
Abstract
Mechanistic insight into the catalytic production of ammonia from dinitrogen is needed to improve the synthesis of this vital molecule. Here we study the use of samarium diiodide (SmI2) and water in the presence of molybdenum complexes that bear PCP-type pincer ligands to synthesize ammonia. The proton-coupled electron transfer during the formation of a N–H bond on the molybdenum imide complex was found to be the rate-determining step at high catalyst concentrations. Additionally, the dimerization step of the catalyst became the rate-determining step at low catalyst concentrations. We designed PCP-type pincer ligands with various substituents at the 5- and 6-positions and observed that electron-withdrawing groups promoted the reaction rate, as predicted by density functional theory calculations. A molybdenum trichloride complex that bears a trifluoromethyl group functioned as the most effective catalyst and produced up to 60,000 equiv. ammonia based on the molybdenum atom of the catalyst, with a molybdenum turnover frequency of up to 800 equiv. min−1. The findings reported here can contribute to the development of an environmentally friendly next-generation nitrogen-fixation system.

Similar content being viewed by others
Main
Ammonia plays an essential role globally as a raw material to produce fertilisers and nitrogen-containing materials, which include pharmaceuticals, plastics, textiles and explosives. Presently, the Haber–Bosch process, with which ~144 metric tonnes of nitrogen are converted into ammonia annually, accounts for the main method to synthesize ammonia at the industrial level1. However, the conversions of dinitrogen and dihydrogen gases into ammonia via the Haber–Bosch process require a high temperature and pressure. Additionally, the preparation of dihydrogen feedstocks consumes a substantial amount of fossil fuels and is accompanied by considerable carbon dioxide (CO2) emission2,3. Ammonia recently attracted attention as a candidate transporter of sustainable energy because of its ease of liquefication, which is suitable for storage and transportation; moreover, only water and dinitrogen gas are emitted via combustion4,5,6. Thus, the past decade witnessed a demand for an environmentally friendly and tractable production of ammonia7,8. Notably, extensive studies were conducted on nitrogen fixation catalysed by heterogeneous catalysts to achieve the production of ammonia at a low temperature and pressure9,10,11,12.
Since the breakthrough reported by Yandulov and Schrock, the development of nitrogen fixation under mild reaction conditions in the presence of transition metal complexes (as homogeneous catalysts) has progressed exceptionally13. Employing the homogenous catalytic reaction system, dinitrogen gas can be converted into ammonia by reactions with reductants and proton sources (as the chemical reagents) in the presence of a catalytic amount of transition metal complexes as the catalysts at atmospheric pressure or room temperature or lower, for example, −78 °C (refs. 14,15,16,17). At the early stage of these reactions, the system often exhibited a limited catalytic activity owing to the deactivation of the catalysts and/or formation of dihydrogen as a side product from the high reactivity of the reductants, as well as the proton sources13,18,19,20,21,22,23,24,25,26,27,28. Very recently, we reported the catalytic formation of ammonia from dinitrogen gas by combining samarium diiodide (SmI2) with water (as the one-electron reductant and the proton source, respectively) in the presence of molybdenum complexes that bear a N-heterocyclic carbene (NHC)-based PCP-type pincer ligand under ambient reaction conditions29 (Fig. 1a)30. Our reaction system exhibited a high catalytic activity, and demonstrated a high production of ammonia; the amount of ammonia produced reached 4,350 equiv. based on the molybdenum atom of the catalysts (the turnover frequency (TOF) was ~120 min–1).

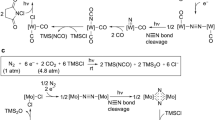
a, Previously reported catalytic reaction30. b, Stoichiometric reduction of 1a with an excess amount of SmI2 in a dinitrogen atmosphere. c, Stoichiometric reaction of 2a with excess amounts of SmI2 and H2O in an argon atmosphere. d, Catalytic reduction of dinitrogen with large amounts of SmI2 and H2O in the presence of the molybdenum complexes as the catalysts. e, Plausible reaction pathway for 1a and 2a. aData are the mean of the multiple individual experiments (a minimum of two), and the error bars represent the standard deviation (s.d.). r.t., room temperature.
Here we designed molybdenum complexes that bear different substituted PCP-type pincer ligands based on experiments, which included the isolation of nitride complexes as key reactive intermediates, as well as kinetic and theoretical studies of the catalytic reaction. Thereafter, we investigated the catalytic activity of the molybdenum complexes in the production of ammonia under ambient reaction conditions with SmI2 and water as the reductant and the proton source, respectively. Ultimately, our system dramatically increased the quantity of ammonia produced, as well as the reaction rate, compared with the results of our previous study30.
