
Abstract
Atherosclerotic cardiovascular disease is a major cause of disability and death worldwide. Most therapeutic approaches target traditional risk factors but ignore the fundamental role of the immune system. This is a huge unmet need. Recent evidence indicates that reducing inflammation may limit cardiovascular events. However, the concomitant increase in the risk of life-threatening infections is a major drawback. In this context, targeting adaptive immunity could constitute a highly effective and safer approach. In this Review, we address the why and how of the immuno-cardiovascular unit, in health and in atherosclerotic disease. We review and discuss fundamental mechanisms that ensure immune tolerance to cardiovascular tissue, and examine how their disruption promotes disease progression. We identify promising strategies to manipulate the adaptive immune system for patient benefit, including novel biologics and RNA-based vaccination strategies. Finally, we advocate for establishing a molecular classification of atherosclerosis as an important milestone in our quest to radically change the understanding and treatment of atherosclerotic disease.
Similar content being viewed by others
Main
Dyslipidemia and elevated low-density lipoprotein (LDL) cholesterol levels are causally involved in the chronic inflammatory response of the vascular wall1. LDL oxidation in the intima and the sterile vascular inflammation associated with it engage adaptive immune responses that profoundly modulate atherosclerosis, from lesion initiation and progression to the occurrence of clinical events2 (Box 1). Accumulating evidence also implicates adaptive immunity in the response to ischemic heart injury3 as a modulator of both post-ischemic cardiac remodeling and atherosclerosis progression4.
Recent studies have provided proof of concept that targeting inflammation may limit the occurrence of cardiovascular (CV) events5,6. Here, we consider the intimate and complex interactions of the immune system with the heart and the vasculature in health and atherosclerotic disease, with a particular focus on adaptive immune mechanisms and their selective manipulation for patient benefit.
Evidence for adaptive immunity in ischemic heart disease
The association of adaptive immune responses with human atherosclerotic cardiovascular disease (ACVD) dates back to the 1980s and the discovery that abundant cells in atherosclerotic lesions expressed HLA-DR in association with activated T lymphocytes7 and immunoglobulins. This finding was further supported by the local presence of oxidized LDL (oxLDL), along with antibodies specific to it and T cells that recognize it8,9. Subsequently, an overwhelming body of evidence has implicated the adaptive immune system in the development and progression of atherosclerosis2,10. More recent studies harnessing the latest developments in mass cytometry and single-cell omics technologies have provided further support for a role of adaptive immunity in ACVD11.
There is ample epidemiological evidence supporting a role for adaptive immunity in ACVD, as people with autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis and other rheumatic diseases, are at substantially increased risk of atherosclerosis and CV events. The levels of autoantibodies often associate with a higher CV risk, suggesting a detrimental role. Moreover, induction of autoimmunity in mice enhances atherosclerosis12, indicating a causal role.
Genome-wide association studies (GWASs)13 also highlight the causal role of inflammation in promoting ACVD14, and genome-wide network-driven systems-biology approaches have identified genes involved in B cell activation as potential key drivers13. Intriguingly, HLA genotypes show only weak association with ACVD15. However, this should not be interpreted as evidence for the lack of a causal role of autoimmune responses. It is likely that people with autoimmune disease are severely under-represented in ACVD case–control cohorts. Furthermore, as discussed in this Review, atherosclerosis does not manifest as a classic autoimmune disease with early breakdown of tolerance to self antigens, but instead involves autoimmune responses to altered or modified self antigens or stress-induced neo-self antigens. Breakdown of tolerance (Box 2), if it occurs, is a secondary and relatively late event in atherosclerosis. More generally, inflammatory responses are highly modulated by environmental factors and are likely to be more important in rapid ACVD acceleration than in linear slow progression, making it difficult to capture their causal role using GWASs16. Moreover, most GWASs have included participants with ACVD with a relatively high lifetime CV-risk burden, particularly a relatively high cholesterol load. Preclinical data indicate that T cells and B cells are much stronger drivers of atherogenesis in mice with low cholesterol levels than in mice with high cholesterol levels17. Thus, it is likely that in cohorts in which high cholesterol levels are not a strong driver, more immune-related variants could be identified. The growing understanding of a fragmentation of large disease entities into smaller ones with different molecular profiles suggests that individual patho-mechanistic differences may also be responsible for the residual CV risk despite optimal treatment of classic CV risk factors. In this context, a deeper understanding of the interplay of adaptive immunity with the major CV risk factors on an individual level is needed. Finally, the substantially increased risk of cardiovascular inflammation, atherosclerosis progression and CV events after immune-checkpoint inhibitor therapy18 is an important reminder of the major role of adaptive immunity in cardiovascular homeostasis.
Why and how adaptive immunity is involved in atherosclerosis
The immuno-vascular unit in health
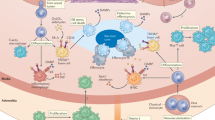
Studies employing two-photon intravital microscopy, mass cytometry and single-cell transcriptomics have revealed the presence of several types of resident immune cells of lymphoid origin (T cells, B cells, natural killer cells and innate lymphoid cells) and myeloid origin (macrophages, dendritic cells (DCs) and mast cells) in the intima and adventitia of normal arteries11 (Fig. 1). The adventitia is also a site of neuro–immune–vascular cell interactions19. The presence of such a vascular-associated lymphoid tissue (VALT) in healthy arteries20 suggests that it may play a role in immune surveillance, and recent work indicates that it is also involved in maintaining vascular homeostasis and adaptation to hemodynamic cues.

VALT is present in normal, healthy arteries and comprises a variety of immune cells that serve essential roles in immune surveillance and vascular homeostasis. Crawling monocytes (Gr1dim in mice, CD14dimCD16+ in humans) sense nucleic acids, and those released after EC damage scavenge debris and microvesicles (MVs) and ensure EC protection. Intimal macrophages (MACAIR) accumulate at sites of intimal thickening (upper right, normal artery) and are the first myeloid cells that scavenge oxLDL in the initial stages of atherosclerosis (middle left, atherosclerotic plaque). This further drives vascular inflammation and leads to the recruitment of inflammatory monocytes (Gr1hi in mice; CD14++CD16− in humans) that give rise to a variety of macrophage subsets. Adventitial macrophages, particularly Lyve1+ macrophages (upper right, normal artery), regulate arterial stiffness, in part through the production of MMP9 and FXIIIA. Their specific contribution to atherosclerosis is currently unknown. The adventitia and perivascular adipose tissue form a permissive niche for the accumulation of atheroprotective ILC2 and innate-like B cells, organized in FALCs (upper middle, normal artery). ILC2-derived type 2 cytokines (for example, IL-5 and IL-13) promote B1 cell activation and the production of natural IgM antibodies, and contribute to an anti-inflammatory macrophage phenotype. Most DCs and T cells of normal arteries are found in the adventitia. T cells of normal arteries are likely to be enriched for a regulatory phenotype146, instructed by DCs presenting vascular-associated antigens (in draining lymphoid organs) and maintaining peripheral tolerance. The inflammatory milieu of developing lesions promotes DC maturation, which favors the generation of TH cells, Teff cells and TEM cells (mostly in draining lymphoid organs) and their recruitment into both the intima and adventitia of atherosclerotic arteries (bottom left). Sustained stimulation of autoreactive Treg cells may downregulate Foxp3, promoting their conversion into TH1 cells and Teff cells. Some of the Treg cells and Teff cells acquire a resident memory phenotype. TH1 cells, Teff cells and TEM cells predominate in advanced lesions (bottom right), and activated cytotoxic CD8+ T cells may acquire an exhausted phenotype. GC activation in secondary lymphoid organs leads to the production of affinity-matured class-switched (IgG, IgE) antibodies, which accumulate in lesions. Medial VSMCs of advanced lesions produce CXCL13 and CCL21 and may adopt features of lymphoid tissue organizer-like cells, leading to ATLO formation (bottom right). ATLOs are conducive to the generation of Treg cells, which serve a counter-regulatory, atheroprotective role. The diseased adventitia also establishes neuro–immune vascular interactions, which affect lesion progression.
Monocytes, macrophages and DCs
Circulating Gr1dim (mouse) and CD14dimCD16+ (human) monocytes that crawl on endothelial cells (ECs) and patrol the vessels for the presence of danger- or damage-associated molecular patterns (DAMPs) fulfill an immune-surveillance program21 (Fig. 1). The number of patrolling monocytes increases in hypercholesterolemia, and their absence is generally associated with increased atherosclerosis22, suggesting a protective role.
Resident arterial macrophages originate from embryonic CX3CR1+ precursors and, postnatally, from definitive hematopoiesis, and are maintained through local proliferation. They accumulate preferentially at branch points characterized by intimal thickening, suggesting a role for hemodynamic stresses. Their location shapes their resident phenotype and leads to distinct gene signatures for intimal versus adventitial macrophages23. The latter maintain arterial tone and regulate arterial remodeling in response to blood flow, in part through MMP9- and FXIIIA-dependent regulation of collagen accumulation24,25 (Fig. 1). Intimal macrophages maintain a non-thrombogenic intravascular state26 and are the earliest foam cells that accumulate in plaques23. In mice, intimal macrophages have previously been confounded with vascular-associated DCs, but it is unclear whether this holds true for human vascular DCs. Resting immature adventitial DCs have been described in healthy human arteries27 and may have a role in maintaining immune tolerance (Fig. 1). Spatial single-cell sequencing will be useful for characterizing the various macrophage/DC subtypes and examining whether different subtypes populate distinct vascular beds and perform specific functions.
