
Abstract
Human induced pluripotent stem cell (hiPSC) to cardiomyocyte (CM) differentiation has reshaped approaches to studying cardiac development and disease. In this study, we employed a genome-wide CRISPR screen in a hiPSC to CM differentiation system and reveal here that BRD4, a member of the bromodomain and extraterminal (BET) family, regulates CM differentiation. Chemical inhibition of BET proteins in mouse embryonic stem cell (mESC)-derived or hiPSC-derived cardiac progenitor cells (CPCs) results in decreased CM differentiation and persistence of cells expressing progenitor markers. In vivo, BRD4 deletion in second heart field (SHF) CPCs results in embryonic or early postnatal lethality, with mutants demonstrating myocardial hypoplasia and an increase in CPCs. Single-cell transcriptomics identified a subpopulation of SHF CPCs that is sensitive to BRD4 loss and associated with attenuated CM lineage-specific gene programs. These results highlight a previously unrecognized role for BRD4 in CM fate determination during development and a heterogenous requirement for BRD4 among SHF CPCs.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
All data supporting the findings of this study are included in the main article and associated files, and Source Data have been provided with this manuscript. All transcriptomic and epigenomic data are available in the Gene Expression Omnibus database under accession number GSE184922, which is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184922.
References
Van Der Linde, D. et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 58, 2241–2247 (2011).
Jin, S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601 (2017).
Pierpont, M. E. et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation 138, e653–e711 (2018).
de Soysa, T. Y. et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 572, 120–124 (2019).
Homsy, J. et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015).
Olley, G. et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange–like syndrome. Nat. Genet. 50, 329–332 (2018).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Sapp, V. et al. Genome-wide CRISPR/Cas9 screening in human iPS derived cardiomyocytes uncovers novel mediators of doxorubicin cardiotoxicity. Sci Rep. 11, 13866 (2021).
Chen, Y. et al. Caveolin-1 plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells into cardiomyocytes. Cardiology 136, 40–48 (2017).
Guo, X. et al. Cardiomyocyte differentiation of mesenchymal stem cells from bone marrow: new regulators and its implications. Stem Cell Res. Ther. 9, 44 (2018).
Singh, A. M. et al. Chibby, an antagonist of the Wnt/β-catenin pathway, facilitates cardiomyocyte differentiation of murine embryonic stem cells. Circulation 115, 617–626 (2007).
Kim, M.-S. et al. Activin-A and Bmp4 levels modulate cell type specification during CHIR-induced cardiomyogenesis. PLoS ONE 10, e0118670 (2015).
Morton, S. U., Quiat, D., Seidman, J. G. & Seidman, C. E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 19, 26–42 (2021).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Krumm, N. et al. Excess of rare, inherited truncating mutations in autism. Nat. Genet. 47, 582–588 (2015).
Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12, 599–606 (1998).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Han, P., Hang, C. T., Yang, J. & Chang, C.-P. Chromatin remodeling in cardiovascular development and physiology. Circ. Res. 108, 378–396 (2011).
Padmanabhan, A. & Haldar, S. M. Drugging transcription in heart failure. J. Physiol. 598, 3005–3014 (2020).
Lovén, J. et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334 (2013).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Lee, J.-E. et al. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 8, 2217 (2017).
Zengerle, M., Chan, K.-H. & Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015).
Yang, L. et al. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development 133, 1575–1585 (2006).
Padmanabhan, A. et al. BRD4 (bromodomain-containing protein 4) interacts with GATA4 (GATA binding protein 4) to govern mitochondrial homeostasis in adult cardiomyocytes. Circulation 142, 2338–2355 (2020).
Linares-Saldana, R. et al. BRD4 orchestrates genome folding to promote neural crest differentiation. Nat. Genet. 53, 1480–1492 (2021).
Jain, R. et al. Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science 348, aaa6071 (2015).
Cai, C.-L. et al. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 5, 877–889 (2003).
Verzi, M. P., McCulley, D. J., De Val, S., Dodou, E. & Black, B. L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 287, 134–145 (2005).
