies have shown that the antibacterial activity of a drug molecule is restored in efflux pump deficient/mutated bacteria.
In this scenario, the development of efflux pump inhibitors was initiated to rehabilitate the activity of antibiotics in resistant strains. Among the different ways of inhibiting the activity of efflux pumps, two possible ways are by blocking the synthesis of ATP via stopping the formation of the electrochemical proton gradient, or by preparing antibiotic-competing compounds to prevent their excretion. Examples of competitive RND efflux pump inhibitors comprise the dipeptidyl amide phenylalanine-arginine-β-naphthylamide (PAβN), pyridopyrimidines, and arylpiperazines. Among the arylpiperazine inhibitors, 1-(1-naphthylmethyl)-piperazine (NMP) is the most effective62. Yoshida and colleagues developed the derivative D13–9001, which specifically inhibits the MexAB-OprM pump and displays good in vitro activity against P. aeruginosa63. Similarly, hydantoin derivatives were found to restore antibacterial potency of antibiotics like nalidixic acid and chloramphenicol against Enterobacter aerogenes by inhibiting the AcrAB–TolC system64.
To date, a number of efflux pump inhibitors have been studied and synthesized to potentiate antibiotics against MDR Gram-negative bacterial pathogens65. Although efflux pump inhibitors are an attractive strategy to increase drug accumulation, their development faces several roadblocks. Commercial production of naturally derived efflux pump inhibitors finds difficulty in synthesizing molecules because of their complex structures, while synthetic inhibitors face poor solubility and toxicity issues. Moreover, being used as a combinational therapy, pharmacokinetic compatibility of both the drugs is required. Additionally, resistance mechanisms other than efflux limit the implication of antibiotic-efflux pump inhibitor combinational therapy. Generally, the efflux pump inhibitor binds at a specific site on target, thus limiting the number of substrates it acts upon. This was observed with PAβN which potentiates only a limited set of antibiotics while being ineffective for others, thus, limiting the use of this combinational therapy. Therefore, antibiotic-efflux pump inhibitor combinations require more studies to develop effective therapy strategies against drug-resistant bacterial pathogens66.
Synergy: a cooperative way of transporting a compound over the OM
The development of membrane disruptive agents offers a synergistic effect with Gram-positive selective antibiotics and potentiates them towards Gram-negative bacteria. In the field of synergy, a fractional inhibitory concentration index (FICI) below 0.5 is considered as a benchmark for the synergistic activity67.
Polymyxin B was the first and very effective membrane-disrupting lipopeptide against Gram-negative bacteria. However, nephrotoxicity associated with polymyxin B restricts its use68. Polymyxin-resistant strains show changes in the LPS layer of the OM, a reduced membrane negative charge and a reduced number of porins69. Therefore, Polymyxin B nonapeptide (PMBN), the first and most effective membrane-disruptive synergist against Gram-negative bacteria, was designed by removing the lipid moiety of polymyxin B and is used as a benchmark for synergistic activity. The compound itself does not possess any antibacterial activity, but effectively potentiates the entry of other antibiotics22. After the discovery of PMBN as a potent permeabilizing synergist of Gram-negative bacterial OM, a vast amount of research has been done on synergists that being inactive alone, can potentiate other antibiotics against Gram-negative bacteria9.
Peptide-based synergists
Apart from PMBN, several truncated derivatives of polymyxin B, including deacylpolymyxin B (DAPB), polymyxin B octapeptide (PMBO) and polymyxin B heptapeptide (PMBH), were found to elicit synergy against Gram-negative bacteria with limited toxicity22. SPR741 (formerly, NAB741), a PMBN derivative with only three positive charges and no lipophilic tail was found to display a potent synergistic effect70. The compound has successfully completed a phase I clinical trial and is now being investigated further for its in vivo efficacy. Additionally, it has been reported that increasing the lipid chain diminishes its anti-bacterial potency but enhances its synergistic potential9. Examples of such compounds are NAB739 and NAB7061, where NAB739 is a very dominant synergist but has less inherent antimicrobial activity than NAB706157,71,72. Several endogenous peptides in humans and other mammals associated with natural innate immunity are found to target the OM of Gram-negative bacteria. Therefore, a large number of derivatives targeting OM of Gram-negatives were designed based on natural human peptides such as cathelicidins, lactoferrin, thrombin, novicidin (sheep), bactenectin (bovine) and indolicidine (bovine). These peptides can be used in combination with drugs that can naturally not be transported in GNB9.
A similar membrane targeting effect was seen for S-(3,4-dichlorobenzyl) isothiourea (A22) and its analogs, that target the bacterial cytoskeletal protein MreB, thereby resulting in increased membrane permeability73.
In the field of peptide-based synergy, lipopeptide-based compounds have been studied extensively. Although they do not possess any antibacterial activity, they can enhance the activity of other antibiotics. They carry a few unnatural and D-amino acids and are designed based on peptides produced by Paenibacillus sp. strain OSY-N. These lipopeptides show low hemolytic activity, exhibit potent synergy by possessing OM-disrupting activity and are found to potentiate clarithromycin and rifampin against polymyxin-resistant E. coli in a murine thigh infection model. Another example of a lipopeptide, paenipeptin-inspired synergist (SLAP-S25), was found to boost the activity of rifampicin and vancomycin against E. coli. SLAP-S25 is established to have disruptive effects on both outer and inner membranes, which is proven by its binding to LPS and phosphatidylglycerol (PG)74.
Dilipid ultrashort cationic lipopeptides (dUSCLs) are lysine-rich tetrapeptides with various lipids at the N-terminal of the peptides. Such dUSCLs are found to have the capability of enhancing the activity of clinically used antibiotics against Gram-negative bacteria75.
Besides, a series of ultrashort tetrabasic lipopeptides with three basic amino acids separated by a molecular scaffold bis(3-aminopropyl)glycin, along with simple fatty acids is also reported to be capable of potentiating clinically used antibiotics against Gram-negative bacteria, which are previously selective for Gram-positive bacteria only76.
Antibiotic-adjuvant combination approach
An adjuvant is a bioactive helper molecule that enhances the activity of antibiotics. It can be an efflux pump inhibitor, a membrane permeabilizer or an enzyme inhibitor. An antibiotic-adjuvant combination approach is one of the most successful forms of treatment therapy where an antibiotic is given in combination with a helper molecule. The activity of this kind of synergist such as a β-lactam–β-lactamase inhibitor combination (e.g., amoxicillin and clavulanic acid, ceftolozane-tazobactam, ceftazidime-avibactam and meropenem-vaborbactam, Imipenem-cilastatin-relebactam triple combination, Aspergillomarasmine A-meropenem) was explored and was found to overcome antibiotic-resistance mechanisms, as reviewed previously77.
Some antibiotic-derived synergists are also explored where antibacterial compounds having OM disruptive activity are conjugated with antibiotics that are otherwise inactive towards Gram-negatives. Aminoglycosides like tobramycin have OM disruptive capability via binding to LPS, thereby causing membrane depolarization. A series of tobramycin-fluoroquinolone conjugates such as tobramycin-moxifloxacin78 and tobramycin-ciprofloxacin79 conjugates were explored and found to enhance the antibiotic activity of several reported drugs, including rifampicin, erythromycin, novobiocin and vancomycin against Gram-negative bacteria. Tobramycin was also found to improve the efficacy of lysine-based amphiphiles by permeabilizing the bacterial outer membrane80,81. These lysine-based amphiphiles include, amongst others, erythromycin and vancomycin. A combination of novobiocin and a tobramycin homodimer has shown efficacy against A. baumannii in a wax moth larvae model82. Tobramycin was also explored after coupling with small-molecule efflux pump inhibitors such as 1-(1-naphthylmethyl)piperazine (NMP) and paroxetine. These inhibitors were found to potentiate its activity against P. aeruginosa83. These synergists are also found to cause OM disruption and inner membrane depolarization. A novel hybrid of levofloxacin and a polybasic peptide was found to exhibit strong activity against MDR clinical P. aeruginosa, E. coli, K. pneumoniae and A. baumannii84.
Recently, two generations of P. aeruginosa biofilm-targeted conjugates of fluoroquinolone antibiotics and glycomimetic lectin binders were disclosed. Introduction of a peptide-based prodrug mechanism reduced the toxic potential of the attached drugs while retaining full antibiotic activity after self-destructive release by P. aeruginosa85,86.
Antibiotic hybrid approach
The antibiotic hybrid approach fuses two or more antibacterial agents into one heteromeric entity, ideally retaining the activity of the individual constituent fragments. This linkage of fragments can be cleavable (prodrug approach) or non-cleavable (hybrid drug approach) and occur between two antibiotics or antibiotic and adjuvant77. For example, an antibiotic hybrid was generated by linking a cefamandole (cephalosporin) derivative to omadine (a metal chelator and bacterial ATP synthesis inhibitor) to form a hybrid prodrug87. Most of the antibiotic hybrid prodrugs are cephalosporin-based. Some other examples of antibiotic hybrids are desacetylcephalothin linked to the alanine racemase inhibitor chloroalanyl dipeptide88,89, desacetylcephalothin linked to triclosan-NB200190, and desacetylcefotaxime linked to fleroxacin-Ro 23–942491. These hybrid prodrugs displayed potent activity against a panel of Gram-negative bacterial pathogens.