T cells, B cells and fat-associated lymphoid clusters
In human arteries, T cells occur mainly at sites of intimal thickening, whereas adventitial T cells are found both in human arteries and in mouse arteries and are thought to home constitutively to the adventitia, in part through an L-selectin-dependent mechanism28 (Fig. 1). Most of these T cells are αβ CD4+ cells, along with a few γδ CD4+ cells, as well as CD8+ T cells. Some express CD25, suggesting that they are in an activated state. However, their characterization remains incomplete.
B cells are largely absent from the intima but are present in the adventitia of both healthy and atherosclerotic arteries (Fig. 1). The fact that adoptively transferred splenic B cells also home to the adventitia of recipient non-atherosclerotic aortas28 further underscores the artery wall as a physiological niche for B cells (Fig. 1). Arterial homing of B cells also depends in part on L-selectin28. A substantial proportion of T cells and B cells is located in the perivascular adipose tissue (PVAT) and may be organized within fat-associated lymphoid clusters (FALCs) around stromal cells. Unlike the formation of secondary lymphoid organs (SLOs), FALC formation is independent of lymphoid tissue-inducer cells and the lymphotoxin-β receptor pathway, but requires TNFR signaling and the presence of commensal flora. PVAT and FALCs contain a substantial amount of innate B1 cells that secrete natural immunoglobulin M (IgM)29, including IgM with specificity for oxidation-specific epitopes (OSEs)30. Part of this B1 cell activation may be driven by innate lymphoid cells type 2 (ILC2)31, which are major producers of atheroprotective interleukin 5 (IL-5) and IL-13 (refs. 31,32,33) (Box 2 and Fig. 1). The fundamental triggers that initiate immune-cell accumulation in normal arteries remain poorly understood (Box 3).
Medial immunoprivilege revisited
The medial layer of the artery wall is highly protected from invasion by effector immune cells. Several mechanisms of medial immunoprivilege have been proposed34. Passive mechanisms include the presence of elastic laminae and the avascular nature of the media. However, these mechanisms are not entirely consistent with the sustained medial immunoprivilege of advanced plaques, even in the presence of extensive neoangiogenesis, abnormal lymphangiogenesis and disrupted elastic laminae. Proposed active mechanisms include the relatively low expression of major histocompatibility complex (MHC) and costimulatory molecules, the high expression of coinhibitory molecules and the production of immunosuppressive factors, mainly IDO1 and transforming growth factor-β (TGF-β), by medial vascular smooth muscle cells (VSMCs). However, VSMCs express high levels of MHC molecules and inflammatory mediators during atherosclerosis, and medial immunoprivilege is preserved in mice with IDO1 deficiency or disruption of TGF-β signaling. Thus, medial immunoprivilege remains a mystery.
Peripheral lymphoid stromal cells, particularly follicular dendritic cells (FDCs), have a critical role in peripheral tolerance. Their expression of self antigen induces both deletional T cell tolerance and non-deletional self-tolerance that maintains antigen-specific regulatory T cells (Treg cells)35. Self-antigen expression by FDCs also controls the elimination of self-reactive B cells, particularly those generated during secondary diversification in the germinal center (GC)36. Interestingly, these stromal cells share the same perivascular precursor with VSMCs and thus express many similar transcripts, both during differentiation and at the mature stage37. Here, we propose that the common ontogenic origin of VSMCs and lymphoid stromal cells is the primary mechanism that ensures medial immunoprivilege. A breakdown in lymphoid stromal-cell-dependent tolerance or a substantial alteration or loss of VSMCs would be required to disrupt the immunoprivilege of the arterial media.
The immuno-vascular unit in atherosclerotic disease
Given the immunoprivilege of the media, it is unlikely that pathogenic adaptive immune cells are activated in response to arterial media components. Intimal and adventitial T cell accumulation most likely indicates an adaptive response toward altered components of, or present in, these arterial layers. The oligoclonal T cell repertoire, as well as the limited sets of hypermutated variable heavy-chain regions and the inverted κ/λ light-chain ratio of B cells found in atherosclerotic lesions38,39, suggest an active expansion of antigen-specific T cell and B cell clones. The autoimmune responses most studied in this context are those directed toward lipoprotein components trapped in the artery wall2,40 and toward proteins expressed by stressed ECs20.
Adaptive T cell immunity in atherosclerosis
How do activated T cells accumulate in inflamed arteries? Naive CD4+ T cell priming in the artery wall41 is probably a marginal phenomenon in the early stages of atherosclerosis. Adventitial resident DCs are more likely than intimal macrophages to migrate to the draining lymph nodes (LNs) for T cell activation. Therefore, activated T cells in early lesions are mostly likely recruited as pre-activated (in draining LNs and SLOs) effector memory T cells (TEM cells) or Treg cells, transmigrating through activated (and potentially antigen-presenting) ECs. Chemokine–chemokine receptor pairs involved in TEM cell recruitment to the inflamed vasculature (for example, CX3CL1–CX3CR1) are likely to be different from those involved in Treg cell recruitment (for example, CCL5–CCR5 (ref. 42) and CCL1–CCR8 (ref. 43)) (Fig. 1). Once in the artery wall, some of these T cells may adopt a resident memory phenotype with low levels of S1PR1 and high expression of CD103 and CD69 (ref. 41). The latter binds oxLDL, limiting type 17 helper T cell (TH17 cell) differentiation while favoring atheroprotective Treg cells44.
LDL particles can also reach SLOs systemically and, in the absence of systemic inflammation (that is, early atherosclerosis), apolipoprotein B (ApoB) peptides are likely to be presented in a tolerogenic manner, generating antigen-specific Treg cells rather than effector T cells (Teff cells) (Fig. 1). This would explain why most ApoB-specific T cells display a regulatory phenotype in the absence of overt atherosclerosis45. It also provides a plausible explanation for the initial surge in Treg cells rather than Teff cells in response to hypercholesterolemia in mice46. This scenario suggests that the first (ApoB-specific) T cells that accumulate in lesions are likely to be Treg cells, which may subsequently switch to helper and effector-memory phenotypes with the increased burden of local and systemic inflammation47. It is also plausible that ApoB-specific Treg cells are instructed in the liver, where ApoB is produced48.
Different scenarios may arise with different antigens, either sequentially or concomitantly. Among these scenarios is the example of autoimmunity to heat-shock proteins (HSPs), whereby pre-existing immunity to ubiquitous HSP60 (a highly immunogenic microbial antigen) could lead to cross-reactivity with autologous HSP60 (overexpressed on stressed ECs), triggering intimal inflammation20. Wick et al. have shown that T cells from early human atherosclerotic lesions are interferon-γ (IFNγ)-producing memory effector CD4+ cells and respond to HSP60 and HSP60-derived peptides in vitro49. This recall response was associated with the generation of pathogenic anti-HSP60 antibodies. However, the early lesions assessed in that study were from individuals with ‘inflammatory storm’ conditions. The latter may have induced an activated T cell phenotype, and therefore it remains unclear whether HSP-specific Teff cells are truly predominant in early atherosclerosis. Furthermore, most of these studies have focussed on intimal T cells, and the antigens that trigger Teff cell responses in the adventitia remain to be identified.
A shift from regulatory to pathogenic T cell immunity
Depletion of CD4+ T cells accelerates early atherosclerosis in mice50, supporting the concept that early CD4+ T cell responses are mostly regulatory and atheroprotective. So, what shifts T cell responses from regulatory to effector? There is not a particular type of antigen-presenting cell (APC) that has been shown to selectively promote a Teff cell phenotype versus a Treg cell phenotype in atherosclerosis51,52. Thus, the activation state of the APC and the microenvironment of antigen presentation are paramount. Sustained local inflammation certainly has a role, activating macrophages and DCs and changing their metabolism and signaling pathways to upregulate costimulatory molecules (for example, CD40) and proinflammatory cytokines (for example, IL-1, IL-6) and chemokines (for example, CCL17) while downregulating coinhibitory molecules (for example, PDL-1) and anti-inflammatory mediators (for example, IL-10 and TGF-β)53,54,55,56. In addition, changes in T cell metabolism, driven by hypercholesterolemia, alterations in lipid-synthesis pathways, hypoxia and other microenvironmental cues, may substantially alter the Treg cells’ suppressive phenotype57,58. Interestingly, sustained activation of Treg cells by self antigens in an inflammatory environment affects the demethylated region in the Foxp3 locus, downregulating Foxp3 expression and destabilizing Treg cells59. These mechanisms could account for the increased plasticity of Treg cells60 and their progressive decline during disease progression, as well as the enrichment for TH1- and TH17-related phenotypes among ApoB-specific T cells of people with atherosclerosis compared with their abundance in those free of ACVD47 (Fig. 1).