Kwon, C. et al. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc. Natl Acad. Sci. USA 104, 10894–10899 (2007).
Gessert, S. & Kühl, M. The multiple phases and faces of Wnt signaling during cardiac differentiation and development. Circ. Res. 107, 186–199 (2010).
Cohen, E. D. et al. Wnt/β-catenin signaling promotes expansion of Isl-1-positive cardiac progenitor cells through regulation of FGF signaling. J. Clin. Invest. 117, 1794–1804 (2007).
Jho, E.-H. et al. Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183 (2002).
Zhao, M.-T. et al. Cell type-specific chromatin signatures underline regulatory DNA elements in human induced pluripotent stem cells and somatic cells. Circ. Res. 121, 1237–1250 (2017).
Devine, W. P., Wythe, J. D., George, M., Koshiba-Takeuchi, K. & Bruneau, B. G. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife 3, e03848 (2014).
Lescroart, F. et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 359, 1177–1181 (2018).
Nomaru, H. et al. Single cell multi-omic analysis identifies a Tbx1-dependent multilineage primed population in the murine cardiopharyngeal mesoderm. Nat. Commun. 12, 6645 (2021).
Chen, Y.-H., Ishii, M., Sun, J., Sucov, H. M. & Maxson, R. E. Jr. Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev. Biol. 308, 421–437 (2007).
Song, L. et al. Lrp6-mediated canonical Wnt signaling is required for lip formation and fusion. Development 136, 3161–3171 (2009).
Rao, J. et al. Stepwise clearance of repressive roadblocks drives cardiac induction in human ESCs. Cell Stem Cell 18, 341–353 (2016).
Gonzalez-Teran, B. et al. Integration of protein interactome networks with congenital heart disease variants reveals candidate disease genes. Preprint at bioRxiv https://doi.org/10.1101/2021.01.05.423837 (2021).
Dawson, M. A. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152 (2017).
Dey, A. et al. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 38, e100293 (2019).
Chatfield, K. C. et al. Congenital heart disease in Cornelia de Lange syndrome: phenotype and genotype analysis. Am. J. Med. Genet. A 158A, 2499–2505 (2012).
Li, F.-F. et al. Characterization of transcriptional repressor gene MSX1 variations for possible associations with congenital heart diseases. PLoS ONE 10, e0142666 (2015).
Lahm, H. et al. Congenital heart disease risk loci identified by genome-wide association study in European patients. J. Clin. Invest. 131, e141837 (2021).
Ivanovitch, K. et al. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. 19, e3001200 (2021).
Boogerd, C. J. J., Moorman, A. F. M. & Barnett, P. Expression of muscle segment homeobox genes in the developing myocardium. Anat. Rec. 293, 998–1001 (2010).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007).
Yang, W. et al. Generation of iPSCs as a pooled culture using magnetic activated cell sorting of newly reprogrammed cells. PLoS ONE 10, e0134995 (2015).
Kreitzer, F. R. et al. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cells 2, 119–131 (2013).
Nagy A., Gertsenstein, M., Vintersten, K. & Behringer, R. (eds) Manipulating the Mouse Embryo: A Laboratory Manual 3rd edn (Cold Spring Harbor Laboratory Press, 2003).
Poleshko, A. et al. Genome-nuclear lamina interactions regulate cardiac stem cell lineage restriction. Cell 171, 573–587 (2017).
Christoforou, N. et al. Mouse ES cell–derived cardiac precursor cells are multipotent and facilitate identification of novel cardiac genes. J. Clin. Invest. 118, 894–903 (2008).
Kattman, S. J. et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8, 228–240 (2011).
Shah, P. P. et al. Pathogenic LMNA variants disrupt cardiac lamina-chromatin interactions and de-repress alternative fate genes. Cell Stem Cell 28, 938–954 (2021).
Lian, X. et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl Acad. Sci. USA 109, E1848–E1857 (2012).