Tobramycin-based hybrids with fluoroquinolones were developed to promote the entry of antibiotics into the periplasm of Gram-negative bacterial cells by self-promoting the uptake mechanism of aminoglycosides (see above). These hybrids displayed antimicrobial activity against P. aeruginosa77. Antibiotic hybrids are thus an interesting approach to drug discovery, but require certain points into consideration: 1. Linking of two moieties should be at non-pharmacophoric regions to ensure the retention of biological activity of both compounds. 2. Spatial length of linker should be optimized77. Structures of cefamandole-omadine conjugate and tobramycin-ciprofloxacin conjugate are depicted in Fig. 4.

A Cefamandole-omadine conjugate, a cephalosporin and 2-mercaptopyridine-N-oxide or pyrithione (metal chelator and ATP synthesis inhibitor) conjugate and B ciprofloxacin‑tobramycin conjugate that effectively targets Gram-negative bacteria via dual mode of action.
Chelating agents as OM-disrupting synergists
OM-disrupting molecules such as chelators or polymyxins compete with divalent cations cross-bridging neighboring LPS molecules. Displacement of these ions leads to enhanced lateral diffusion of molecules in the bacterial membrane6. The potential of the chelating agent EDTA to synergize with other antibiotics against Gram-negative bacteria is well known92. Additionally, Ayres and Russell have highlighted the ability of sodium polyphosphates as potent synergists with several antibiotics93. Citric acid is also found to exhibit synergistic activity with antibiotics including erythromycin, novobiocin, rifampicin, methicillin, and gentamicin93. Moreover, 2,3-dimercaptosuccinic acid, an effective treatment of lead intoxication, was also found to potentiate hydrophobic antibiotics against Gram-negative bacteria by permeabilizing OM of bacteria, as depicted by increased N-phenylnaphthylamine (NPN) uptake9. Despite the excellent activity of chelating agents, polymyxin-resistant strains of S. typhimurium and E. coli were also reported with 75% lower binding to polymyxins compared to the parent strains. Studies conducted on the LPS layer of these bacteria revealed increased levels of aminoarabinose and phosphoethanolamine formation due to the esterification of lipid A phosphates. This lowered negative charge significantly reduces the binding of polymyxin and other chelating agents6.
Terpene-based synergists
Some natural products possess antibacterial activity due to their membrane permeabilizing ability and show synergistic effects with antibiotics against MDR Gram-negative bacteria. For example, some terpenoids such as eugenol, citronellol, carvacrol, geraniol, menthol, myrcene, and thymol show synergistic effects with penicillin against E. coli. Eugenol, a compound found in clove oil from Eugenia aromatic functions as membrane permeabilizer via inhibiting an ATPase and has synergistic effect with ampicillin, penicillin, oxacillin, erythromycin, norfloxacin, tetracycline, chloramphenicol, vancomycin, rifampin, and polymyxin B against MDR Gram-negative bacteria, following different mechanisms. Among these synergized antibiotics, penicillin, ampicillin, oxacillin, vancomycin, and polymyxin B affect the cell membrane via different targets, norfloxacin damages the cell membrane by removing divalent cations from LPS-binding sites, while erythromycin, rifampin, tetracycline, and chloramphenicol attack the ribosome of bacteria94.
Entry via siderophores and their mimetics
Iron, under physiological conditions, is scarcely present in its free form (10–9 M) due to its rapid oxidation from Fe2+ to Fe3+, subsequently forming insoluble hydroxides. The uptake of iron in this form is a challenge for organisms relying on this naturally available iron. To circumvent iron shortage, bacteria display certain receptor proteins that bind a variety of iron-containing molecules. Additionally, they produce and secrete high-affinity iron-chelating compounds called siderophores. Upon binding to bacterial receptors, they are translocated across the membrane via ABC importers95. A natural Trojan horse mechanism of delivering antibiotics along with siderophores was identified in bacteria through the identification of albomycins (ferrichrome siderophore analog conjugated with thioribosyl pyrimidine antibiotic), ferrimycins, salmycins and some microcins. These siderophores were found to show antibacterial effects against several Gram-negatives. For example, albomycin (secreted by Streptomyces strains) displays activity against certain GNB including E. coli. Similarly, microcin E492 (MccE492) produced by K. pneumoniae RYC492 displayed activity against E. coli, S. enteritidis and S. typhimurium96. Naturally occurring hydroxamate-type siderophores are covalently attached to the antibiotic moiety to formulate these sideromycins. Considering this transport mechanism, efforts were made to increase antibiotic-loaded sideromycin uptake in pathogenic bacteria. Generally, conjugation involves catechol/hydroxamate siderophore analogs covalently attached to a β-lactam drug97.
Zahner and group reported the first synthetic siderophore antibiotic conjugate in which desferrioxamine B (DFO B) with a sulfonamide was ligated to varying cephalosporines, fluoroquinolones, and other antibiotics. However, none of them was found to target Gram-negative bacteria98. Later, Miller and coworkers designed tris-catechol siderophore derivatives, containing an enterobactin analog linked to amipicillin/amoxycillin, that displayed potent antimicrobial activity against P. aeruginosa99.
The siderophore monosulfactam BAL30072 has demonstrated antibacterial potency against MDR Gram-negative bacteria in vitro and in vivo. BAL30072 is a combination of a siderophore-mimicking small molecule, hydroxypyridone and a monocyclic β-lactam antibiotic linked via an oxime, which enables access to the bacterial cell through the iron uptake system. Under in vitro conditions, it has displayed its inhibitory effect against MDR strains of Burkholderiapseudomallei, P. aeruginosa and A. baumannii, including strains producing class C carbapenemases. In an in vivo rat soft-tissue infection model, the conjugate potently inhibited 80% of A. baumannii strains tested. Additionally, the combination of BAL30072 and carbapenems was found effective against MDR Gram-negative bacteria, particularly against Enterobacteriaceae and P. aeruginosa in vitro. BAL30072 has been tested in a phase I study in human subjects but the current development status is unknown97.
Shionogi & Co has developed a novel siderophore cephalosporin conjugate, cefiderocol (brand name Fetroja), for treating UTI and adult pneumonia caused by MDR Gram-negative bacteria. Structurally, cefiderocol is similar to ceftazidime (third-generation cephalosporin, having a pyrrolidinium group at position 3) and cefepime (fourth-generation cephalosporins, having an oxime group at position 7) along with a catechol group on the side chain at position 3 which differs from both ceftazidime and cefepime. Its high stability made the compound active against a variety of Ambler class A, B, C, and D β-lactamases. Under in vitro conditions, it was found more potent than both ceftazidime-avibactam and meropenem against MDR strains of A. baumannii, K. pneumoniae, carbapenemase (KPC)-producing Enterobacterales, MDR P. aeruginosa and Stenotrophomonas maltophilia. Under in vivo conditions, cefiderocol potently inhibited MDR and XDR Gram-negative bacterial pathogens in mouse and rat infection models100. The compound acts by binding to penicillin-binding protein-3, thereby, inhibiting bacterial cell wall synthesis. The compound has proven its efficacy in clinical trials and was approved by FDA in 2020 for the treatment of HABP and VABP28. The structure of cephalosporin conjugate cefiderocol (Fetroja) and monosulfactam BAL30072 is shown in Fig. 5.

A Cephalosporin conjugate cefiderocol (Fetroja), an FDA approved siderophore cephalosporin conjugate for the treatment of hospital acquired bacterial pneumonia (HABP) and ventilator-associated bacterial pneumonia (VABP). B Monosulfactam BAL30072, a monocyclic beta-lactam antibiotic with potent activity against drug-resistant Gram-negative pathogens.
Adding to the list, artificial siderophores based on the 1,3,5-N,N′,N′′-tris-(2,3-dihydroxybenzoyl)-triaminomethylbenzene (MECAM) core were studied in conjugation with β-lactam drugs and daptomycin against Gram-positive bacteria and Gram-negative bacteria. MECAM–aminopenicillin conjugates were found to display some activity against Gram-positive and Gram-negative pathogens101. A variety of synthetic siderophores such as linear Orn-derived tris-hydroxamate, isocyanuric acid-based tri-Orn hydroxamate, biscatecholate-monohydroxamate, etc. have been designed to date. These siderophores share structural similarities with naturally occurring catechol and hydroxamate siderophores. Ligation of these synthetic or semisynthetic siderophores with antibiotic warheads can be an effective bullet targeting GNB98. However, designing siderophore-antibiotic conjugates is limited by their extracellular stability, uptake pathways, receptor binding properties, and its transport across the membrane96.