The role of the various CD4+ T cell subsets has been extensively reviewed2. Foxp3+ Treg cells and IL-10-producing type 1 regulatory T cells are protective, whereas TH1 cells promote disease development. The roles of TH2 and TH17 cells appear to be contextual and most likely depend on the relative abundance of pro- and anti-atherogenic cytokines produced by these T cells2,61. It is important to note that a clear distinction between the various TH cell archetypes may not be relevant in vivo, and it is highly likely that most plaque T cells display a continuum of helper phenotypes. Indeed, recent single-cell studies in advanced human plaques described an enrichment for chronically activated TEM cells displaying a resident phenotype that are enriched for interferon pathways, but TH cell-specific transcription factors could not be linked to specific clusters62,63. There is genetic and experimental evidence for a significant role of IL-1, IL-6 and IL-18 pathways in ACVD. Intriguingly, IL-1 and IL-6 signaling pathways were more likely to be activated in CD4+ T cells of asymptomatic carotid plaques than in those of symptomatic carotid plaques in humans62. The therapeutic implications of this finding remain unclear.
Many other T cell subtypes have been suggested to modulate atherosclerosis, but these subsets, and in particular their antigens, remain poorly characterized. The preferential distribution of γδ CD4+ T cells, natural killer T cells64 and mucosal-associated invariant T cells in the gut and liver suggests that these subsets are more likely to have a role in atherosclerosis in the context of metabolic syndrome, through their immune and metabolic activities65. The role of CD8+ T cells will require further attention. These cells are enriched in advanced human lesions and display several phenotypes62,63, including central memory, effector memory and cytotoxic phenotypes, with exhausted CD8+ T cells being enriched in symptomatic carotid plaques relative to their abundance in asymptomatic carotid plaques62. CD8+ T cells are suggested to promote atherosclerosis through target-cell lysis or induction of monopoiesis66. Their antigen specificity is still largely unknown, although some CD8+ T cells recognize ApoB100-derived peptides67. A Qa-1-restricted regulatory CD8+ T cell subset has been shown to limit atherosclerosis in mice through the control of the follicular helper T cell (TFH cell) GC B cell response (discussed below)68. Its relevance to human atherosclerosis remains unexplored.
Adaptive B cell immunity in atherosclerosis
Follicular B2 cells represent the major part of B cells in secondary lymphoid organs and the circulating blood. B2 cells are involved mainly in T cell-dependent responses, as they differentiate into GC B cells undergoing class-switching and affinity maturation aided by TFH cells. Analyses of atherosclerotic arteries support a relative expansion of B2-like over B1-like cells, and the accumulation of activated plasmablasts11,39. In advanced disease, some VSMCs and other local stromal cells express high levels of CXCL13 and CCL21 and adopt features of lymphoid tissue organizer-like cells. B cells in the adventitia become organized in artery tertiary lymphoid organs (ATLOs)69 that represent a site of recruitment to the artery wall and contain different subsets of B cells that participate in GC responses70 (Fig. 1). Despite the documented presence of B cells in the vascular wall, their direct local contribution to atherogenesis is largely unknown. ATLOs are atheroprotective in mice69. However, it is unclear whether this is due to protective local B cell functions or to the predominance of induced immunosuppressive Treg cells in ATLOs69. Moreover, effector functions associated with antibody production do not require local B cells, as specific antibodies produced in the bone marrow or the spleen can reach plaques via the circulation71,72.
The activation of B2 cells is responsible for the production of class-switched antibodies against modified lipoproteins and other self components. The help of TFH cells, found mainly in B cell follicles, is required for the generation of memory B cells and long-lived plasma cells that secrete affinity-matured class-switched antibodies. TFH cell deficiency appears to limit atherogenesis58. However, this requires further exploration, because TFH cells located outside the GC may be required for both switched and unswitched extrafollicular responses, with potentially different effects on atherosclerosis. Of note, activation of marginal zone (MZ) B cells impairs TFH cells via upregulation of PD-L1, thereby limiting proatherogenic T cell-dependent responses73.
B2 cells have antibody-mediated atherogenic functions
Follicular B cells are proatherogenic; their preferential depletion with CD20-targeted antibodies or by genetic targeting of the BAFFR pathway reduces atherosclerosis74,75,76. Because follicular B cells participate in GC reactions, the likely explanation for their proatherogenic properties is that they contribute to the generation of pathogenic high-affinity IgG antibodies. Hyperlipidemia in mice promotes a GC response with increased serum levels of IgG2b and IgG2c, as well as OSE-specific IgG antibodies and several classical autoantibodies77. Of note, Apoe–/– mice that are unable to make IgG antibodies due to a deficiency of the activation-induced deaminase, which is critical for class-switch recombination, displayed increased total and malondialdehyde (MDA)-specific IgM levels and developed less atherosclerosis78. Genetic ablation of GC-derived antibodies greatly reduced lesion size in Apoe–/– mice71,79. Moreover, administration of an IgG preparation from plasma of atherosclerotic Apoe–/– mice aggravated disease71. These data indicate that GC-derived antibodies have the capacity to promote disease. However, few pathogenic IgG antibody responses have been described, such as anti-HSP60/65 IgG antibodies that bind HSP60 in stressed ECs and promote atherogenesis by antibody-dependent cell-mediated cytotoxicity20, and IgG autoantibodies to the 78-kDa glucose-regulated protein, which has been described as another endothelium-derived autoantigen80, that trigger EC activation. Thus, there is still a great need to identify the antigens of GC-derived pathogenic antibodies (Box 4).
Antibody-mediated atheroprotective functions of B2 cells
Beyond the atheroprotective properties of B1 cell- and MZ B cell-derived OSE-specific IgM antibodies10,81 (Box 1), GC-derived IgG antibodies may also have protective functions50, such as promoting the proliferation of smooth muscle cells and plaque stability79. Passive immunization of Ldlr–/– mice with a recently characterized autoantigen, ALDH4A1, resulted in reduced lipid levels and decreased atherosclerosis82. T cell-dependent IgG antibodies with reactivity to ApoB were also shown to protect T cell receptor (TCR)-transgenic mice from atherosclerosis by promoting LDL clearance and reducing serum cholesterol levels83. Moreover, despite in vitro studies demonstrating proinflammatory effects of anti-oxLDL IgG in macrophages84 and several epidemiological studies demonstrating an association of anti-oxLDL IgG titers with ACVD85, direct experimental evidence of a proatherogenic role in vivo is missing. In contrast, numerous immunization studies in mice and rabbits, which initially aimed at establishing a proatherogenic role for anti-oxLDL immune responses, demonstrated a robust induction of T cell-dependent IgG antibodies and reduced atherosclerosis10. For example, immunization with models of OSE generally induced a TH2-biased response, for which IgG1 is a surrogate marker32,86. Although other mechanisms may be at play, there is also direct evidence for an atheroprotective effect of OSE-specific IgG antibodies. Indeed, infusion of monoclonal human IgG1 against the OSEs MDA-LDL and phosphocholine (PC) protect mice from atherosclerosis87,88. Owing to the human origin of the infused IgG antibodies, the contribution of the IgG subclass cannot be deducted, but it is probably important for the protective capacity of these IgG antibodies. Blockade of the many proinflammatory properties of oxLDL may be part of the protective effect, as mice expressing a single-chain version of the anti-PC IgM, E06, developed reduced inflammation and decreased atherosclerosis89.
Fc receptor-mediated effects of antibodies
To fully understand the contribution of class-switched antibody responses in atherosclerosis, not only antigen specificity but also the different effector functions of antibody subclasses need to be considered81. Key effector functions that also need to be explored in context of the bound antigens are neutralization and clearance of antigens (for example, of oxLDL and self antigens), antibody-dependent cell-mediated cytotoxicity (for example, of HSP60-expressing ECs), complement activation and inflammation, as well as cellular activation. For the latter, the role of Fcγ-receptors has been studied in a series of settings, and in general indicated an overall pathogenic role for activating Fcγ receptors and protective effects of the inhibitory FcγRIIB10,90. However, these effects are complicated by sexual dimorphism, disease-stage-specific effects, cell-type-specific differences and potential non-canonical roles of these receptors10,90. Future studies need to further dissect the involvement of Fcγ receptors in mediating IgG effector functions independent of immunomodulatory roles and in an antigen-dependent manner. A clear proatherogenic role has been found for IgE antibodies, which trigger mast-cell and macrophage activation through FcεR in the artery wall91,92. Whether these effects involve the recognition of specific antigens remains to be shown.
Other effector functions of B cells
B cells also contribute to atherosclerosis through cytokine secretion. For example, expression of TNF by B cells may be in part responsible for the proatherogenic effects of B2 cells93, and innate response activator B cells are GM-CSF-secreting B cells that promote atherogenesis through GM-CSF-mediated DC activation94. The role of B cell-derived IL-10 is still unclear and requires further studies10. For all these subsets, it remains uncertain whether their effects are mediated as simple bystander activities or are associated with certain B cell receptor identities.
Finally, recent studies have indicated that the peripheral nervous system is involved in an artery–brain cross-talk that affects local neuro–immune–vascular interactions in the adventitia, with important consequences for atherosclerosis progression in mice19. The implication of this circuit for human atherosclerosis is currently unknown.