Results and discussion
Preparation and reactivity of the molybdenum–nitride complex
In our previous study, which was based on the stoichiometric and catalytic reactions of molybdenum(III) trihalide complexes that bear a pyridine-based PNP-type pincer ligand, [MoX3(PNP)] (X = Cl, Br and I)—in which the corresponding molybdenum(IV)–nitride complex [Mo(≡N)I(PNP)] was a key reactive intermediate in the catalytic formation of ammonia under ambient reaction conditions—the molybdenum(III) trichloride complex that bears the NHC-based PCP-type pincer ligand [MoCl3(PCP)] (1a; PCP = 1,3-bis((di-tert-butylphosphino)methyl)benzimidazol-2-ylidene) exhibited an excellent catalytic activity30. However, the corresponding molybdenum(IV)–nitride complex that bears the NHC-based PCP-type pincer ligand [Mo(≡N)I(PCP)] (2a) was not prepared.
Therefore, we first prepared 2a in this study following the same procedure as that to prepare [Mo(≡N)I(PNP)]. The reaction of 1a with 5 equiv. SmI2 in atmospheric dinitrogen and THF for five minutes at room temperature (25 °C) afforded 2a in a 72% NMR yield (Fig. 1b). Next, via the reaction of [MoI3(PCP)] with KC8 as a reductant, we isolated the nitride complex (2a) as a pure form. The detailed molecular structure of 2a was confirmed via X-ray analysis (Supplementary Fig. 7). This experimental result, which is illustrated in Fig. 1b, indicated that the transformation of 1a into 2a proceeded via the N≡N bond cleavage of a dinitrogen-bridged Mo(I)–N≡N–Mo(I) complex (I) after a two-electron reduction and ligand exchanges of 1a. We previously carried out a kinetic study on the stoichiometric reaction of [MoI3(PNP)] with SmI2 to give [Mo(≡N)I(PNP)] via a dinitrogen-bridged dimolybdenum(I) complex [MoI(PNP)]–N≡N–[MoI(PNP)] (ref. 30). This experimental result indicates that the stoichiometric transformation of [MoI3(PNP)] into [Mo(≡N)I(PNP)] proceeded via a dinuclear complex, such as the dinitrogen-bridged dimolybdenum complex as a key reactive intermediate31. In addition, we more recently investigated cycling between molybdenum(I)–dinitrogen and molybdenum(IV)–nitride complexes to support our proposed reaction pathway for the catalytic formation of ammonia from dinitrogen, together with density functional theory (DFT) calculations32. Based on our previous results, here we quantitatively confirmed the conversion of the nitride ligand in 2a into ammonia via the reaction of 2a with 5 equiv. SmI2 and H2O in an argon atmosphere and THF for 15 minutes at 25 °C (Fig. 1c). These experimental results indicated that dinitrogen can be transformed into ammonia employing 2a as a reactive intermediate.
Next, we compared the catalytic activities of 1a and 2a in ammonia formation under the optimized reaction conditions (Fig. 1d). The reaction of dinitrogen (1 atm) with 28,800 equiv. SmI2 (the reductant) and 28,800 equiv. H2O (the proton source) in the presence of a catalytic amount (25 nmol) of 1a in THF for four hours at 25 °C yielded 8,410 equiv. ammonia based on the molybdenum atom of the catalyst (88% yield based on SmI2). The catalytic reaction that employed 2a rather than 1a as the catalyst exhibited almost the same catalytic activity. Based on these stoichiometric and catalytic reactions, we expected that the molybdenum(IV)–nitride complex (2a) would function as a key reactive intermediate to effectively promote the catalytic nitrogen.
Based on our previous30,31,32 and present findings, we proposed the following reaction pathway for the production of ammonia employing 1a as the precatalyst and 2a as key reactive intermediate (Fig. 1e): first, the two-electron reduction of 1a with 2 equiv. SmI2, as well as the ligand exchange of chloride with the iodide obtained from SmI2, yields the corresponding dinitrogen-bridged dimolybdenum(I) complex (A). Afterwards, the cleavage of the bridged dinitrogen ligand in A affords the corresponding molybdenum(IV)–nitride complex (2a). Subsequently, 2a is converted into the corresponding molybdenum(I)–ammonia complex (B) via three reduction and protonation steps that employed imide and amide complexes. The proton-coupled electron transfer (PCET) process33,34,35 in the reaction of dinitrogen with SmI2 and H2O is generally proposed as the key steps36,37,38. After the formation of B, its subsequent dimerization facilitates the formation of a six-coordinated A (C). Finally, the elimination of the ammonia ligand from C regenerates A, and thereby completes the catalytic cycle. As pointed out in the previous paragraph, this proposed reaction pathway was supported by DFT calculations in the reaction that employed the corresponding molybdenum complexes that bear the PNP-type pincer ligand32. We previously investigated the stoichiometric reactions of the molybdenum–nitride complex that bear the PNP-type pincer ligand, [Mo(≡N)I(PNP)], as well as their kinetic isotope effect (KIE)30. These experimental results revealed that the transformation of 2a into B also proceeded via PCET.