Although the transport of antibiotics via siderophore has proven to be an attractive antibacterial combination, its efficacy is compromised by downregulation of iron transport receptors in response to the competition between siderophore-antibiotic conjugates and native siderophore production. For example, deletion of TonB-dependent receptors PiuA and PirA in A. baumannii, frameshift mutations in inner membrane protein complex in exbD3 or tonB3 genes, overexpression of proteins involved in regulation of iron transport proteins (e.g., FecIRA operon) were found to affect the efficacy of siderophore conjugates including BAL30072 and MC-128. Additionally, several mechanisms of bacteria confering resistance to cefiderocol have now been studied and beautifully reviewed by Karakonstantis and colleagues. These mechanisms include expression of β-lactamase enzymes (including NDM, KPC, and AmpC, OXA-427, and PER- and SHV-type ESBLs), porin mutations (OmpK35, OmpK36 and OmpK37 in K. pneumonia, OprD in P. aeruginosa and OmpC, OmpF in Enterobacter spp.), modification in siderophore receptors (PiuA/PiuD and PirA in P. aeruginosa, CirA and/or Fiu in Enterobacterales, pirA and piuA in A. baumannii and cir A in K. pneumoniae), over-expression of efflux pumps (sugE and chrA in K. pneumoniae, MexAB–OprM in P. Aeruginosa, smeDEF in S. maltophilia and AxyABM in Achromobacterxylosoxidans), and target (PBP-3) modifications102,103.
A study conducted from 2018 to 2019 across China highlighted the involvement of NDM-5, 4 amino-acid insertions (YRIN/YRIK) at position 333 of penicillin-binding protein 3 (PBP3) and expression of premature siderophore receptor cirAin conferring resistant against cefiderocol104.
Delivery systems of colicin-like bacteriocin
Some bacteria, such as E. coli and related species produce and release small antimicrobial peptides called colicins that show their lethal effect against other susceptible bacteria (e.g., Citrobacter freundii releases colicin A or Shigella boydii releases colicin U). These peptides bind to specific receptors present on OM for entry of specific nutrients and translocate these nutrients through OM and periplasm by utilizing the Tol or TonB systems. Their mechanism of action involves either the formation of a voltage-dependent channel into the inner membrane or utilizing their endonuclease activity on DNA (e.g., ColE2), rRNA, or tRNA (RNase colicins (E3, E4, E5, E6, and D)). Several other bacteria producing such bacteriocins include Pseudomonas pyogenes (pyocins), Enterobacteriaceae such as E. cloacae (cloacins), Yersinia pestis (pesticins), Klebsiella species (klebicins or klebocins), Serratia marcescens (marcescins, colicins L and 24), Photorhabdusluminescens (lumicins), and Bacillus megaterium (megacins)105.
Delivering a drug molecule through this pathway was studied by Biswas and colleagues. They assessed the synergistic activity of Lactobacillus bacteriocin extract alone and in combination with antibiotics cefotaxime, polymyxin B, imipenem, and tigecycline. Purified bacteriocin from Lactobacillus, Pediococcus, and Enterococcus potentially displayed significantly higher inhibition in combination with antibiotics, as compared to antibiotics alone against MDR clinical pathogens E. coli (GN9, IB9, GN13), K. pneumoniae KP7, and A. baumannii AB13775. The combination was even effective in inhibiting the growth of clinical pathogens harboringβ-lactamases106.
Additionally, lectin-like bacteriocins, such as LlpAs, were identified as midsize bacteriotoxins that include tandem B-lectin domains followed by a short non-conserved carboxy-terminal extension. These bacteriotoxins are unable to penetrate the outer membrane due to their large size but act by targeting the outer membrane protein BamA, thereby impairing the BAM machinery of Gram-negative bacteria107. Several bacteriocins (class II bacteriocins, nisin, other lantipeptides) are reported to be noncytotoxic on different eukaryotic cell lines even at higher concentrations. The small size, non-immunogenic nature, biocompatibility, and biodegradability of bacteriocins make them an ideal candidate to be studied further as a replacement strategy for antibiotics108.
Charge-based lipid rearrangements
Interaction of positively charged ions with negatively charged LPS results in the rearrangement of lipid molecules, creating a temporary entry pathway for compounds into the periplasmic space. Octenidine (OCT, N,Nʹ-(1,10 decanediyldi-1[4H]-pyridinyl-4-ylidene)-bis-(1-octanamine) dihydrochloride) enters Gram-negative bacteria via this mechanism. OCT is a synthetic antimicrobial molecule deployed in several countries for skin, mucous membrane, and wound anti-sepsis. OCT was found to display a broad-spectrum antibacterial activity against both MDR Gram-positive and Gram-negative bacteria. The compound displayed its high efficacy at a low concentration within a short contact time, even in the presence of interfering substances such as blood or mucin. MOA studies revealed that the compound interacts with the OM of Gram-negative bacteria, resulting in chaotic lipid rearrangement. This membrane disruption leads to the entry of the compound into the periplasmic space109.
Translocation through nanopores
An additional pathway of entry of specific antibiotics into bacterial cells is through bacterial nanopores. Bacterial cells utilize these nanopores to transport ions and other substrates into the cells. The conduction through these nanopores depends largely on parameters such as ion current, temperature, electrostatic and steric effects110. Several studies have been conducted to study the transport of cations and anions through the matrix porins OmpF, osmoporin OmpK36, phosphoporinPhoE by Brownian dynamics simulations and highlight the effect of change in electrostatic potential at pore constriction site on ion transport111.
Jiajun and group studied the effect of divalent ions (like magnesium) on fluoroquinolones entry into bacterial cells via OM nanopores. The study revealed that divalent ions bind to the negatively charged residues inside the porins. By doing so, they reverse the ion selectivity of the pore, thus allowing the entry of molecules which else were unable to enter. Moreover, divalent ions can also chelate with antibiotic molecules like fluoroquinolone and produce differences in dipole moment of both the compounds involved, thereby affecting entry via these pores112.
Liposome-mediated entry
Another promising strategy for delivering a drug molecule into a bacterial cell is lipid-based delivery (nano)systems. The strategy involves encapsulation of a drug molecule in liposomes, which offers stability and safety to the drugs by accumulating it at infection site, minimizing drug toxicity, and protecting it from peripheral degradation. Liposomes are vesicular concentric bilayer structures surrounding aqueous compartments or units. Their unique architecture which is capable of encapsulating hydrophilic drugs within the aqueous compartment and/or carrying hydrophobic drugs inside their lipid bilayer makes them different from other nanoparticles. Liposomes vary according to size, number of lamellae, lipid composition, charge of the bilayer (anionic, cationic, or neutral), and surface functionalization with polymers or ligands. Liposomes act on Gram-negative bacteria by fusing with their OM, leading to its structural disruption and potentially reversing its low permeability. A number of antibiotic-fused liposomes have been reported that have potentiated the otherwise inactive molecules against clinically important Gram-negative bacterial pathogens. For example, aminoglycoside-loaded liposomes targeting resistant clinical strains of P. aeruginosawere reported by Mugabe et al. These liposomes resulted in an increased antimicrobial susceptibility of the bacteria as compared to free drug113. Liposomal formulations of aminoglycosides are in clinical use. Nacucchio et al. studied the potency of liposome-encapsulated antibiotics to address resistance associated with enzymatic hydrolysis. Phosphatidylcholine and cholesterol-containing liposomes with piperacillin were able to overcome hydrolysis by exogenous staphylococcal β-lactamases114. Adding to the advantage, liposomes encapsulating rifabutin was found effective even against MDR intracellular pathogens like Mycobacterium tuberculosis. Moreover, Polymyxin B encapsulated in Chitosan–dipalmitoylphosphatidylcholine:distearoylphosphatidylethanolamine:cholesterol was found effective in eliminating biofilm-producing bacteria115. Thus, the property of liposome-encapsulated drugs to overcome AMR due to impermeable OM, efflux mechanisms, enzymatic degradation, and biofilm formation make it an ideal strategy to restore treatment options against clinically important MDR bacterial pathogens. However, specificity of liposomes to target bacterial cells only needs to be optimized115. To this end, P. aeruginosa targeted liposomes with a glycomimetic-decorated surface have recently been reported116.
Dendrimers
Dendrimers (Ds) have emerged as an innovative approach targeting MDR Gram-negative pathogens. Generally, dendrimers are branched macromolecules made up of a surface functional group, a core unit, and a branching unit attached to a core. Different families of Ds with different mechanism of action have been studied including glycodendrimers (resist toxin binding to eukaryotic cells, interfere with bacterial adhesion), cationic dendrimers (disrupts bacterial membranes), anionic dendrimers (detergent-like action) and peptidic dendrimers (disrupt bacterial membrane or interact with their intracellular components)117. Cationic Ds as drug delivery system play a significant role in the field of nanomedicine. Some cationic Ds show synergy with commonly used antibiotics and are found to have antibiofilm effects or antimicrobial effects against MDR bacteria118. Cationic Ds such as polyamidoamine (PAMAM) Ds and propylene imine (PPI) Ds carry a large number of peripheral ionizable primary and tertiary amine groups, which give them a high positive charge and correspondingly high antibacterial activity. Most representative cationic PAMAM Ds and PPI Ds developed include levofloxacin (0.1 μg/mL) co-administered with highly maltose-coated 3GPPIs (PPI-G3-DS-Mal), C16-DABCO-loaded mannose-terminated 4G PAMAM Ds, G7 PAMAM-D, self-assembly poly (aryl ether)-PAMAM-based amphiphilic Ds, CdS/Ag2S (QDs)-loaded PAMAM Ds/MWCNTs, G1-G4 NO-releasing-alkyl(QA)-PAMAM Ds, 1G NO-releasing octyl- and dodecyl-modified PAMAM Ds119. Besides, the synergistic effect of PPI glycodendrimers with a dense maltose shell (PPI-G3-DS-Mal) with levofloxacin is reported to reduce the growth of E. coli and the same dendrimer when combined with amoxicillin shows a synergistic effect against Gram-negative bacteria by potentiating the antimicrobial activity of amoxicillin118.