Adaptive immunity in response to cardiac ischemic injury
Resident DCs and T cells in normal hearts
The normal heart harbors several populations of resident macrophages and several populations of DCs, including monocyte-derived DCs, cDC1 and cDC2 (ref. 95). The precise spatial distribution of DC subsets is poorly characterized. However, these DCs, namely IRF8-dependent cDC1, drive the generation of autoreactive Treg cells that are specific for cardiac self antigens, particularly α-myosin heavy chain96. This is an important homeostatic mechanism that ensures peripheral non-deletional tolerance to a self antigen that escapes central negative selection97 (Fig. 2a). T cells in normal hearts are enriched for selective TRBV-J rearrangements and TRBV gene segments compared with those in peripheral blood, indicating that they may recognize and tolerize against tissue-specific antigens98. Single-cell TCR sequencing will facilitate the characterization of these T cells and their antigen specificities. It will also be important to dissect the different steps involved in maintaining peripheral tolerance to cardiac-specific antigens. A well-developed and functional cardiac lymphatic network could facilitate this immune-surveillance task. Biomechanically induced IL-33 may promote and/or maintain immunosuppressive and reparative ST2+ cardiac Treg cells99. Furthermore, the heart produces hepatocyte growth factor, which is known to induce immune-regulatory DCs and instructs T cell cardiotropism by binding to its receptor c-Met in draining LNs, inducing the release of β-chemokines and promoting T cell recruitment through CCR5 (ref. 100). This mechanism may be required for cardiac Treg cell accumulation at steady state, and is consistent with the role of CCR5 in promoting Treg cell recruitment to the heart after myocardial infarction (MI), suppressing inflammation and reducing adverse remodeling101 (Fig. 2a).

The heart and its adjacent pericardial and adipose tissue harbor several types of immune cells. a, At steady state, conventional DCs (cDCs), particularly cDC1, home to draining LNs and instruct the generation of tissue-specific (for example, myosin heavy chain-α) Treg cells146, which establish peripheral tolerance. Heart-derived HGF binds to DCs and induces an immune-regulatory phenotype. HGF also promotes chemokine production (for example, of CCL5) by c-MET-expressing T cells, which drives CCR5-dependent recruitment and cardiac Treg cell accumulation. Treg cell immunosuppressive function is further promoted through a local (cardiac) paracrine/autocrine adenosinergic loop, involving CD39/CD73 and adenosine A2A receptor (A2AR), or by engaging ST2 signaling with heart-derived IL-33. Pericardial innate lymphoid cells type 2 (ILC2)-derived IL-5 and IL-13 activate innate-like B cells to produce natural IgM antibodies and anti-inflammatory IL-10, which contribute to an anti-inflammatory and reparative macrophage phenotype. b, During ischemic injury, DCs acquire an inflammatory phenotype that provides instructions for the development of TH cells, Teff cells and TEM cells, as well as CD8+ cytotoxic T cells. Enhanced production of GM-CSF by pericardial innate-like B cells further promotes DC activation and the generation of TH cells and Teff cells. These T cells upregulate CX3CR1 and CXCR3, and their recruitment into the ischemic heart is facilitated by heart-derived CX3CL1 and CXCL10. GC reactions develop in the spleen and draining LNs, leading to the production of (heart-specific) autoantibodies, with potential detrimental consequences. Splenic MZ B cells are activated by the release of DAMPs, including in the form of mitochondria-containing microvesicles, which activate Toll-like receptors (TLRs), leading to CCL7 production. The latter promotes inflammatory monocyte mobilization from the bone marrow, enhancing the accumulation of inflammatory macrophages within the ischemic myocardium. In most cases, the ischemic heart is able to maintain the presence of a sufficient number of Treg cells to prevent the occurrence of overt full-blown cardiac autoimmunity.
Adaptive T cell immunity in response to MI
Ischemic injury releases cardiac self antigens in an inflammatory milieu that promotes maturation and activation of APCs96 (Fig. 2b). The infiltrating monocytes facilitate cardiac self-antigen trafficking to draining LNs, promoting the development of pathogenic, autoreactive T cells102. With the exception of pDCs, all other DC subtypes were shown to license pathogenic autoreactive TH cells and/or Teff cells96,98,103 (Fig. 2b). The mechanisms responsible for the recruitment, retention and further activation of pathogenic CD4+ T cells post-MI remain poorly understood. CX3CR1 expression on human CD4+ T cells post-MI was associated with their decline in the circulation, potentially attracted to the injured myocardium by increased production of CX3CL1 (ref. 104). This is consistent with the mechanisms of tissue recruitment of TEM cells, but additional mechanisms are likely at play (Fig. 2b). CD4+ T cells in the ischemic heart show a preferential expansion of TRBV gene segments and TRBV-J rearrangements relative to their abundance in circulating T cells98. It is still unclear, however, whether these clones are different from those found in non-ischemic hearts, or whether the same clones that are present before ischemia switch from naive and regulatory to effector memory and TH1/TH17 phenotypes. This early CD4+ T cell activation post-MI is detrimental overall and contributes to increased infarct size and reduced heart function105. CD8+ T cells are also activated and accumulate in the ischemic heart98, producing IFNγ and displaying cytotoxicity toward cardiomyocytes106 (Fig. 2b). Although their antigen specificity is still unknown, their granzyme B-dependent106 detrimental effect107 requires the recognition of self antigens in an MHC class I-restricted manner106. This is consistent with the detrimental role of CLEC9A-expressing cDC1s, which promote CD8+ T cell cross-priming107. As in CD4+ T cells, a role for the CX3CR1–CX3CL1 pathway has been proposed in myocardial homing of CD8+ T cells and their detrimental role104. The same group described accelerated senescence of circulating effector memory TEMRA CD8+ T cells108. However, the implication of this finding to post-MI cardiac remodeling is unclear. The role of other T cell subsets requires further investigation. The TCR repertoire of γδ T cells becomes significantly restricted in people with acute MI and is associated with increased expression of IL-17A109. CD4−CD8− (double-negative) T cells also accumulate in large numbers in ischemic hearts107, but their significance and role remain unexplored.
The appearance of pathogenic, autoreactive T cells post-MI may constitute a threat to immune homeostasis. However, in most circumstances and in the absence of an autoimmune-prone background, overt cardiac autoimmunity does not develop. This may be explained, in part, by the concomitant increase in cardiac self-antigen-specific Treg cells3,99,110. The ischemic heart sustains the recruitment of circulating Treg cells and their local proliferation and expansion99, and induces the conversion of recruited conventional T cells into Treg cells3. However, the mechanisms responsible for these effects are poorly understood. Enhanced release of IL-33 from stressed necrotic cells may have a role. Other data point to the presence of a local paracrine–autocrine adenosinergic loop enhancing Treg cell immunosuppressive effects105,111. Several additional hypotheses merit exploration. The hypoxic environment could maintain a tolerogenic DC phenotype. Furthermore, the site of T cell priming outside the heart may play a role. Autoreactive T cell priming by DCs in the spleen after MI has been shown to promote Treg cell generation112, whereas DC-dependent priming of T cells in draining LNs, supported by high levels of hepatocyte growth factor (HGF) and CXCL10, may drive a sustained recruitment of pathogenic T cells100 (Fig. 2b).
Although CD4+ T cell depletion is protective in the acute phase of MI105, total CD4+ T cell deficiency in mice impairs long-term cardiac remodeling and the recovery of heart function, suggesting that CD4+ T cells have an overall protective role113. Indeed, a series of experimental studies point to a protective role of Treg cells in experimental MI114,115, and low levels of circulating CD4+Foxp3+ Treg cells in humans correlate with increased risk of acute coronary events at follow-up116. Cardioprotective effects of Treg cells are diverse and include the regulation of adaptive and innate immune responses, their impact on stromal cells, modulating fibroblast activation and matrix deposition99,115, and their regulation of cardiomyocyte apoptosis and proliferation117. Despite the relative preservation of Treg cells at the acute phase of MI, a subset of them may become dysfunctional and potentially pathogenic over time118. Those cells appear to have lost their suppressive properties, upregulated the expression of proinflammatory mediators (TNF, TNFR1, IFNγ) and acquired anti-angiogenic properties118. The molecular pathways responsible for these alterations require further investigation.
B cell responses to cardiac ischemic injury
B cells, including B1-like and B2-like cells, represent a large portion of resident leukocytes in the myocardium119,120. They represent circulating cells that slowly transit through the myocardium to support cardiac homeostasis. Additional resident B1-like cells are found in the pericardial adipose tissue as part of FALCs121,122 (Fig. 2a). Intravascular myocardial B cells appear to support the expression of MHC class II on resident cardiac macrophages123. However, the (patho)physiological relevance of this finding is currently unclear. Studies in mice and rats have shown that within 7 days following MI, more B cells accumulate in the myocardium and pericardial FALCs121,124. Pericardial B cells produce GM-CSF post-MI and promote CCR7-mediated DC migration and T cell activation in pericardial adipose tissue, with detrimental consequences on post-MI remodeling and recovery of heart function121 (Fig. 2b). Systemic B cells, and more particularly MZ B cells, also become activated early after MI, and their depletion improves cardiac remodeling and the recovery of heart function124,125. The detrimental effect of mature B cells is attributed to their proinflammatory role, producing CCL7 and promoting monocyte mobilization and recruitment124,125 (Fig. 2b). Except for the production of certain natural IgM antibodies against non-myosin heavy chain II that promote myocardial damage126, the immediate effects of B cells are unlikely to depend on specific antibodies. However, following the acute phase, myocardial ischemia triggers GC formation in draining mediastinal LNs4 (Fig. 2b). Such newly induced autoantibodies, such as those against cardiac myosin, have been associated with post-MI heart failure127. Thus, it is possible that MI leads to the emergence of autoantigens released during myocardial damage, which in turn trigger autoimmune responses and memory B cells that worsen cardiac function and may even accelerate atherosclerosis post-MI4. The latter is particularly relevant in the setting of secondary prevention. However, not all B cells are detrimental. IL-10-producing B cells of the pericardial adipose tissue may help in inflammation resolution and heart recovery post-MI122 (Fig. 2).