Luo, B. et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl Acad. Sci. USA 105, 20380–20385 (2008).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z. & Zhang, B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205 (2019).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Robinson, M., McCarthy, D. J., Chen, Y. & Smyth, G. K. Package ‘edger’. Preprint at http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.367.3149&rep=rep1&type=pdf (2012).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Gonzalez-Teran, B. et al. Transcription factor protein interactomes reveal genetic determinants in heart disease. Cell 185, 794–814 (2022).
Cheshire, C. et al. nf-core/cutandrun: nf-core/cutandrun v3.2 Tin Albatross. https://doi.org/10.5281/zenodo.8305872 (2023).
Acknowledgements
The authors thank the Srivastava and Jain laboratories for critical discussions and feedback and K. Ozato (National Institutes of Health) for experimental reagents. We are grateful to V. Vedantham (University of California, San Francisco (UCSF)), S. Hota (Gladstone Institutes), I. Kathiriya (UCSF) and M. Costa (Gladstone Institutes) for thoughtful commentary on the manuscript. We thank B. Taylor (Gladstone Institutes) and K. Claiborn (Gladstone Institutes) for editorial assistance as well as A. Silva (Ana Silva Illustrations) and G. Maki (Gladstone Institutes) for assistance with illustrations. We thank the University of Pennsylvania iPSC core for technical assistance. This work was supported by the NIH (R35 HL166663 and R01 HL139783; F31 HL147416 to R.L-S.; K08HL157700 to A.P.; P01 HL146366, R01 HL057181, R01 HL127240 and R01 HL015100 to D.S.), the Burroughs Wellcome Foundation Career Award for Medical Scientists (R.J.), the Allen Foundation (R.J.), the American Heart Association (R.J. and S.M.), the National Science Foundation (15-48571 to R.J.), the Swiss National Science Foundation (P400PM_186704 and P2LAP3_178056 to M.A.), the Japan Society for the Promotion of Science Overseas Research Fellowship (T.N.), the Sarnoff Cardiovascular Research Foundation (A.P.), the Michael Antonov Charitable Foundation (A.P.), the Frank A. Campini Foundation (A.P.), the Tobacco‐Related Disease Research Program (578649 to A.P.), the A. P. Giannini Foundation (P0527061 to A.P.), the Roddenberry Foundation (D.S.), the L. K. Whittier Foundation (D.S.), Dario and Irina Sattui (D.S.) and the Younger Family Fund (D.S.).
Author information
Authors and Affiliations
Contributions
A.P., R.J. and D.S. conceived and designed the study. A.P., Y.d.S., V.S., P.P.S., Q.W., L.L., C.Y.L., N.S., A. Poleshko, N.B., R.L.-S., T.N. and L.Y. performed all the experiments. A.P., Y.d.S., V.S., A. Pelonero, S.U.M., M.J., R.Y., A.K., L.Y. and R.J. analyzed the data. M.A., S.M.H. and D.S. assisted with data interpretation. A.P. and R.J. wrote the manuscript. D.S. and R.J. supervised the project. All authors edited and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.S. is a scientific co-founder, shareholder and director of Tenaya Therapeutics. S.M.H. is an executive, officer and shareholder of Amgen and is a scientific co-founder and shareholder of Tenaya Therapeutics. M.J. is founder, shareholder and executive of Sapient Bioanalytics, LLC. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Bernice Morrow, John Hinson, Michael A. Burke, Lijie Shi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Integrating a CM differentiation screen and CHD variants.