Schito and colleagues investigated the antimicrobial activity of three non-toxic, 5th generation polyester-based cationic dendrimers G5Ds:G5H, G5K, and G5HK against several MDR clinical strains. All the dendrimers were found to potentially target MDR non-fermenting GNB P. aeruginosa, S. maltophilia, and A. baumannii with MICs from 0.5 to 33 µM. Time kill and turbidimetric studies of G5K revealed a rapid, non-lytic bactericidal activity. Additionally, their non-cytotoxic nature makes them an attractive target to be investigated further for their drug-like properties119. Reymond et al. investigated the antimicrobial efficacy of a peptidic dendrimer, G3KL. The structure of the molecule contains leucine and branched chain lysine residues. Activity of G3KL was tested against 32 strains of A. baumannii and 35 strains of P. aeruginosa. G3KL displayed a MIC of 8 µg/mL against all tested strains120. However, its large size and issues in its synthesis were the matter of concern. Therefore, the authors designed a new antimicrobial peptide dendrimer G2KL by reducing the number of amino acids. But the modification diminished its antimicrobial efficacy against E. coli, P. aeruginosa, and Bacillus subtilis. Considering this diminished activity, they modified G2KL by adding a short fatty acid chain, formulating TNS18, which displayed good activity against MDR strains of A. baumannii, P. aeruginosa, and E. coli with a MIC of 4–8 µg/ml121. Despite excellent antibacterial efficacy, dendrimers call for careful optimization for their low biodegradability, susceptibility to opsonization, toxicity to mammalian cells, including hemolytic toxicity, cytotoxicity, and hematological toxicity, as well as fast clearance117.
Fragment-based drug discovery (FBDD)
Another innovative approach of developing a drug molecule is based on fragment-based drug design. This strategy involves finding a new drug/scaffold based on the screening of compound libraries with MW < 300 g/mol. FBDD utilizes strategies such as differential scanning fluorimetry, isothermal titration microcalorimetry, nuclear magnetic resonance, surface plasmon resonance, and X-ray crystallography. Binders are selected for their further synthetic remodeling into drug molecules by fragment growing or fragment merging. This FBDD process gains attention in the field of target-based drug discovery because of its interesting advantages such as minimal experimental costs and offering diverse hits122. Moreau and colleagues screened ~25,000 fragments from Novartis fourth-generation core fragment library targeting the bacterial phosphopantetheine-adenylyl-transferase (PPAT). The enzyme is involved in CoA biosynthesis, but interestingly, does not show high sequence homology with its human analog and thus can be an attractive target of drug molecules. From 25,000 fragments, 631 hits showing good binding affinities with the target (i.e., displaying ≥30% inhibition) were studied further. Triazolopyrimidinone and azabenzimidazole were identified to bind bacterial PPAT. However, the antibacterial activity was against efflux pump-deficient strains123. Similarly, Mansbach et al. screened a fragment library in silico to target OM of Gram-negative bacteria and 43 fragments were predicted to cross the OM. These fragments were then experimentally studied for their OM permeabilizing efficacy and the eight compounds depicting highest OM permeability based on a computational algorithm were studied for their antibacterial activity against P. aeruginosa and A. baumannii, of which five compounds displayed potent activity (µP∆6Pore/µP∆6 > 0.8) against P. aeruginosa124.
Conclusion
Gram-negative bacteria are amongst the most difficult-to-treat bacteria. This primarily results from their uniquely complex cell envelope, which not only acts as a barrier for the entry of antibiotics into the cell but also depletes accumulating drugs by efflux pumps. New antibiotics targeting intracellular processes are being developed, but transporting these drugs across the highly impermeable OM is the major hurdle. Antibiotics targeting the OM, such as outer membrane permeabilizers, OM protein targeting compounds, and compounds entering through porins are currently available.
However, MDR Gram-negative bacteria have evolved resistance mechanisms to escape from the action of these drugs by mutations or the action of drug-targeting enzymes. Unfortunately, resistance against drugs currently in clinical trials is also already observed. Taken together, the emergence of AMR in drugs is inevitable and multiple resistances can accumulate in single pathogens. Therefore, we must prepare for the threat of total drug resistance21.
Physicochemical parameters of antimicrobials have been shown to play an important role for accumulation into Gram-negative bacteria. Correlating these requires both high-throughput and structure-agnostic accumulation assays38. This review highlights the recent ways of targeting the highly selective OM of Gram-negative bacteria by tuning physicochemical properties of drugs or other innovative approaches to overcome the OM. Since we are in the antimicrobial resistance era, we must take further action to develop more effective antibiotics with strong resistance-defeating capabilities.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Bassetti, M. & Garau, J. Current and future perspectives in the treatment of multidrug-resistant Gram-negative infections. J Antimicrob. Chemother. 76, iv23–iv37. https://doi.org/10.1093/jac/dkab352 (2021).
Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
O’Neill, J. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. Grande-Bretagne https://amrreview.org/sites/default/files/AMR%20Review%20Paper%20%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (2014).
Miethke, M. et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 5, 726–749 (2021).
Cochrane, S. A. et al. Antimicrobial lipopeptide tridecaptin A1 selectively binds to Gram-negative lipid II. Proc. Natl Acad. Sci. USA 113, 11561–11566 (2016).
Delcour, A. H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794, 808–816 (2009).
Boucher, H. W. Bad bugs, no drugs 2002-2020: progress, challenges, and call to action. Trans. Am. Clin. Climatol. Assoc. 131, 65–71 (2020).
Choi, U. & Lee, C. R. Distinct roles of outer membrane porins in antibiotic resistance and membrane integrity in Escherichia coli. Front. Microbiol. 10, 953 (2019).
Wesseling, C. M. J. & Martin, N. I. Synergy by perturbing the Gram-Negative outer membrane: opening the door for Gram-positive specific antibiotics. ACS Infect. Dis. 8, 1731–1757 (2022).
Raetz, C. R. H., Reynolds, C. M., Trent, M. S. & Bishop, R. E. Lipid a modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 76, 295–329 (2007).
Tiz, D. B., Kikelj, D. & Zidar, N. Expert opinion on drug discovery overcoming problems of poor drug penetration into bacteria: challenges and strategies for medicinal chemists. Expert Opin. Drug Discov. 00, 1–11 (2018).
Amaral, L., Martins, A., Spengler, G. & Molnar, J. Efflux pumps of Gram-negative bacteria: what they do, how they do it, with what and how to deal with them. Front. Pharmacol. 4, 168 (2014).
McMurry, L., Petrucci, R. E. J. & Levy, S. B. Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc. Natl Acad. Sci. USA 77, 3974–3977 (1980).
Alegun, O. Mechanism of antibiotic permeability through the Gram-negative bacterial envelope https://uknowledge.uky.edu/chemistry_etds/156/.
Ramirez, M. S. & Tolmasky, M. E. Aminoglycoside modifying enzymes. Drug Resist. Updates 13, 151–171 (2010).
Egorov, A. M., Ulyashova, M. M. & Rubtsova, M. Y. Bacterial enzymes and antibiotic resistance. Acta Nat. 10, 33–48 (2018).
Darby, E. M. et al. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 21, 280–295 (2023).
MacNair, C. R. & Tan, M. W. The role of bacterial membrane vesicles in antibiotic resistance. Ann. N.Y. Acad. Sci. 1519, 63–73 (2023).
Silver, L. L. A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem. 24, 6379–6389 (2016).
Cama, J., Henney, A. M. & Winterhalter, M. Breaching the barrier: quantifying antibiotic permeability across gram-negative bacterial membranes. J. Mol. Biol. 431, 3531–3546 (2019).
Payne, D. J., Gwynn, M. N., Holmes, D. J. & Pompliano, D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40 (2007).
Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 56, 395–411 (1992).
Alcaraz, A., Nestorovich, E. M., Aguilella-Arzo, M., Aguilella, V. M. & Bezrukov, S. M. Salting out the ionic selectivity of a wide channel: the asymmetry of OmpF. Biophys. J. 87, 943–957 (2004).
Cohen, S. P. et al. Endogenous active efflux of norfloxacin in susceptible Escherichia coli. Antimicrob. Agents Chemother. 32, 1187–1191 (1988).
Chevalier, J., Malléa, M. & Pagès, J. M. Comparative aspects of the diffusion of norfloxacin, cefepime and spermine through the F porin channel of Enterobacter cloacae. Biochem. J. 348, 223–227 (2000).
Charrel, R. N., Pages, J. M., De Micco, P. & Mallea, M. Prevalence of outer membrane porin alteration in beta-lactam-antibiotic-resistant Enterobacter aerogenes. Antimicrob. Agents Chemother. 40, 2854–2858 (1996).