Targeting adaptive immune responses for patient benefit
A few important points should be considered regardless of which therapeutic strategy is pursued to target the immune response in ACVD (Box 5).
Harnessing the adaptive immune system for patient benefit could be pursued through various modalities. The therapeutic objective is to break the vicious circle of pathogenic adaptive immune activation and promote regulatory homeostatic immunity. Here, we will highlight a few immunomodulatory approaches in development (Fig. 3).

The figure, which is not exhaustive, highlights some of the most promising therapeutic strategies to manipulate the adaptive immune system (through neutralizing pathogenic or promoting regulatory immunity) for the benefit of individuals with, or at risk of, ischemic cardiovascular diseases. Current strategies tested in the clinic include the use of a monoclonal antibody (Ab) against CD20 (that is, rituximab) to deplete mature B cells, and the use of recombinant (rec) low-dose IL-2 to promote Treg cells. Promising strategies in development include the development of CD8-depleting monoclonal antibodies, neutralizing antibodies to block proinflammatory OSEs, such as anti-PC and anti-MDA antibodies, CAR-Treg cells targeting OSEs, RNA-based immunogenic vaccines targeting OSEs and RNA-based tolerogenic vaccines using ApoB100-derived peptides. FO B, follicular B cell; GCB, GC B cell.
Deleting/neutralizing proatherogenic immunity
Targeting pathogenic B cells
CD20-mediated B cell depletion reduces atherosclerosis, MI remodeling and post-MI accelerated atherosclerosis in experimental models4,74,75,124. CD20 antibodies preferentially deplete B2 cells and preserve atheroprotective B1 cells. The RITA-MI phase 1/2a trial tested the safety and tolerability of a single intravenous injection of rituximab in people with acute ST-elevation MI (STEMI)128. The treatment led to a rapid and dose-dependent reduction in circulating B cells, and echocardiographic data suggested that LV-ejection fraction was improved after 6 months. This impact on cardiac remodeling is now being tested in the phase 2b RITA-MI2. A phase 2 trial of rituximab in people with systolic heart failure is also underway (NCT03332888).
Belimumab is approved for treatment of SLE. It targets BAFF, a cytokine of the BAFF–APRIL system that is essential for B2 cell survival. Although targeting of BAFFR was atheroprotective in mice76, BAFF neutralization was proatherogenic owing to non-canonical atheroprotective effects of BAFF signaling in myeloid cells that inhibit TLR9–IRF7-dependent inflammatory responses129. Thus, caution is warranted regarding this specific B cell-depletion approach.
Several other B cell-depleting agents are being developed (some, for example, target CD22 and CD19), but they may also deplete protective subsets, and data in experimental atherosclerosis are lacking.
Monoclonal anti-OSE antibodies
Neutralizing different OSEs, such as MDA or PC, with specific humanized monoclonal antibodies has been successful in preclinical ACVD models87,88. The efficacy of anti-MDA antibodies in humans has been tested in the GLACIER trial, which assessed aortic 18F-FDG-PET uptake in people receiving orticumab (human anti-MDA-ApoB100 IgG1) over a period of 12 weeks130. Although this trial did not show an effect, two randomized double-blind placebo-controlled phase 2 trials with different study designs are underway. NCT04776629 is investigating the effect of orticumab on coronary plaque burden in individuals with psoriasis who have increased CVD risk, and NCT03991143 is investigating the effect of a human IgG1 against PC (ATH3G10) on CV outcomes in participants with STEMI. Future approaches could employ RNA-based technologies to produce sustained levels of protective antibodies.
Immunogenic vaccination strategies
A range of vaccination strategies that raise anti-OSE antibodies are atheroprotective in animal models40,86,131,132. In particular, the molecular mimicry between PC and the capsular polysaccharide of Streptococcus pneumoniae has been exploited to trigger high levels of atheroprotective anti-PC IgM antibodies in Ldlr–/– mice by immunizing them with pneumococcal extracts131. This prompted the idea to translate these findings and evaluate whether vaccination with approved polyvalent pneumococcal vaccines also induces anti-PC antibodies and reduces cardiovascular events. However, PC is not a major constituent of pneumococcal vaccine preparations, and small studies using the 13-valent conjugate pneumococcal vaccine Prevnar 13 failed to show a robust induction of anti-oxLDL or anti-PC antibodies, and special vaccination protocols may be required133,134. The effect of the 23-valent vaccine Pneumovax 23 on cardiovascular outcomes (acute coronary syndrome (ACS), ischemic stroke) is currently being tested in the randomized placebo-controlled AUSPICE trial135. It will be interesting if this study confirms the cardioprotective signals from a meta-analysis of several observational studies136.
Re-establishing immune homeostasis
IL-2-based therapeutics
The concept of low-dose IL-2 therapy in CVD137 builds on the essential role of IL-2 in promoting Treg cells and the exquisite sensitivity of these cells to ultra-low doses of IL-2. IL-2 also limits the switch of autoreactive Treg cells to Teff cells that could result from TCR overactivation59. A first proof-of-concept safety and biological efficacy trial, LILACS, was completed. Low-dose IL-2 significantly expanded Treg cells in people with stable and unstable ischemic heart disease without adverse events of major concern138. A dose of 1.5 × 106 IU was selected and is currently being tested in IVORY (NCT04241601), a randomized, double-blind, placebo-controlled trial testing the superiority of low-dose IL-2 in reducing vascular inflammation in individuals with ACS. Low-dose IL-2 therapy may have protective effects beyond Treg cells. In LILCAS, low-dose IL-2 increased the activation of ILC2 (ref. 139), an immune-cell population with protective roles in atherosclerosis31 and cardiac remodeling post-MI139.
Other IL-2 biologics
More targeted approaches to enhance IL-2 selectivity are being developed; for example, an antibody–IL-2 conjugate that induces a conformational change resulting in lower affinity for IL-2Rα and competitive advantage for Treg cells140. Other approaches include IL-2 muteins with enhanced Treg cell selectivity141, some of which are being tested in clinical trials (NCT03422627; NCT03451422; NCT03410056). Cell-therapy approaches are also in development using engineered T cells that express defined ligand (normal or mutated IL-2)–receptor (mutated versions of the IL-2R subunits) complexes, and that signal exclusively together142.
Cell-therapy-based approaches
Chimeric antigen receptor (CAR) expression renders a population of T cells uniformly specific for a defined antigen and has revolutionized T cell therapy. Engineered CAR-Treg cells that express scFv antibodies directed against atherosclerosis-relevant epitopes, such as OSEs, are expected to preferentially home to disease sites and suppress local inflammation. Such CAR-Treg cells may also be engineered to express their own IL-2, further stabilizing them and enhancing their efficacy. Technological breakthroughs allowing for the use of allogeneic cells would overcome major hurdles in translating this technology to the clinic.
Tolerogenic vaccination strategies
The discovery that Treg cells are atheroprotective143 opened the possibility for therapeutic strategies that promote antigen-specific Treg cells144. Several tolerogenic strategies have been tested, particularly using MHC class II-restricted ApoB peptides, and these were shown to reduce atherogenesis, likely in a Treg cell-dependent manner45,145. Unfortunately, translation of this knowledge to the clinic has been very slow. This is due to several factors, including but not limited to insufficient knowledge and characterization of the adaptive immune response in human atherosclerosis and the need for correct HLA class II matching for peptide-based vaccines, as well as uncertainty about the vaccination route and the type of adjuvant needed to induce protective autoantibodies (while preventing the generation of pathogenic T cells and autoantibodies), or required to elicit a tolerogenic Treg cell response. Increasing knowledge of the adaptive immune response of human atherosclerosis, boosted by the advent of single-cell multiomics technologies, along with the revolution in vaccine design and delivery, as shown by the exceptional efficacy of combining RNA and lipid nanoparticle technologies to combat COVID-19, will help overcome these hurdles and will accelerate the development of various immunogenic and tolerogenic vaccines to prevent and treat ACVD.
Perspectives and conclusion
Despite unequivocal evidence for a fundamental role of the immune system in ACVD, newly developed anti-inflammatory treatments have been shown to be incremental or ineffective5. We believe that two major areas with fundamental gaps in knowledge deserve particular attention.
Despite substantial evidence from GWASs for a major role of inflammation in human atherosclerosis, there is a huge gap in characterizing the causal, mechanistic pathways. We operate at a distance from biology, and we desperately lack the comprehensive cell-type and disease-specific phenotyping information that will enable us to produce useful knowledge.