(a) Top 1000 genes ranked by enrichment or depletion. (b, c) Categorization of top 200 genes enriched (b) or depleted (c) in cardiac myocytes compared to undifferentiated hiPSCs by biological groups. (d, e) Gene ontology (GO) analysis of top enriched (d) or depleted (e) hits (see Methods for details). (f-g) Venn diagrams demonstrating the number of CHD (f) or Non-CHD (g) probands with predicted damaging DNVs in hits identified in our screen as enriching hiPSC:CM differentiation or depleting hiPSC:CM differentiation. Arrows depict Venn diagrams representing the number of probands from each cohort with predicted damaging DNVs identified in our screen that have mutations in known dominant CHD genes or where these candidate CHD genes may potentially be causative. (h) Venn diagram demonstrating the number of CHD probands with (purple) or without (brown) damaging DNVs in hits identified in our screen highlighting no enrichment for extracardiac anomalies (p = 0.43) or neurodevelopmental delay (NDD; p = 0.23) and a slight enrichment for conotruncal CHD (p = 0.04). (i) TNNT2+ cells quantified by flow cytometry at day 10 of hiPSC to CM differentiation in WTC11 cells treated with 100 nM JQ1 starting at day 6 (n = 3 biologically independent samples). (j) MZ3 treatment (500 nM for 0, 3.5, 7, and 16 hours) effectively degrades BRD4 in SV20 hiPSCs as assessed by immunoblot analysis for BRD4 with β-actin expression as a loading control. In all graphs, error bars represent ±1 SEM. * represents p = 0.0271 (two-tailed unpaired t test).
Extended Data Fig. 2 Inhibition or deletion of BRD4 inhibits mESC cardiac differentiation.
(a) Expression of Myh6, Nkx2-5, and Tnnt2 at day 7 of mESCs cardiac differentiation treated with JQ1 (100 nM) starting day 5 of differentiation (n = 3 biologically independent samples). (b) TNNT2+ cells quantified by flow cytometry at d9 of mESC differentiation in CMV-CreERT2;Brd4flox/flox cells treated with 4-hydroxytamoxifen (TAM), JQ1 (250 nM) or MZ3 (500 nM) starting at day 5 (n = 3 biologically independent samples; FACS gating strategy in Supplementary Fig. 1). (c-d) Immunofluorescence of TNNT2 at day 9 of mESC to CM differentiation in wild type cells treated with JQ1 (100 nM) or vehicle starting at day 5. (e-f) Immunofluorescence of BRD4 in vehicle (ethanol, e-e′′) and TAM (f-f′′) treated undifferentiated mESCs. (g) Volcano plot showing RNA-seq from CMV-CreERT2;Brd4flox/flox (TAM vs. vehicle [VEH]) mESCs and gene ontology analysis of downregulated and upregulated genes (see Methods for details). (h) Heatmap showing expression of select transcription factor and muscle structural protein genes from RNA-seq in CMV-CreERT2;Brd4flox/flox (TAM vs. VEH) mESC-derived cardiac tissues (day 10; TAM or VEH added at day 5). In all graphs, error bars represent ±1 SEM. For a, all comparisons are made relative to 0 nM compound for each gene; * represents p < 0.0493, ** represents p < 0.0085 (two-tailed unpaired t test). For b, all comparisons are made relative to VEH for each condition; * represents p < 0.0188 (two-tailed unpaired t test). Scale Bars = 100 µm (c, d, e, e′, e′′, f, f′, f′′).
Extended Data Fig. 3 Loss of BRD4 in Isl1Cre-SHF progenitors in vivo results in myocardial hypoplasia.