Chopra, I. Molecular mechanisms involved in the transport of antibiotics into bacteria. Parasitology 96, S25–S44 (1988).
McCreary, E. K., Heil, E. L. & Tamma, P. D. New perspectives on antimicrobial agents: cefiderocol. Antimicrob. Agents Chemother. 65, e0217120 (2021).
Kaye, K. S. et al. Fosfomycin for injection (ZTI-01) versus piperacillin-tazobactam for the treatment of complicated urinary tract infection including acute pyelonephritis: ZEUS, a phase 2/3 randomized trial. Clin. Infect. Dis. 69, 2045–2056 (2019).
Avent, M. L. et al. Fosfomycin: what was old is new again. Intern. Med. J. 48, 1425–1429 (2018).
Hawkey, P. M. et al. Treatment of infections caused by multidrug-resistant Gram-negative bacteria: report of the British Society for Antimicrobial Chemotherapy/Healthcare Infection Society/British Infection Association Joint Working Party. J. Antimicrob. Chemother. 73, iii2–iii78 (2018).
US Food and Drug Administration. Tygacil (tigecycline): prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021821s049lbl.pdf.
Chastain, D. B., White, B. P., Cretella, D. A. & Bland, C. M. Is it time to rethink the notion of carbapenem-sparing therapy against extended-spectrum β-lactamase-producing Enterobacteriaceae bloodstream infections? A critical review. Ann. Pharmacother. 52, 484–492 (2018).
Iskandar, K. et al. Antibiotic discovery and resistance: the chase and the race. Antibiotics 11, 1–38 (2022).
Leusmann, S., Ménová, P., Shanin, E., Titz, A. & Rademacher, C. Glycomimetics for the inhibition and modulation of lectins.Chem. Soc. Rev. 52, 3663–3740 (2023). https://pubs.rsc.org/en/content/articlelanding/2023/cs/d2cs00954d.
Lakemeyer, M., Zhao, W., Mandl, F. A., Hammann, P. & Sieber, S. A. Thinking outside the box-novel antibacterials to tackle the resistance crisis. Angew. Chem. (Int. Ed. Engl.) 57, 14440–14475 (2018).
Mohapatra, S. S., Dwibedy, S. K. & Padhy, I. Polymyxins, the last-resort antibiotics: mode of action, resistance emergence, and potential solutions. J. Biosci. 46, 85 (2021).
Zhao, S. et al. Defining new chemical space for drug penetration into Gram-negative bacteria. Nat. Chem. Biol. 16, 1293–1302 (2020).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26 (2001).
O’Shea, R. & Moser, H. E. Physicochemical properties of antibacterial compounds: implications for drug discovery. J. Med. Chem. 51, 2871–2878 (2008).
Muñoz, K. A. & Hergenrother, P. J. Facilitating compound entry as a means to discover antibiotics for Gram-negative bacteria. Acc. Chem. Res. 54, 1322–1333 (2021).
Scott, L. J. Delafloxacin: a review in acute bacterial skin and skin structure infections. Drugs 80, 1247–1258 (2020).
Intravenous Pentamidine for Pneumocystis jirovecii Pneumonia https://clinicaltrials.gov/ct2/show/NCT02669706?term=pentamidine&draw=2&rank=1.
Brown, K. S., Reed, M. D., Dalal, J. & Makii, M. D. Tolerability of aerosolized versus intravenous pentamidine for Pneumocystis jirovecii pneumonia prophylaxis in immunosuppressed pediatric, adolescent, and young adult patients. J. Pediatr. Pharmacol. Ther. 25, 111–116 (2020).
Dorlo, T. P. C. & Kager, P. A. Pentamidine dosage: a base/salt confusion. PLoS Negl. Trop. Dis. 2, e225 (2008).
Libman, M. D., Miller, M. A. & Richards, G. K. Antistaphylococcal activity of pentamidine. Antimicrob. Agents Chemother. 34, 1795–1796 (1990).
Maciejewska, D. et al. In vitro screening of pentamidine analogs against bacterial and fungal strains. Bioorg. Med. Chem. Lett. 24, 2918–2923 (2014).
Stokes, J. M. et al. Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol. 2, 17028 (2017).
Amos, H. & Vollmayer, E. Effect of pentamidine on the growth of Escherichia coli. J. Bacteriol. 73, 172–177 (1957).
Wesseling, C. M. J., Slingerland, C. J., Veraar, S., Lok, S. & Martin, N. I. Structure-activity studies with bis-amidines that potentiate Gram-positive specific antibiotics against Gram-negative pathogens. ACS Infect. Dis. 7, 3314–3335 (2021).
Zhao, R. et al. Effectiveness of ertapenem for treatment of infections in children: an evidence mapping and meta-analysis. Front. Pediatr. 10, 982179 (2022).
Counter, F. T. et al. Synthesis and antimicrobial evaluation of dirithromycin (AS-E 136; LY237216), a new macrolide antibiotic derived from erythromycin. Antimicrob. Agents Chemother. 35, 1116–1126 (1991).
Massey, E. H., Kitchell, B. S., Martin, L. D. & Gerzon, K. Antibacterial activity of 9(S)-erythromycylamine-aldehyde condensation products. J. Med. Chem. 17, 105–107 (1974).
Farmer, S., Li, Z. S. & Hancock, R. E. Influence of outer membrane mutations on susceptibility of Escherichia coli to the dibasic macrolide azithromycin. J. Antimicrob. Chemother. 29, 27–33 (1992).
Ropponen, H. K. et al. Assessment of the rules related to gaining activity against Gram-negative bacteria. RSC Med. Chem. 12, 593–601 (2021).
Macnair, C. R., Tsai, C. N. & Brown, E. D. Creative targeting of the Gram-negative outer membrane in antibiotic discovery. Ann. N.Y. Acad. Sci. 1459, 69–85 (2020).
Jenkins, R. J. & Dotson, G. D. Dual targeting antibacterial peptide inhibitor of early lipid A biosynthesis. ACS Chem. Biol. 7, 1170–1177 (2012).
García-Quintanilla, M. et al. Inhibition of LpxC Increases antibiotic susceptibility in Acinetobacter baumannii. Antimicrob. Agents Chemother. 60, 5076–5079 (2016).
Vinuesa, V., Cruces, R., Nonnoi, F. & McConnell, M. J. Inhibition of LpxC increases the activity of iron chelators and gallium nitrate in multidrug-resistant Acinetobacter baumannii. Antibiotics 10, 609 (2011).
Imai, Y. et al. A new antibiotic selectively kills Gram-negative pathogens. Nature 576, 459–464 (2019).
Seyfert, C. E. et al. Darobactins exhibiting superior antibiotic activity by cryo-em structure-guided biosynthetic engineering. Angew. Chem. (Int. Ed. Engl.). 62, e202214094 (2023).
Bohnert, J. A. & Kern, W. V. Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob. Agents Chemother. 49, 849–852 (2015).
Yoshida, K.-I. et al. MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 15, 7087–7097 (2007).
Otręebska-Machaj, E. et al. Efflux pump blockers in Gram-negative bacteria: the new generation of hydantoin based-modulators to improve antibiotic activity. Front. Microbiol. 7, 622 (2016).
Benedetto Tiz, D., Kikelj, D. & Zidar, N. Overcoming problems of poor drug penetration into bacteria: challenges and strategies for medicinal chemists. Expert Opin. Drug Discov. 13, 497–507 (2018).
AlMatar, M., Albarri, O., Makky, E. A. & Köksal, F. Efflux pump inhibitors: new updates. Pharmacol. Rep. 73, 1–16 (2021).
Odds, F. C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52, 1 (2003).
Zavascki, A. P. & Nation, R. L. Nephrotoxicity of polymyxins: is there any difference between colistimethate and polymyxin B? Antimicrob. Agents Chemother. 61, e02319–16 (2017).
Olaitan, A. O., Morand, S. & Rolain, J. M. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front. Microbiol. 5, 643 (2014).
French, S. et al. Potentiation of antibiotics against Gram-negative bacteria by polymyxin B analogue SPR741 from unique perturbation of the outer membrane. ACS Infect. Dis. 6, 1405–1412 (2020).
MacNair, C. R. et al. Overcoming mcr-1 mediated colistin resistance with colistin in combination with other antibiotics. Nat. Commun. 9, 458 (2018).
Tyrrell, J. M., Aboklaish, A. F., Walsh, T. R., Vaara, T. & Vaara, M. The polymyxin derivative NAB739 is synergistic with several antibiotics against polymyxin-resistant strains of Escherichia coli, Klebsiella pneumoniae and Acinetobacter baumannii. Peptides 112, 149–153 (2019).
Klobucar, K. et al. Genetic and chemical screening reveals targets and compounds to potentiate gram-positive antibiotics against gram-negative bacteria. ACS Infect. Dis. 8, 2187–2197 (2022).
Moon, S. H. et al. Novel linear lipopeptide paenipeptins with potential for eradicating biofilms and sensitizing Gram-negative bacteria to rifampicin and clarithromycin. J. Med. Chem. 60, 9630–9640 (2017).