The second gap is due to our reliance on an outdated morphological classification of atherosclerosis. We need to move toward the medicine of the future and establish a molecular classification of atherosclerosis. This will require comprehensive insight at single-cell resolution by studying healthy and diseased arteries, at all major arterial sites and across space and time. Deep characterization of antigen-specific T cells and B cells, along with their antigens and antibodies, must be conducted in a systematic way, at different anatomical sites (arteries, SLOs, ATLOs and blood), in large numbers of individuals and at various time points of the disease process. This effort will be facilitated by the continuous development of advanced single-cell technologies and computational analysis, coupled with powerful antigen-screening libraries and advanced proteomics, allowing for the prioritization of atherosclerosis-specific immune receptors and their relevant antigens and antibodies. These can then be harnessed for the development of transformative diagnostic and therapeutic strategies. The exceptional recent advances in RNA-based and cell-based therapies are expected to further facilitate and accelerate the translation of this knowledge into benefit for patients.
References
Boren, J. et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 41, 2313–2330 (2020).
Roy, P., Orecchioni, M. & Ley, K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-021-00584-1 (2021).
Rieckmann, M. et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J. Clin. Invest. 129, 4922–4936 (2019).
Kyaw, T. et al. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur. Heart J. 42, 938–947 (2021). Mouse study demonstrating the role of GC B cells and antibodies in accelerated atherosclerosis post-MI, with potential implications for secondary prevention.
Zhao, T. X. & Mallat, Z. Targeting the immune system in atherosclerosis: JACC state-of-the-art review. J. Am. Coll. Cardiol. 73, 1691–1706 (2019).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Hansson, G. K., Holm, J. & Jonasson, L. Detection of activated T lymphocytes in the human atherosclerotic plaque. Am. J. Pathol. 135, 169–175 (1989).
Palinski, W. et al. Low density lipoprotein undergoes oxidative modification in vivo. Proc. Natl Acad. Sci. USA 86, 1372–1376 (1989).
Stemme, S. et al. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc. Natl Acad. Sci. USA 92, 3893–3897 (1995).
Sage, A. P., Tsiantoulas, D., Binder, C. J. & Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 16, 180–196 (2019).
Zernecke, A. et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 127, 402–426 (2020).
Wade, N. S. & Major, A. S. The problem of accelerated atherosclerosis in systemic lupus erythematosus: insights into a complex co-morbidity. Thromb. Haemost. 106, 849–857 (2011).
Huan, T. et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 33, 1427–1434 (2013).
Mauersberger, C., Schunkert, H. & Sager, H. B. Inflammation-related risk loci in genome-wide association studies of coronary artery disease. Cells 10, 440 (2021).
Bjorkbacka, H. et al. Weak associations between human leucocyte antigen genotype and acute myocardial infarction. J. Intern. Med. 268, 50–58 (2010).
Bjorkegren, J. L. M., Kovacic, J. C., Dudley, J. T. & Schadt, E. E. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J. Am. Coll. Cardiol. 65, 830–845 (2015).
Dansky, H. M., Charlton, S. A., Harper, M. M. & Smith, J. D. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc. Natl Acad. Sci. USA 94, 4642–4646 (1997).
Drobni, Z. D. et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 142, 2299–2311 (2020).
Mohanta, S. K. et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature (in the press). Detailed description of neuro–immune–vascular interactions in the adventitia and outer media of large arteries, establishing an artery–brain crosstalk and the impact thereof on maintenance of artery tertiary lymphoid organs and the progression of atherosclerosis.
Wick, G., Jakic, B., Buszko, M., Wick, M. C. & Grundtman, C. The role of heat shock proteins in atherosclerosis. Nat. Rev. Cardiol. 11, 516–529 (2014).
Cros, J. et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity 33, 375–386 (2010).
Narasimhan, P. B., Marcovecchio, P., Hamers, A. A. J. & Hedrick, C. C. Nonclassical monocytes in health and disease. Annu. Rev. Immunol. 37, 439–456 (2019).
Williams, J. W. et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 21, 1194–1204 (2020).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 1191 (2018).
Zhou, J. et al. CXCR3-dependent accumulation and activation of perivascular macrophages is necessary for homeostatic arterial remodeling to hemodynamic stresses. J. Exp. Med. 207, 1951–1966 (2010).
Hernandez, G. E. et al. Aortic intimal resident macrophages are essential for maintenance of the non-thrombogenic intravascular state. Nat. Cardiovasc. Res. 1, 67–84 (2022).
Ma-Krupa, W. et al. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J. Exp. Med. 199, 173–183 (2004).
Galkina, E. et al. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 203, 1273–1282 (2006).
Jackson-Jones, L. H. et al. Fat-associated lymphoid clusters control local IgM secretion during pleural infection and lung inflammation. Nat. Commun. 7, 12651 (2016).
Srikakulapu, P. et al. Perivascular adipose tissue harbors atheroprotective IgM-producing B cells. Front. Physiol. 8, 719 (2017).
Newland, S. A. et al. Type-2 innate lymphoid cells control the development of atherosclerosis in mice. Nat. Commun. 8, 15781 (2017).
Binder, C. J. et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J. Clin. Invest. 114, 427–437 (2004).
Cardilo-Reis, L. et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 4, 1072–1086 (2012).
Tellides, G. & Pober, J. S. Inflammatory and immune responses in the arterial media. Circ. Res. 116, 312–322 (2015).
Roozendaal, R. & Mebius, R. E. Stromal cell–immune cell interactions. Annu Rev Immunol 29, 23–43 (2011).
Chan, T. D. et al. Elimination of germinal-center-derived self-reactive B cells is governed by the location and concentration of self-antigen. Immunity 37, 893–904 (2012).
Krautler, N. J. et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell 150, 194–206 (2012).
Lin, Z. et al. Deep sequencing of the T cell receptor beta repertoire reveals signature patterns and clonal drift in atherosclerotic plaques and patients. Oncotarget 8, 99312–99322 (2017).
Hamze, M. et al. Characterization of resident B cells of vascular walls in human atherosclerotic patients. J. Immunol. 191, 3006–3016 (2013).
Nilsson, J. & Hansson, G. K. Vaccination strategies and immune modulation of atherosclerosis. Circ. Res. 126, 1281–1296 (2020).
MacRitchie, N. et al. The aorta can act as a site of naive CD4+ T-cell priming. Cardiovasc. Res. 116, 306–316 (2020).
Li, J. et al. CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ. Res. 118, 1540–1552 (2016).
Vila-Caballer, M. et al. Disruption of the CCL1–CCR8 axis inhibits vascular Treg recruitment and function and promotes atherosclerosis in mice. J. Mol. Cell. Cardiol. 132, 154–163 (2019).
Tsilingiri, K. et al. Oxidized low-density lipoprotein receptor in lymphocytes prevents atherosclerosis and predicts subclinical disease. Circulation 139, 243–255 (2019).
Kimura, T. et al. Regulatory CD4+ T cells recognize major histocompatibility complex class II molecule-restricted peptide epitopes of apolipoprotein B. Circulation 138, 1130–1143 (2018). Identification of an ApoB peptide as the first Treg cell epitope in human and mouse atherosclerosis.
Maganto-Garcia, E., Tarrio, M. L., Grabie, N., Bu, D. X. & Lichtman, A. H. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation 124, 185–195 (2011).
Wolf, D. et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B100-reactive CD+ T-regulatory cells. Circulation 142, 1279–1293 (2020).
Mailer, R. K. W., Gistera, A., Polyzos, K. A., Ketelhuth, D. F. J. & Hansson, G. K. Hypercholesterolemia induces differentiation of regulatory T cells in the liver. Circ. Res. 120, 1740–1753 (2017).
Almanzar, G. et al. Autoreactive HSP60 epitope-specific T-cells in early human atherosclerotic lesions. J Autoimmun 39, 441–450 (2012).
Sage, A. P. et al. X-Box binding protein-1 dependent plasma cell responses limit the development of atherosclerosis. Circ. Res. 121, 270–281 (2017).
Sage, A. P. et al. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation 130, 1363–1373 (2014).
Clement, M. et al. Deletion of IRF8 (interferon regulatory factor 8)-dependent dendritic cells abrogates proatherogenic adaptive immunity. Circ. Res. 122, 813–820 (2018).
Zernecke, A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 35, 763–770 (2015).
Bonacina, F. et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat. Commun. 9, 3083 (2018).
Clement, M. et al. Impaired autophagy in CD11b+ dendritic cells expands CD4+ regulatory T cells and limits atherosclerosis in mice. Circ. Res. 125, 1019–1034 (2019).
Lacy, M. et al. Cell-specific and divergent roles of the CD40L–CD40 axis in atherosclerotic vascular disease. Nat. Commun. 12, 3754 (2021).
Baardman, J. & Lutgens, E. Regulatory T cell metabolism in atherosclerosis. Metabolites 10, 279 (2020).
Gaddis, D. E. et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat. Commun. 9, 1095 (2018).
Bailey-Bucktrout, S. L. et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 39, 949–962 (2013).
Butcher, M. J. et al. Atherosclerosis-driven Treg plasticity results in formation of a dysfunctional subset of plastic IFNγ+ TH1/Tregs. Circ. Res. 119, 1190–1203 (2016).