(a) Lineage tracing of Isl1Cre/+;Brd4flox/+ and Isl1Cre/+;Brd4flox/flox with R26mTmG/+ allele. Immunohistochemistry of ISL1-derived cells (GFP) and TNNT2 or BRD4 (red) in heart and quantitative assessment of right ventricular myocardial wall thickness in indicated genotypes at E14 (5 sections from n = 2 control embryos and 11 sections from n = 4 mutant embryos). (b) Hematoxylin and eosin staining of a section through outflow tract and RV of Isl1Cre/+;Brd4flox/+ and Isl1Cre/+;Brd4flox/flox embryos at E12.5 and quantitative assessment of right ventricular myocardial wall thickness in indicated genotypes (10 sections from n = 2 control embryos and 11 sections from n = 2 mutant embryos). (c) Quantification of percentage phospho-histone H3-, cleaved caspase 3-, and TUNEL-positive cells in the RV of E12.5 and E14.5 embryos of indicated genotypes (n = 2 biologically independent samples per genotype at E12.5; n = 3-4 biologically independent samples per genotype at E14.5). (d) Hematoxylin and eosin staining of a section through outflow tract and RV of Isl1Cre/+;Brd4flox/+ and Isl1Cre/+;Brd4flox/flox embryos at E10 and quantitative assessment of right ventricular myocardial wall thickness in indicated genotypes (9 sections from n = 3 control embryos and 8 sections from n = 3 mutant embryos). (e) Quantification of percentage phospho-histone H3- and TUNEL-positive cells in the RV of E10 control (Isl1Cre/+;Brd4flox/+ or Brd4flox/flox) and Isl1Cre/+;Brd4flox/flox embryos (n = 4 biologically independent samples per condition). (f) BRD4 and TNNT2 immunohistochemistry along with lineage tracing with R26mTmG/+ allele in Isl1Cre/+;Brd4flox/+ and Isl1Cre/+;Brd4flox/flox E9.5 embryos at level of right ventricle. Error bars represent ±1 SEM. All comparisons are made as indicated; * represents p = 0.0091, ** represents p = 0.0021, **** represents p < 0.0001 (two-tailed unpaired t test). RV, right ventricle; LV, left ventricle; OT, outflow tract. Scale Bars = 100 µm (a, b, d, f).
Extended Data Fig. 4 Loss of BRD4 in Mef2c-AHF-Cre-SHF CPCs in vivo results in right ventricular thinning.
(a) Hematoxylin and eosin staining of a section through outflow tract and RV of Mef2c-AHF-Cre;Brd4flox/+ and Mef2c-AHF-Cre;Brd4flox/flox embryos at E13.5 and quantitative assessment of right ventricular myocardial wall thickness in indicated genotypes (19 sections from n = 3 control embryos and 18 sections from n = 3 mutant embryos). (b) Quantification of percentage phospho-histone H3- and cleaved caspase 3-positive cells in the RV of E13.5 Mef2c-AHF-Cre;Brd4flox/+ and Mef2c-AHF-Cre;Brd4flox/flox embryos (n = 3-4 biologically independent samples per genotype). (c) Hematoxylin and eosin staining of a section through outflow tract and RV of Mef2c-AHF-Cre;Brd4flox/+ and Mef2c-AHF-Cre;Brd4flox/flox embryos at E10.5 and quantitative assessment of right ventricular myocardial wall thickness in indicated genotypes (6 sections from n = 3 control embryos and 6 sections from n = 3 mutant embryos). (d) Quantification of percentage phospho-histone H3- and cleaved caspase 3-positive cells in the RV of E10.5 Mef2c-AHF-Cre;Brd4flox/+ and Mef2c-AHF-Cre;Brd4flox/flox embryos (n = 3 biologically independent samples per genotype). Error bars represent ±1 SEM. All comparisons are made as indicated; * represents p = 0.0489, ** represents p = 0.0146, **** represents p < 0.0001 (two-tailed unpaired t test). RV, right ventricle; LV, left ventricle. Scale Bars = 100 µm (a, c).
Extended Data Fig. 5 Wnt signaling is dysregulated upon BRD4 depletion in CPCs.