Domalaon, R. et al. Dilipid ultrashort cationic lipopeptides as adjuvants for chloramphenicol and other conventional antibiotics against Gram-negative bacteria. Amino Acids 51, 383–393 (2019).
Schweizer, L., Ramirez, D. & Schweizer, F. Effects of lysine N-ζ-methylation in ultrashort tetrabasic lipopeptides (UTBLPs) on the potentiation of rifampicin, novobiocin, and niclosamide in Gram-negative bacteria. Antibiotics 11, 335 (2022).
Domalaon, R., Idowu, T., Zhanel, G. G. & Schweizer, F. Antibiotic hybrids: the next generation of agents and adjuvants against Gram-negative pathogens? Clin Microbiol. Rev. 31, e00077–17 (2018).
Gorityala, B. K. et al. Hybrid antibiotic overcomes resistance in P. aeruginosa by enhancing outer membrane penetration and reducing efflux. J. Med. Chem. 59, 8441–8455 (2016).
Gorityala, B. K. et al. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew. Chem. (Int. Ed. in Engl.) 55, 555–559 (2016).
Lyu, Y. et al. Amphiphilic tobramycin-lysine conjugates sensitize multidrug resistant Gram-negative bacteria to rifampicin and minocycline. J. Med. Chem. 60, 3684–3702 (2017).
Ghosh, C. et al. Small molecular antibacterial peptoid mimics: the simpler the better! J. Med. Chem. 57, 1428–1436 (2014).
Idowu, T., Ammeter, D., Rossong, H., Zhanel, G. G. & Schweizer, F. Homodimeric tobramycin adjuvant repurposes novobiocin as an effective antibacterial agent against Gram-negative bacteria. J. Med. Chem. 62, 9103–9115 (2019).
Yang, X., Domalaon, R., Lyu, Y., Zhanel, G. G. & Schweizer, F. Tobramycin-linked efflux pump inhibitor conjugates synergize fluoroquinolones, rifampicin and fosfomycin against multidrug-resistant Pseudomonas aeruginosa. J. Clin. Med. 7, 158 (2018).
Berry, L., Domalaon, R., Brizuela, M., Zhanel, G. G. & Schweizer, F. Polybasic peptide-levofloxacin conjugates potentiate fluoroquinolones and other classes of antibiotics against multidrug-resistant Gram-negative bacteria. MedChemComm 10, 517–527 (2019).
Meiers, J., Rox, K. & Titz, A. Lectin-targeted prodrugs activated by Pseudomonas aeruginosa for self-destructive antibiotic release. J Med. Chem. 65, 13988–14014 (2022).
Meiers, J. et al. Directing drugs to bugs: antibiotic-carbohydrate conjugates targeting biofilm-associated lectins of Pseudomonas aeruginosa. J. Med. Chem. 63, 11707–11724 (2020).
O’Callaghan, C. H., Sykes, R. B. & Staniforth, S. E. A new cephalosporin with a dual mode of action. Antimicrob. Agents Chemother. 10, 245–248 (1976).
Mobashery, S. & Johnston, M. Inactivation of alanine racemase by beta-chloro-L-alanine released enzymatically from amino acid and peptide C10-esters of deacetylcephalothin. Biochemistry. 26, 5878–5884 (1987).
Mobashery, S., Lerner, S. A. & Johnston, M. Conscripting .beta.-lactamase for use in drug delivery. Synthesis and biological activity of a cephalosporin C10-ester of an antibiotic dipeptide. J. Am. Chem. Soc. 108, 1685–1686 (1986).
Li, Qing et al. NB2001, a novel antibacterial agent with broad-spectrum activity and enhanced potency against beta-lactamase-producing strains. Antimicrob. Agents Chemother. 46, 1262–1268 (2002).
Jones, R. N., Barry, A. L. & Thornsberry, C. Antimicrobial activity of Ro 23-9424, a novel ester-linked codrug of fleroxacin and desacetylcefotaxime. Antimicrob. Agents Chemother. 33, 944–950 (1989).
Russell, A. D. Effect of magnesium ions and ethylenediamine tetra-acetic acid on the activity of vancomycin against Escherichia coli and Staphylococcus aureus. J. Appl. Bacteriol. 30, 395–401 (1967).
Ayres, H. M., Furr, J. R. & Russell, A. D. Effect of permeabilizers on antibiotic sensitivity of Pseudomonas aeruginosa. Lett. Appl. Microbiol. 28, 13–16 (1999).
Hemaiswarya, S. & Doble, M. Ã. Synergistic interaction of eugenol with antibiotics against Gram-negative bacteria. Phytomedicine. 16, 997–1005 (2009).
Faraldo-Gómez, J. D. & Sansom, M. S. P. Acquisition of siderophores in Gram-negative bacteria. Nat. Rev. Mol. Cell Biol. 4, 105–116 (2003).
Mislin, G. L. A. & Schalk, I. J. Siderophore-dependent iron uptake systems as gates for antibiotic Trojan horse strategies against Pseudomonas aeruginosa. Metall. Integr. Biometal Sci. 6, 408–420 (2014).
de Carvalho, C. C. C. R. & Fernandes, P. Siderophores as “Trojan Horses”: tackling multidrug resistance? Front. Microbiol. 5, 290 (2014).
Al Shaer, D., Al Musaimi, O., de la Torre, B. G. & Albericio, F. Hydroxamate siderophores: natural occurrence, chemical synthesis, iron binding affinity and use as Trojan horses against pathogens. Eur. J. Med. Chem. 208, 112791 (2020).
Ji, C., Miller, P. A. & Miller, M. J. Iron transport-mediated drug delivery: practical syntheses and in vitro antibacterial studies of tris-catecholate siderophore-aminopenicillin conjugates reveals selectively potent antipseudomonal activity. J. Am. Chem. Soc. 134, 9898–9901 (2012).
Zhanel, G. G. et al. Cefiderocol: a siderophore cephalosporin with activity against carbapenem-resistant and multidrug-resistant Gram-Negative Bacilli. Drugs 79, 271–289 (2019).
Pinkert, L. et al. Antibiotic conjugates with an artificial MECAM-based siderophore are potent agents against Gram-Positive and Gram-Negative bacterial pathogens. J. Med. Chem. 64, 15440–15460 (2021).
Karakonstantis, S., Rousaki, M. & Kritsotakis, E. I. Cefiderocol: systematic review of mechanisms of resistance, heteroresistance and in vivo emergence of resistance. Antibiotics 11, 723 (2022).
Jousset, A. B. et al. Rapid selection of a cefiderocol-resistant Escherichia coli producing NDM-5 associated with a single amino acid substitution in the CirA siderophore receptor. J. Antimicrob. Chemother. 78, 1125–1127 (2023).
Wang, Q. et al. occurrence of high levels of cefiderocol resistance in carbapenem-resistant Escherichia coli before its approval in China: a report from China CRE-Network. Microbiol. Spectr. 10, e0267021 (2022).
Cascales, E. et al. Colicin biology. Microbiol. Mol. Biol. Rev. 71, 158–229 (2007).
Biswas, K., Upadhayay, S., Rapsang, G. F. & Joshi, S. R. Antibacterial and synergistic activity against β-lactamase-producing nosocomial bacteria by bacteriocin of LAB isolated from lesser known traditionally fermented products of India. HAYATI J. Biosci. 24, 87–95 (2017).
Ghequire, M. G. K., Swings, T., Michiels, J., Buchanan, S. K. & De Mot, R. Hitting with a BAM: selective killing by lectin-like bacteriocins. MBio. 9, e02138–17 (2018).
Gradisteanu Pircalabioru, G. et al. Bacteriocins in the era of antibiotic resistance: Rising to the challenge. Pharmaceutics 13, 196 (2021).
Malanovic, N., Ön, A., Pabst, G., Zellner, A. & Lohner, K. Octenidine: Novel insights into the detailed killing mechanism of Gram-negative bacteria at a cellular and molecular level. Int. J. Antimicrob. Agents 56, 106146 (2020).
Perez Sirkin, Y. A., Tagliazucchi, M. & Szleifer, I. Transport in nanopores and nanochannels: some fundamental challenges and nature-inspired solutions. Mater. Today Adv. 5, 100047 (2020).
Schirmer, T. & Phale, P. S. Brownian dynamics simulation of ion flow through porin channels. J. Mol. Biol. 294, 1159–1167 (1999).
Wang, J., Prajapati, J. D., Kleinekathöfer, U. & Winterhalter, M. Dynamic interaction of fluoroquinolones with magnesium ions monitored using bacterial outer membrane nanopores. Chem. Sci. 11, 10344–10353 (2020).
Mugabe, C., Halwani, M., Azghani, A. O., Lafrenie, R. M. & Omri, A. Mechanism of enhanced activity of liposome-entrapped aminoglycosides against resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50, 2016–2022 (2006).
Nacucchio, M. C., Bellora, M. J., Sordelli, D. O. & D’Aquino, M. Enhanced liposome-mediated activity of piperacillin against staphylococci. Antimicrob. Agents Chemother. 27, 137–139 (1985).
Ferreira, M. et al. Liposomes as antibiotic delivery systems: a promising nanotechnological strategy against antimicrobial resistance. Molecules 26, 2047 (2021).