Taleb, S., Tedgui, A. & Mallat, Z. IL-17 and TH17 cells in atherosclerosis: subtle and contextual roles. Arterioscler Thromb. Vasc. Biol. 35, 258–264 (2015).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019). Single-cell analyses of human carotid plaques, demonstrating the complexity and heterogeneity of infiltrating adaptive immune cells and their activation in symptomatic disease.
Depuydt, M. A. C. et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res. 127, 1437–1455 (2020).
Getz, G. S. & Reardon, C. A. Natural killer T cells in atherosclerosis. Nat. Rev. Cardiol. 14, 304–314 (2017).
He, S. et al. Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature 566, 115–119 (2019).
Schafer, S. & Zernecke, A. CD8+ T cells in atherosclerosis. Cells 10, 37 (2020).
Dimayuga, P. C. et al. Identification of apoB-100 peptide-specific CD8+ T cells in atherosclerosis. J. Am. Heart Assoc. 6, e005318 (2017).
Clement, M. et al. Control of the T follicular helper-germinal center B-cell axis by CD8+ regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 131, 560–570 (2015).
Hu, D. et al. Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin beta receptors. Immunity 42, 1100–1115 (2015).
Srikakulapu, P. et al. Artery tertiary lymphoid organs control multilayered territorialized atherosclerosis B-cell responses in aged ApoE–/– mice. Arterioscler. Thromb. Vasc. Biol. 36, 1174–1185 (2016).
Tay, C. et al. Follicular B cells promote atherosclerosis via T cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler. Thromb. Vasc. Biol. 38, e71–e84 (2018).
Chou, M. Y. et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J. Clin. Invest. 119, 1335–1349 (2009).
Nus, M. et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 23, 601–610 (2017).
Ait-Oufella, H. et al. B cell depletion reduces the development of atherosclerosis in mice. J. Exp. Med. 207, 1579–1587 (2010).
Kyaw, T. et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 185, 4410–4419 (2010).
Sage, A. P. et al. BAFF receptor deficiency reduces the development of atherosclerosis in mice—brief report. Arterioscler. Thromb. Vasc. Biol. 32, 1573–1576 (2012).
Centa, M. et al. Acute loss of apolipoprotein E triggers an autoimmune response that accelerates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 38, e145–e158 (2018).
Hutchinson, M. A. et al. Auto-antibody production during experimental atherosclerosis in ApoE–/– mice. Front. Immunol. 12, 695220 (2021).
Centa, M. et al. Germinal center-derived antibodies promote atherosclerosis plaque size and stability. Circulation 139, 2466–2482 (2019). Demonstration of the effect of germinal-center-derived antibodies in promoting atherosclerotic lesion size and modulating plaque stability in mice.
Crane, E. D. et al. Anti-GRP78 autoantibodies induce endothelial cell activation and accelerate the development of atherosclerotic lesions. JCI Insight 3, e99363 (2018).
Porsch, F., Mallat, Z. & Binder, C. J. Humoral immunity in atherosclerosis and myocardial infarction: from B cells to antibodies. Cardiovasc. Res. 117, 2544–2562 (2021).
Lorenzo, C. et al. ALDH4A1 is an atherosclerosis auto-antigen targeted by protective antibodies. Nature 589, 287–292 (2021). An elegant high-throughput approach for the identification and evaluation of novel B cell antigens in atherosclerosis using single-cell analyses, mass spectrometry and recombinant technology.
Gistera, A. et al. Low-density lipoprotein-reactive T cells regulate plasma cholesterol levels and development of atherosclerosis in humanized hypercholesterolemic mice. Circulation 138, 2513–2526 (2018).
Rhoads, J. P. et al. Oxidized low-density lipoprotein immune complex priming of the Nlrp3 inflammasome involves TLR and FcγR cooperation and is dependent on CARD9. J. Immunol. 198, 2105–2114 (2017).
van den Berg, V. J. et al. Anti-Oxidized LDL antibodies and coronary artery disease: a systematic review. Antioxidants 8, 84 (2019).
Papac-Milicevic, N., Busch, C. J. & Binder, C. J. Malondialdehyde epitopes as targets of immunity and the implications for atherosclerosis. Adv. Immunol. 131, 1–59 (2016).
Schiopu, A. et al. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1–/–/low-density lipoprotein receptor–/– mice. J. Am. Coll. Cardiol. 50, 2313–2318 (2007).
de Vries, M. R. et al. Identification of IgG1 isotype phosphorylcholine antibodies for the treatment of inflammatory cardiovascular diseases. J. Intern. Med. 290, 141–156 (2021).
Que, X. et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 558, 301–306 (2018). Overexpression of a single-chain variable fragment of E06, which binds to the phosphocholine headgroup, reduces systemic inflammation, atherosclerosis progression, aortic stenosis and hepatic steatosis in mice.
Bagchi-Chakraborty, J. et al. B cell Fcγ receptor IIb modulates atherosclerosis in male and female mice by controlling adaptive germinal center and innate B-1-cell responses. Arterioscler. Thromb. Vasc. Biol. 39, 1379–1389 (2019).
Tsiantoulas, D. et al. Increased plasma IgE accelerate atherosclerosis in secreted IgM deficiency. Circ. Res. 120, 78–84 (2017).
Zhang, X. et al. IgE contributes to atherosclerosis and obesity by affecting macrophage polarization, macrophage protein network, and foam cell formation. Arterioscler. Thromb. Vasc. Biol. 40, 597–610 (2020).
Tay, C. et al. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc. Res. 111, 385–397 (2016).
Hilgendorf, I. et al. Innate response activator B cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation 129, 1677–1687 (2014).
Lavine, K. J. et al. The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (part 4). J. Am. Coll. Cardiol. 72, 2213–2230 (2018).
Van der Borght, K. et al. Myocardial infarction primes autoreactive T cells through activation of dendritic cells. Cell Rep. 18, 3005–3017 (2017). This study shows that cardiac cDC1s drive the proliferation and differentiation of cardiac α-myosin-specific Treg cells at steady state, while cDC2s drive the proliferation of autoreactive T cells and their differentiation into effector cells after myocardial infarction.
Lv, H. et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J. Clin. Invest. 121, 1561–1573 (2011).
Tang, T. T. et al. Pathologic T-cell response in ischaemic failing hearts elucidated by T-cell receptor sequencing and phenotypic characterization. Eur. Heart J. 40, 3924–3933 (2019). Bulk TCR sequencing on heart-infiltrating T cells reveals TCR clonotypes shared between ischemic failing hearts of several patients, with a dominance of CD4+ TH1 cells and cytotoxic CD8+ T cells.
Xia, N. et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation 142, 1956–1973 (2020).
Komarowska, I. et al. Hepatocyte growth factor receptor c-Met instructs T cell cardiotropism and promotes T cell migration to the heart via autocrine chemokine release. Immunity 42, 1087–1099 (2015).
Dobaczewski, M., Xia, Y., Bujak, M., Gonzalez-Quesada, C. & Frangogiannis, N. G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am. J. Pathol. 176, 2177–2187 (2010).
DeBerge, M. et al. Monocytes prime autoreactive T cells after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 318, H116–H123 (2020).
Lee, J. S. et al. Conventional dendritic cells impair recovery after myocardial infarction. J. Immunol. 201, 1784–1798 (2018).
Boag, S. E. et al. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J. Clin. Invest. 125, 3063–3076 (2015).
Yang, Z. et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 114, 2056–2064 (2006).
Santos-Zas, I. et al. Cytotoxic CD8+ T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 12, 1483 (2021). Evidence for a deleterious role of CD8+ T cells following acute MI in mice and pigs through the production of granzyme B, with circulating levels of the latter being predictive of 1-year mortality in people with acute MI.
Forte, E. et al. Cross-priming dendritic cells exacerbate immunopathology after ischemic tissue damage in the heart. Circulation 143, 821–836 (2021).
Hoffmann, J. et al. Myocardial ischemia and reperfusion leads to transient CD8 immune deficiency and accelerated immunosenescence in CMV-seropositive patients. Circ. Res. 116, 87–98 (2015).
Chen, X. M. et al. Gene expression pattern of TCR repertoire and alteration expression of IL-17A gene of γδ T cells in patients with acute myocardial infarction. J. Transl. Med. 16, 189 (2018).
Klingenberg, R. et al. Clonal restriction and predominance of regulatory T cells in coronary thrombi of patients with acute coronary syndromes. Eur. Heart J. 36, 1041–1048 (2015).
Xia, N. et al. Activated regulatory T-cells attenuate myocardial ischaemia/reperfusion injury through a CD39-dependent mechanism. Clin. Sci. 128, 679–693 (2015).
Wang, Y. et al. C-X-C motif chemokine receptor 4 blockade promotes tissue repair after myocardial infarction by enhancing regulatory T cell mobilization and immune-regulatory function. Circulation 139, 1798–1812 (2019).
Hofmann, U. et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663 (2012).
Matsumoto, K. et al. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 52, 382–387 (2011).
Weirather, J. et al. Foxp3+CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115, 55–67 (2014).
Wigren, M. et al. Low levels of circulating CD4+FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler. Thromb. Vasc. Biol. 32, 2000–2004 (2012).
Zacchigna, S. et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat. Commun. 9, 2432 (2018). Evidence for a role of Treg cells in promoting fetal and maternal cardiomyocyte proliferation after MI in mice, with a significant impact on infarct size and cardiac contractility.