(a) Image of Brd4flox/flox embryo appearing in Fig. 2i with region microdissected for bulk RNA-seq highlighted in red. (b) Principal component analysis of RNA-seq from Isl1Cre/+;Brd4flox/+ (blue) and Isl1Cre/+;Brd4flox/flox (red) E9.5 embryos. (c) Volcano plots of E9.5 Isl1Cre/+;Brd4flox/+ vs. Isl1Cre/+;Brd4flox/flox embryonic hearts (same data appearing in Fig. 4) with a subset of cardiac and Wnt-related genes annotated (see Methods for details). Brd4flox/flox(d, f) and Isl1Cre/+;Brd4flox/flox (e, g) E9.5 embryos at level of right ventricle stained with ISL1 (red, d-e), BRD4 (green, d-e), or AXIN2 (green, f-g). Note the expansion of ISL1- (arrow heads) and AXIN2- (dotted line) expressing cells into the RV from distal outflow tract in mutant embryos. (h-m) ISL1 and AXIN2 Immunohistochemistry of E10.5 Mef2c-AHF-Cre;Brd4flox/+ (h,k) and Mef2c-AHF-Cre;Brd4flox/flox (i,j,l,m) embryos at the level of outflow tract. (h-j, ISL1; k-m, AXIN2). Note the expansion of ISL1- (arrow heads) and AXIN2- (dotted line) expressing cells into the right ventricle from distal outflow tract in mutant embryos. (n-o) AXIN2 RNAscope of Brd4flox/flox (n,n′) and Isl1Cre/+;Brd4flox/flox (o,o′) E9.5 embryos at the level of the right ventricle (n′ and o′ are magnified images of n and o, respectively). (p-q) ISL1 representative immunofluorescence at day 8 of mESC-derived cardiac cultures treated with vehicle (DMSO; VEH) (p) or JQ1 (500 nM) (q) starting at day 5. (r) Isl1 expression in day 8-9 mESC-derived cardiac cultures treated with increasing doses of JQ1 (0–500 nM; JQ1 added at day 5; n = 3 n = 3 biologically independent samples per dose). (s-t) AXIN2 RNAscope of Mef2c-AHF-Cre;Brd4flox/+ (s,s′) and Mef2c-AHF-Cre;Brd4flox/flox (t,t′) E10.5 embryos at level of right ventricle (s′ and t′ are magnified images of s and t, respectively). For r, all comparisons are made relative to 0 nM compound. *** represents p = 0.0007 (two-tailed unpaired t test). RV, right ventricle; OT, outflow tract. Scale Bar = 250 µm (a), 50 µm (d-m, n, o, s, t), 100 µm (p, q, n′, o′, s′, t′).
Extended Data Fig. 6 BRD4 regulates Wnt signaling during CM differentiation.
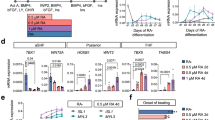
(a) Attenuating Wnt signaling at the CPC stage (day 5) in mESC to CM differentiation by doubling the normal concentration of the small molecule Wnt inhibitor XAV939 concomitant with Brd4 genetic deletion by 4-hydroxytamoxifen treatment (TAM) in CMV-CreERT2;Brd4flox/flox mESCs partially normalizes expression of CPC markers and Msx1/2 by qRT-PCR (n = 3-4 biologically independent samples per genotype). (b-e) Attenuating Wnt signaling at the CPC stage (day 5) in mESC to CM differentiation by doubling the normal concentration of the small molecule Wnt inhibitor XAV939 concomitant with Brd4 genetic deletion by 4-hydroxytamoxifen treatment (TAM) in CMV-CreERT2;Brd4flox/flox mESCs partially normalizes TNNT2 staining by immunofluorescence at day 9 of CM differentiation (for e, n = 3 biologically independent samples per condition). (f, g) Attenuation of Wnt signaling at the CPC stage (day 6) in hiPSC to CM differentiation by addition of the small molecule Wnt inhibitor IWP4 (5 μM for low dose and 10 μM high dose) concomitant with BET inhibition using JQ1 (25 nM for low dose and 50 nM for high dose) increases the number of TNNT2+ cells as assessed by flow cytometry (n = 3 biologically independent samples per condition; gating strategy in Supplementary Fig. 2). Error bars represent ±1 SEM. For a,e,f,g all comparisons are made with p values as indicated (two-way ANOVA with Tukey’s multiple comparisons test).
Extended Data Fig. 7 Generation of BRD4FLAG/FLAG hiPSC line and BRD4 occupancy in cardiac progenitor cells.