Metelkina, O. et al. Targeting extracellular lectins of Pseudomonas aeruginosa with glycomimetic liposomes. J. Mater. Chem. B 10, 537–548 (2022).
Alfei, S., & Schito, A. M. From nanobiotechnology, positively charged biomimetic dendrimers as novel antibacterial agents: a review. Nanomaterials 10 https://doi.org/10.3390/nano10102022 (2020).
Wrońska, N., Majoral, J. P., Appelhans, D., Bryszewska, M. & Lisowska, K. Synergistic effects of anionic/cationic dendrimers and levofloxacin on antibacterial activities. Molecules. 24, 1–11 (2019).
Schito, A. M. & Alfei, S. Antibacterial activity of non-cytotoxic, amino acid-modified polycationic dendrimers against Pseudomonas aeruginosa and other non-fermenting Gram-Negative bacteria. Polymers 12, 1818 (2020).
Pires, J. et al. In vitro activity of the novel antimicrobial peptide dendrimer g3kl against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 59, 7915–7918 (2015).
Siriwardena, T. N. et al. Lipidated peptide dendrimers killing multidrug-resistant bacteria. J. Am. Chem. Soc. 140, 423–432 (2018).
Li, Q. Application of fragment-based drug discovery to versatile targets. Front. Mol. Biosci. 7, 180 (2020).
Moreau, R. J. et al. Fragment-based drug discovery of inhibitors of phosphopantetheine adenylyltransferase from Gram-negative bacteria. J. Med. Chem. 61, 3309–3324 (2018).
Mansbach, R. A. et al. Development of a fragment-based machine learning algorithm for designing hybrid drugs optimized for permeating Gram-negative bacteria. arXiv. 1907.13459 (2019).
Mawal, Y., Critchley, I. A., Riccobene, T. A. & Talley, A. K. Ceftazidime-avibactam for the treatment of complicated urinary tract infections and complicated intra-abdominal infections. Expert Rev. Clin. Pharmacol. 8, 691–707 (2015).
Yaghoubi, S. et al. Tigecycline antibacterial activity, clinical effectiveness, and mechanisms and epidemiology of resistance: narrative review. Eur. J. Clin. Microbiol. Infect. Dis. 41, 1003–1022 (2022).
Patel, T. S., Pogue, J. M., Mills, J. P. & Kaye, K. S. Meropenem-vaborbactam: a new weapon in the war against infections due to resistant Gram-negative bacteria. Future Microbiol. 13, 971–983 (2018).
Kashuba, A. D., Nafziger, A. N., Drusano, G. L. & Bertino, J. S. J. Optimizing aminoglycoside therapy for nosocomial pneumonia caused by Gram-negative bacteria. Antimicrob. Agents Chemother. 43, 623–629 (1999).
Sorbera, M., Chung, E., Ho, C. W. & Marzella, N. Ceftolozane/Tazobactam: a new option in the treatment of complicated Gram-negative infections. J. Formulary Manag. 39, 825–832 (2014).
Harris, P. N. A. et al. Effect of piperacillin-tazobactam vs meropenem on 30-day mortality for patients with E. coli or Klebsiella pneumoniae bloodstream infection and ceftriaxone resistance: a randomized clinical trial. JAMA 320, 984–994 (2018).
Mansour, H., Ouweini, A. E. L., Chahine, E. B. & Karaoui, L. R. Imipenem/cilastatin/relebactam: a new carbapenem β-lactamase inhibitor combination. Am. J. Health-Syst. Pharm. 78, 674–683 (2021).
Giacobbe, D. R. et al. Use of colistin in adult patients: a cross-sectional study. J. Glob. Antimicrob. Resist. 20, 43–49 (2020).
Greenberg, R. N., Reilly, P. M., Weinandt, W. J., Bollinger, M. & Kennedy, D. J. Cefoperazone-sulbactam combination in the treatment of urinary tract infections: efficacy, safety, and effects on coagulation. Clin. Ther. 10, 52–56 (1987).
Alosaimy, S., Abdul-Mutakabbir, J. C., Kebriaei, R., Jorgensen, S. C. J. & Rybak, M. J. Evaluation of eravacycline: a novel fluorocycline. Pharmacotherapy 40, 221–238 (2020).
Wu, J. Y., Srinivas, P. & Pogue, J. M. Cefiderocol: a novel agent for the management of multidrug-resistant Gram-negative organisms. Infect. Dis. Ther. 9, 17–40 (2020).
Goldstein, E. J. et al. Introduction of ertapenem into a hospital formulary: effect on antimicrobial usage and improved in vitro susceptibility of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 5122–5126 (2009).
Zhao, Y. et al. Cooperative membrane damage as a mechanism for pentamidine-antibiotic mutual sensitization. ACS Chem. Biol. 17, 3178–3190 (2022).
Jones, R. N., Huynh, H. K. & Biedenbach, D. J. Activities of doripenem (S-4661) against drug-resistant clinical pathogens. Antimicrob. Agents Chemother. 48, 3136–3140 (2004).
Hilas, O., Ezzo, D. C. & Jodlowski, T. Z. Doripenem (doribax), a new carbapenem antibacterial agent. P & T Peer-Rev. J. Formul. Manag. 33, 134–180 (2008).
Perry, C. M. & Ibbotson, T. Biapenem. Drugs 62, 2221–2234 (2002).
Thakare, R., Singh, S., Dasgupta, A. & Chopra, S. Lascufloxacin hydrochloride to treat bacterial infection. Drugs Today 56, 365–376 (2020).
Hosogaya, N. et al. Evaluation of efficacy and safety of lascufloxacin for nursing and healthcare associated pneumonia: single-arm, open-label clinical trial: a study protocol. Medicine 102, e33092 (2023).
Zhanel, G. G. et al. Lefamulin: a novel oral and intravenous pleuromutilin for the treatment of community-acquired bacterial pneumonia. Drugs 81, 233–256 (2021).
Clark, J. A. & Burgess, D. S. Plazomicin: a new aminoglycoside in the fight against antimicrobial resistance. Ther. Adv. Infect. Dis. 4, 2049936120952604 (2020).
Sutcliffe, J. A., O’Brien, W., Fyfe, C. & Grossman, T. H. Antibacterial activity of eravacycline (TP-434), a novel fluorocycline, against hospital and community pathogens. Antimicrob. Agents Chemother. 57, 5548–5558 (2013).
Basseatti, M., Ginocchio, F. & Mikulska, M. New treatment options against Gram-negative organisms. Crit. Care 15, 215 (2011).
Butler, M. S. et al. Analysis of the clinical pipeline of treatments for drug-resistant bacterial infections: despite progress, more action is needed. Antimicrob. Agents Chemother. 66, e0199121 (2022).
Shi, S. et al. Synergistic effect of the novel β-lactamase inhibitor BLI-489 combined with imipenem or meropenem against diverse carbapenemase-producing carbapenem-resistant Enterobacterales. J. Antimicrob. Chemother. 77, 1301–1305 (2022).
Richter, H. G. et al. Design, synthesis, and evaluation of 2 beta-alkenyl penam sulfone acids as inhibitors of beta-lactamases. J. Med. Chem. 39, 3712–3722 (1996).
Kaur, K., Adediran, S. A., Lan, M. J. K. & Pratt, R. F. Inhibition of beta-lactamases by monocyclic acyl phosph(on)ates. Biochemistry 42, 1529–1536 (2003).
Buynak, J. D., Ghadachanda, V. R., Vogeti, L., Zhang, H. & Chen, H. Synthesis and evaluation of 3-(carboxymethylidene)- and 3-(carboxymethyl)penicillinates as inhibitors of beta-lactamase. J.Organ. Chem. 70, 4510–4513 (2005).
Livermore, D. M., Mushtaq, S. & Warner, M. Activity of BAL30376 (monobactam BAL19764 + BAL29880 + clavulanate) versus Gram-negative bacteria with characterized resistance mechanisms. J. Antimicrob. Chemother. 65, 2382–2395 (2010).
Paukner, S., Hesse, L., Prezelj, A., Solmajer, T. & Urleb, U. In vitro activity of LK-157, a novel tricyclic carbapenem as broad-spectrum {beta}-lactamase inhibitor. Antimicrob. Agents Chemother. 53, 505–511 (2009).
Simpson, I. N. et al. Synthesis and biological activity of AM-112 and related oxapenem analogues. J. Antibiot. 56, 838–847 (2003).
Arixa pharmaceuticals announces acquisition by Pfizer’s hospital business _ business wire. (n.d.). https://www.businesswire.com/news/home/20201022005157/en/Arixa-Pharmaceuticals-Announces-Acquisition-by-Pfizer%E2%80%99s-Hospital-Business.
Sader, H. S., Mendes, R. E., Duncan, L. R., Carvalhaes, C. G. & Castanheria, M. Antimicrobial activity of cefepime/zidebactam (WCK 5222), a β-lactam/β-lactam enhancer combination, against clinical isolates of Gram-negative bacteria collected worldwide (2018-19). J. Antimicrob. Chemother. 77, 2642–2649 (2022).