Bansal, S. S. et al. Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 139, 206–221 (2019).
Adamo, L. et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 5, e134700 (2020). Detailed characterization of B cells in naive mouse hearts and their circulating origin and transit properties through the heart.
Rocha-Resende, C. et al. Developmental changes in myocardial B cells mirror changes in B cells associated with different organs. JCI Insight 5, e139377 (2020).
Horckmans, M. et al. Pericardial adipose tissue regulates granulopoiesis, fibrosis, and cardiac function after myocardial infarction. Circulation 137, 948–960 (2018). Identification of lymphoid cell clusters in human and murine epicardial adipose tissue and their role in regulating cardiac remodeling post-MI.
Wu, L. et al. IL-10-producing B cells are enriched in murine pericardial adipose tissues and ameliorate the outcome of acute myocardial infarction. Proc. Natl Acad. Sci. USA 116, 21673–21684 (2019).
Rocha-Resende, C., Pani, F. & Adamo, L. B cells modulate the expression of MHC-II on cardiac CCR2– macrophages. J. Mol. Cell. Cardiol. 157, 98–103 (2021).
Zouggari, Y. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 19, 1273–1280 (2013).
Sun, Y. et al. Splenic marginal zone B lymphocytes regulate cardiac remodeling after acute myocardial infarction in mice. J. Am. Coll. Cardiol. (in the press). Identification of MZ B cells as mediators of adverse cardiac remodeling post-MI and the contribution of miR21-dependent upregulation of HIF1α to this effect.
Haas, M. S. et al. Blockade of self-reactive IgM significantly reduces injury in a murine model of acute myocardial infarction. Cardiovasc Res 87, 618–627 (2010).
Kaya, Z., Leib, C. & Katus, H. A. Autoantibodies in heart failure and cardiac dysfunction. Circ. Res. 110, 145–158 (2012).
Zhao, T. X. et al. Rituximab in patients with acute ST-elevation myocardial infarction (RITA-MI): an experimental medicine safety study. Cardiovasc. Res. 118, 872–882 (2021). Treatment with rituximab is safe when given in the acute ST-elevation MI setting and substantially alters circulating B cell subsets.
Tsiantoulas, D. et al. B cell-activating factor neutralization aggravates atherosclerosis. Circulation 138, 2263–2273 (2018).
Lehrer-Graiwer, J. et al. FDG-PET imaging for oxidized LDL in stable atherosclerotic disease: a phase II study of safety, tolerability, and anti-inflammatory activity. JACC Cardiovasc. Imaging 8, 493–494 (2015).
Binder, C. J. et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 9, 736–743 (2003).
Binder, C. J., Papac-Milicevic, N. & Witztum, J. L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 16, 485–497 (2016).
Houben, T. et al. Pneumococcal immunization reduces neurological and hepatic symptoms in a mouse model for Niemann–Pick type C1 disease. Front. Immunol. 9, 3089 (2018).
Grievink, H. W. et al. The effect of a 13-valent conjugate pneumococcal vaccine on circulating antibodies against oxidized LDL and phosphorylcholine in man, a randomized placebo-controlled clinical trial. Biology 9, 345 (2020).
Ren, S. et al. Rationale and design of a randomized controlled trial of pneumococcal polysaccharide vaccine for prevention of cardiovascular events: The Australian Study for the Prevention through Immunization of Cardiovascular Events (AUSPICE). Am. Heart J. 177, 58–65 (2016).
Ren, S. et al. Effect of the adult pneumococcal polysaccharide vaccine on cardiovascular disease: a systematic review and meta-analysis. Open Heart 2, e000247 (2015).
Zhao, T. X., Newland, S. A. & Mallat, Z. 2019 ATVB Plenary Lecture: interleukin-2 therapy in cardiovascular disease: the potential to regulate innate and adaptive immunity. Arterioscler. Thromb. Vasc. Biol. 40, 853–864 (2020).
Zhao, T. X. et al. Regulatory T cell response to low-dose interleukin-2 in ischemic heart disease. NEJM Evidence 1, EVIDoa2100009 (2021). In this phase 1b/2a study, low-dose IL-2 expanded Treg cells without adverse events of major concern, and single-cell RNA sequencing demonstrated the engagement of distinct pathways and cell–cell interactions after low-dose IL-2.
Yu, X. et al. Innate lymphoid cells promote recovery of ventricular function after myocardial infarction. J. Am. Coll. Cardiol. 78, 1127–1142 (2021).
Trotta, E. et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat. Med. 24, 1005–1014 (2018).
Khoryati, L. et al. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci. Immunol. 5, eaba5264 (2020).
Sockolosky, J. T. et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine–receptor complexes. Science 359, 1037–1042 (2018).
Ait-Oufella, H. et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 12, 178–180 (2006).
Herbin, O. et al. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 32, 605–612 (2012).
Kimura, T. et al. Atheroprotective vaccination with MHC-II-restricted ApoB peptides induces peritoneal IL-10-producing CD4 T cells. Am. J. Physiol. Heart Circ. Physiol. 312, H781–H790 (2017).
Tiret, L. et al. Genetic analysis of the interleukin-18 system highlights the role of the interleukin-18 gene in cardiovascular disease. Circulation 112, 643–650 (2005).
Sheedy, F. J. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 14, 812–820 (2013).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Pattarabanjird, T., Li, C. & McNamara, C. B cells in atherosclerosis: mechanisms and potential clinical applications. JACC Basic Transl. Sci. 6, 546–563 (2021).
Kyaw, T. et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ. Res. 109, 830–840 (2011).
Rosenfeld, S. M. et al. B-1b cells secrete atheroprotective IgM and attenuate atherosclerosis. Circ. Res. 117, e28–e39 (2015).
Srikakulapu, P. et al. Chemokine receptor-6 promotes B-1 cell trafficking to perivascular adipose tissue, local IgM production and atheroprotection. Front. Immunol. 12, 636013 (2021).
Tsiantoulas, D. et al. Circulating microparticles carry oxidation-specific epitopes and are recognized by natural IgM antibodies. J. Lipid Res. 56, 440–448 (2015).
Obermayer, G. et al. Natural IgM antibodies inhibit microvesicle-driven coagulation and thrombosis. Blood 137, 1406–1415 (2021).
Perry, H. M. et al. Helix-loop-helix factor inhibitor of differentiation 3 regulates interleukin-5 expression and B-1a B cell proliferation. Arterioscler. Thromb. Vasc. Biol. 33, 2771–2779 (2013).
Doring, Y. et al. B-cell-specific CXCR4 protects against atherosclerosis development and increases plasma IgM levels. Circ. Res. 126, 787–788 (2020).
Gruber, S. et al. Sialic acid-binding immunoglobulin-like lectin G promotes atherosclerosis and liver inflammation by suppressing the protective functions of B-1 cells. Cell Rep. 14, 2348–2361 (2016).
Grasset, E. K. et al. Sterile inflammation in the spleen during atherosclerosis provides oxidation-specific epitopes that induce a protective B-cell response. Proc. Natl Acad. Sci. USA 112, E2030–E2038 (2015).
Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1+ precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
Tsiantoulas, D. et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature 597, 92–96 (2021). Identification of a non-canonical function for the B cell cytokine APRIL with critical implications for vascular homeostasis and atherosclerotic cardiovascular disease.
Ryu, H. et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nat. Immunol. 19, 583–593 (2018).
Zhivaki, D. & Kagan, J. C. Innate immune detection of lipid oxidation as a threat assessment strategy. Nat. Rev. Immunol. 5, eaba5264 (2021).
York, A. G. et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163, 1716–1729 (2015).
Ridker, P. M. How common is residual inflammatory risk? Circ. Res. 120, 617–619 (2017).
Acknowledgements
Z.M. is supported by the British Heart Foundation (RCAM/104, RCAM-659, RRCAM.163), the British Heart Foundation Center for Research Excellence (RE/18/1/34212), and the NIHR Cambridge Biomedical Research Centre (RG85315). C.J.B. is supported by grants from the Austrian Science Fund (FWF SFB F54) and the Vienna Science and Technology Fund (LS18-090). Z.M. and C.J.B. are supported by the Leducq Foundation (Transatlantic Network of Excellence; TNE-20CVD03).
Author information
Authors and Affiliations
Contributions
Z. M. and C. J. B. wrote the manuscript and reviewed it for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Jan Nilsson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mallat, Z., Binder, C.J. The why and how of adaptive immune responses in ischemic cardiovascular disease. Nat Cardiovasc Res 1, 431–444 (2022). https://doi.org/10.1038/s44161-022-00049-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44161-022-00049-1
This article is cited by
-
Finding the right balance on the challenging path to clinical translation of anti-inflammatory therapies for ischemic heart disease
Nature Cardiovascular Research (2024)
-
Identifying the sensor elements of regulatory T cells in atherosclerosis
Nature Cardiovascular Research (2024)
-
Mapping the functions of IgM antibodies in atherosclerotic cardiovascular disease
Nature Reviews Cardiology (2023)
-
Control of the post-infarct immune microenvironment through biotherapeutic and biomaterial-based approaches
Drug Delivery and Translational Research (2023)
-
Pleiotropic genetic architecture and novel loci for C-reactive protein levels
Nature Communications (2022)