(a) Targeting strategy to introduce 3XFLAG epitope tag into the N-terminus of the endogenous BRD4 locus. (b) Karyotyping results of BRD4FLAG/FLAG hiPSC line. (c) Western blot analysis of protein lysates collected from BRD4FLAG/FLAG hiPSCs using FLAG antibody demonstrates expression of 3XFLAG-tagged BRD4 isoforms that are degraded upon addition of the PROTAC BET degrader dBET650. (d) Pearson correlation matrices demonstrating high reproducibility between replicate CUT&RUN datasets. (e-g) Track view of indicated loci showing CUT&RUN factor occupancy (FLAG, BRD4, H3K4Me3) or H3K27Ac ChIP-seq enrichment in hiPSC-derived CPCs.
Extended Data Fig. 8 Iterative filtering steps for selection of cells analyzed in scRNA-seq.
(a-c) UMAP plots of all cells (n = 23,592) collected from the microdissected heart and surrounding pharyngeal mesoderm (n = 2 embryos per genotype) (a), labeled by sample identity (b), and number of features (c). (d) Feature plots for expression of example marker genes used to define cell types (for example, Hbb-y for blood cells; Dlx2, Dlx5, and Twist1 for neural crest cells; Lhx2 and Foxc2 for branchiomeric muscle progenitors; Epcam for endoderm). (e-g) UMAP plots demonstrate expression of the Cre transgene (e) occurs in clusters marked by high Mef2c (f) and Isl1 (g) expression, consistent with Cre driven in Mef2c-expressing second heart field cells. (h) Clusters of Cre expressing cells detected at E9.5 in our scRNA-seq dataset selected for further analysis (n = 4,640). (i) Feature plot for Cre expression in a UMAP of cells from (h) following normalization and reclustering. (h’-i’) Cluster 0 cells highlighted in red on UMAP plots from h and i.
Extended Data Fig. 9 Pathway analysis of differentially expressed genes by cluster.
Pathway analysis of differentially expressed genes between mutant (Mef2c-AHF-Cre;Brd4flox/flox) and control (Brd4flox/flox) embryos by cluster for each cellular population identified in our scRNA-seq studies (see Methods for details).
Extended Data Fig. 10 BRD4 loss increases MSX1- and MSX2-positive cells in vivo.
Mef2c-AHF-Cre;Brd4flox/+ (a) and Mef2c-AHF-Cre;Brd4flox/flox (b, c) E10.5 embryos at level of right ventricle stained with MSX1/2 (yellow); inset shows area indicated by arrowheads. RNAscope in Brd4flox/flox (d, e), Isl1Cre/+;Brd4flox/flox (f, g), Mef2c-AHF-Cre;Brd4flox/+ (h, i), and Mef2c-AHF-Cre;Brd4flox/flox (j, k) E9.5-10.5 embryos at level of right ventricle for MSX1 (d,f,h,j) or MSX2 (e,g,i,k). Regions in yellow boxes in d-k are shown in higher magnification in d′-k′. RV, right ventricle; OT, outflow tract. Scale Bars = 50 µm (a-c, d-k), 165 µm (d′, e′, f′, g′), 125 µm (h′, j′), 200 µm (i′, k′).
Supplementary information
Supplementary information
Supplementary Tables 1 and 2, Supplementary Datasets 1 and 2 and Supplementary Figs. 1 and 2
Source data
Source Data Fig. 1d
Statistical Source Data
Source Data Extended Data Fig. 1d
Statistical Source Data
Source Data Extended Data Fig. 2
Statistical Source Data
Source Data Extended Data Fig. 3
Statistical Source Data
Source Data Extended Data Fig. 4
Statistical Source Data
Source Data Extended Data Fig. 5r
Statistical Source Data
Source Data Extended Data Fig. 6
Statistical Source Data
Source Data Extended Data Fig. 1j
Unprocessed western blots
Source Data Extended Data Fig. 7c
Unprocessed western blots
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Padmanabhan, A., de Soysa, T.Y., Pelonero, A. et al. A genome-wide CRISPR screen identifies BRD4 as a regulator of cardiomyocyte differentiation. Nat Cardiovasc Res 3, 317–331 (2024). https://doi.org/10.1038/s44161-024-00431-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44161-024-00431-1