Petropoulou, D., Siopi, M., Vourli, S. & Pournaras, S. Activity of sulbactam-durlobactam and comparators against a national collection of carbapenem-resistant Acinetobacter baumannii isolates from Greece. Front. Cell. Infect. Microbiol. 11, 814530 (2021).
Hamrick, J. C. et al. VNRX-5133 (Taniborbactam), a broad-spectrum inhibitor of serine- and metallo-β-lactamases, restores activity of cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 64, e01963–19 (2020).
Mallalieu, N. L. et al. Safety and pharmacokinetic characterization of nacubactam, a novel β-lactamase inhibitor, alone and in combination with meropenem, in healthy volunteers. Antimicrob. Agents Chemother. 64, e02229–19 (2020).
O’Donnell, J. et al. Pharmacokinetic/pharmacodynamic determination and preclinical pharmacokinetics of the β-lactamase inhibitor ETX1317 and its orally available prodrug ETX0282. ACS Infect. Dis. 6, 1378–1388 (2020).
Karlowsky, J. A., Hackel, M. A. & Sahm, D. F. In vitro activity of ceftibuten/VNRX-5236 against urinary tract infection isolates of antimicrobial-resistant Enterobacterales. Antimicrob. Agents Chemother. 66, e0130421 (2022).
P1 single and multiple ascending dose (SAD_MAD) Study of IV QPX7728 alone and combined with QPX2014 in NHV. https://clinicaltrials.gov/ct2/show/NCT04380207.
Livermore, D. M. et al. Activities of NXL104 combinations with ceftazidime and aztreonam against carbapenemase-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55, 390–394 (2011).
Yamada, K. et al. In vivo efficacy of biapenem with ME1071, a novel metallo-β-lactamase (MBL) inhibitor, in a murine model mimicking ventilator-associated pneumonia caused by MBL-producing Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 42, 238–243 (2013).
Bruss, J. et al. Single- and multiple-ascending-dose study of the safety, tolerability, and pharmacokinetics of the polymyxin derivative SPR206. Antimicrob. Agents Chemother. 65, e0073921 (2021).
Lepak, A. J., Wang, W. & Andes, D. R. Pharmacodynamic evaluation of MRX-8, a novel polymyxin, in the neutropenic mouse thigh and lung infection models against Gram-negative pathogens. Antimicrob. Agents Chemother. 64, e01517–e01520 (2020).
Huband, M. D. et al. In vitro activity of KBP-7072, a novel third-generation tetracycline, against 531 recent geographically diverse and molecularly characterized Acinetobacter baumannii species complex isolates. Antimicrob. Agents Chemother. 64, e02375–19 (2020).
Huband, M. D. et al. Activity of the novel aminomethylcycline KBP-7072 and comparators against 1,057 geographically diverse recent clinical isolates from the SENTRY surveillance program, 2019. Antimicrob. Agents Chemother. 66, e0139721 (2022).
Hennessen, F. et al. Overcomes resistance mechanisms exerted on tetracyclines and natural chelocardin. Antibiotics 9, 619 (2020).
Becker, K. et al. Efficacy of EBL-1003 (apramycin) against Acinetobacter baumannii lung infections in mice. Clin. Microbiol. Infect. 27, 1315–1321 (2021).
Richter, M. F. & Hergenrother, P. J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N.Y. Acad. Sci. 1435, 18–38 (2019).
Acred, P., Brown, D. M., Turner, D. H. & Wilson, M. J. Pharmacology and chemotherapy of ampicillin-a new broad-spectrum penicillin. Brit. J. Pharmacol. Chemother. 18, 356–369 (1962).
Savage, V. J. et al. Biological profiling of novel tricyclic inhibitors of bacterial DNA gyrase and topoisomerase IV. J. Antimicrob. Chemother. 71, 1905–1913 (2016).
Richter, M. F. et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017).
Parkinso, N. E. I. et al. Deoxynybomycins inhibit mutant DNA gyrase and rescue mice infected with fluoroquinolone-resistant bacteria. Nat. Commun. 6, 6947 (2015).
Hiramatsu, K. et al. Curing bacteria of antibiotic resistance: reverse antibiotics, a novel class of antibiotics in nature. Int. J. Antimicrob. Agents 39, 478–485 (2012).
Parker, E. N. et al. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nature Microbiol. 5, 67–75 (2020).
Howe, J. A. et al. Selective small-molecule inhibition of an RNA structural element. Nature 526, 672–67 (2015).
Tari, L. W. et al. Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PloS One 8, e84409 (2013).
Onishi, H. R. et al. Antibacterial agents that inhibit lipid A biosynthesis. Science 274, 980–982 (1996).
A study to assess the safety, tolerability, and pharmacokinetics of ACHN-975 in healthy volunteers. https://clinicaltrials.gov/ct2/show/NCT01597947?term=NCT01597947&draw=2&rank=1.
A multiple dose study to assess the safety, tolerability, and pharmacokinetics of ACHN-975 in healthy volunteers https://clinicaltrials.gov/ct2/show/NCT01870245?term=NCT01870245&draw=1&rank=1.
Birck, M. R., Holler, T. P. & Woodard, R. W. Identification of a slow tight-binding inhibitor of 3-deoxy-D-manno-octulosonic acid 8-phosphate synthase. J. Am. Chem. Soc. 122, 9334–9335 (2000).
Novartis licenses three novel anti-infective programs to Boston Pharmaceuticals https://www.novartis.com/news/media-releases/novartis-licenses-three-novel-anti-infective-programs-boston-pharmaceuticals (2018).
Lehman, K. M. & Grabowicz, M. Countering Gram-negative antibiotic resistance: recent progress in disrupting the outer membrane with novel therapeutics. Antibiotics 8, 163 (2019).
Xiao, Y., Gerth, K., Müller, R. & Wall, D. Myxobacterium-produced antibiotic TA (myxovirescin) inhibits type II signal peptidase. Antimicrob. Agents Chemother. 56, 2014–2021 (2012).
Vogeley, L. et al. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 351, 876–880 (2016).
Ho, H. et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 557, 196–201 (2018).
Mandler, M. D. et al. Novobiocin enhances polymyxin activity by stimulating lipopolysaccharide transport. J. Am. Chem. Soc. 140, 6749–6753 (2018).
Martin-Loeches, I., Dale, G. E. & Torres, A. Murepavadin: a new antibiotic class in the pipeline. Expert Rev. Anti-Infect. Ther. 16, 259–268 (2018).
Srinivas, N. et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327, 1010–1013 (2010).
Vetterli, S. U. et al. Thanatin targets the intermembrane protein complex required for lipopolysaccharide transport in Escherichia coli. Sci. Adv. 4, eaau2634 (2018).
Sherman, D. J., Okuda, S., Denny, W. A. & Kahne, D. Validation of inhibitors of an ABC transporter required to transport lipopolysaccharide to the cell surface in Escherichia coli. Bioorgan. Med. Chem. 21, 4846–4851 (2013).
Nickerson, N. N. et al. A Novel Inhibitor of the LolCDE ABC Transporter Essential for Lipoprotein Trafficking in Gram-Negative Bacteria. Antimicrob. Agents Chemother. 62, e02151–17 (2018).
Pathania, R. et al. Chemical genomics in Escherichia coli identifies an inhibitor of bacterial lipoprotein targeting. Nat. Chem. Biol. 5, 849–856 (2009).
Barker, C. A. et al. Degradation of MAC13243 and studies of the interaction of resulting thiourea compounds with the lipoprotein targeting chaperone LolA. Bioorgan. Med. Chem. Lett. 23, 2426–2431 (2013).
Buss, J. A. et al. Pathway-directed screen for inhibitors of the bacterial cell elongation machinery. Antimicrob. Agents Chemother. 63, e01530–18 (2019).
Hart, E. M. et al. A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier. Proc. Natl Acad. Sci. USA 116, 21748–21757 (2019).
Hagan, C. L., Wzorek, J. S. & Kahne, D. Inhibition of the β-barrel assembly machine by a peptide that binds BamD. Proc. Natl Acad. Sci. USA 112, 2011–2016 (2015).
Sader, H. S. et al. Antimicrobial activity of POL7306 tested against clinical isolates of Gram-negative bacteria collected worldwide. J. Antimicrob. Chemother. 75, 1518–1524 (2020).
Tomasek, D. et al. Structure of a nascent membrane protein as it folds on the BAM complex. Nature 583, 473–478 (2020).
Acknowledgements
We acknowledge funding through an Indo-German collaborative grant to S.V./S.C. (ICMR, AMR/INDO/GER/218/2019-ECD-II) and A.T. (BMBF: SRI12742-H2L-MDRAB. grant no. 01DQ20003), respectively. D.S. thanks UGC while R.M. and R.B. thank DST-INSPIRE.
Author information
Authors and Affiliations
Contributions
D.S., R.M., R.B., M.C., J.M., A.T., S.V. and S.C. conceptualized, wrote, and reviewed/edited the final manuscript. D.S., R.M. and R.B. contributed equally to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saxena, D., Maitra, R., Bormon, R. et al. Tackling the outer membrane: facilitating compound entry into Gram-negative bacterial pathogens. npj Antimicrob Resist 1, 17 (2023). https://doi.org/10.1038/s44259-023-00016-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44259-023-00016-1