Kinetic study of the catalytic reaction
To gain more insight into the reaction pathway of the formation of ammonia (Fig. 1e), we determined the kinetic parameters of the catalytic reduction of dinitrogen via the reaction with SmI2 and H2O in the presence of 1a in THF at room temperature. A THF solution that contained SmI2 (0.12 M) and H2O (0.12 M) was mixed in atmospheric dinitrogen (1 atm) in the presence of 6–0.5 μM 1a at room temperature. The initial rate of producing ammonia (\(v_{{\rm{NH}}_3}\)) was determined, following the ammonia yield from the catalytic reaction that was quenched at the initial stage. The reaction order with respect to 1a was obtained from the slope of the plot of log(\(v_{{\rm{NH}}_3}\)) versus log([1a]), where [1a] is the concentration of 1a (Fig. 2a). The reaction order exhibited two different kinetic regions based on the concentration of the catalyst. One was the first order, which employed the catalyst at high concentrations (region A, represented by the green line), and the other was the second order, which employed the catalyst at low concentrations (region B, represented by the orange line). Similar phenomena were observed in the ruthenium-catalysed oxidation of water in which the dimerization of the ruthenium complexes was the key step39,40.

The green and orange plots and lines represent the data in the presence of high and low concentrations of the catalyst, 1.5 μM < [1a] and [1a] < 1.5 μM, respectively. Data are the average of two or three experiments with error bars based on s.d. a, Plots of log(\(v_{{\rm{NH}}_3}\)) versus log([1a]). b, Plots of log(\(v_{{\rm{NH}}_3}\)) versus log([SmI2]). c, Plots of log(\(v_{{\rm{NH}}_3}\)) versus log[H2O].
To investigate the rate-determining steps of both regions, we determined the rate orders in the presence of SmI2 and H2O at high ([1a] = 4.17 μM) and low ([1a] = 0.7 μM) concentrations of 1a (the catalyst). Thus, employing a high concentration of the catalyst (region A), we estimated that SmI2 and H2O exhibited first-order kinetics (0.95 and 0.76 for SmI2 and H2O, respectively), as estimated for 1a (Fig. 2b,c, green line). These experimental results indicated that the reaction involving the three molecules, 1a, SmI2 and H2O, was the rate-determining step in region A. Such reaction steps were attributable to the reduction and protonation steps in PCET (Fig. 1e). Conversely, the estimated small rate orders in SmI2 and H2O employing a low concentration of the catalyst were not really zeroth order (0.34 and 0.39 for SmI2 and H2O, respectively) (Fig. 2b,c, orange line). These small orders may be due to the result of a background reduction of water into dihydrogen41. Based on these results, we deduced that only the concentration of the molybdenum catalyst affected the second-order reaction rate at a low concentration of the catalyst. Thus, at a low catalyst concentration, the dimerization of the molybdenum complexes accounts for the rate-determining step (Fig. 1e).
Further, we measured the kinetic isotope effect of the catalytic reaction with H2O or D2O in both concentration regions. A kH/kD value of 2.61 was observed in region A; this value confirmed that the PCET process was the rate-determining step36,37,38. Conversely, a kH/kD value of 1.42 was observed in region B, although no kinetic isotope effect was predicted by the kinetic study because of the lack of a relationship between the proton sources and the dimerization step of the catalyst as the rate-determining step. Although the details are unclear, the kH/kD differences between the two regions are consistent with a change in the rate-determining step at low and high catalyst concentrations.
Density functional theory calculations of the bond dissociation free energies
Next, we employed two strategies, based on the kinetic study (Fig. 2), to improve the catalytic activity of the system by tuning the pincer ligand of the molybdenum catalysts. The first strategy involved accelerating the PCET process in the presence of a high concentration of the catalyst, and the other involved accelerating the dimerization process in the presence of a low concentration of the catalyst. Here we focused on adding hydrogen atoms to the nitride complex (2a) to produce an ammine complex (IIIa) via PCET, after which we computationally predicted the influence of introducing the substituents to the pincer ligand of 2a on the thermodynamic stability of the hydrogenated intermediates. We previously reported that the nitrogen-fixing activity of the molybdenum–dinitrogen complexes that bear PNP-type pincer ligands could be tuned by introducing electron-donating and withdrawing groups to the pincer ligands42,43.
First, we performed the DFT calculations at the B3LYP-D3 theory level44,45,46,47,48 to evaluate the bond dissociation free energies (BDFEs) of the N–H bonds in three possible reaction intermediates, beginning with 2a, namely the imide (Ia), amide (IIa) and ammine (IIIa) complexes. BDFEs were calculated according to the reaction [MoI(NHx)(PCP)] → [MoI(NHx−1)(PCP)] + H (x = 1–3); therefore, [MoI(NHx)(PCP)] lost a proton and an electron simultaneously. Chirik and co-workers utilized the BDFEs of N–H bonds to discuss the photoinduced PCET-driven reductions of different transition-metal–nitrogen complexes, such as manganese–nitride49,50, titanium–amide51 and cobalt–imide complexes51. In our previous study on the molybdenum-catalysed nitrogen fixation reaction that employed [Mo(N2)(PMePh2)4], the BDFE of the N–H bond of a molybdenum–diazenide (Mo–NNH) complex was employed to evaluate the hydrogen atom affinity52. Table 1 lists the calculated BDFE(N–H) values of the three intermediates. The calculated BDFE(N–H) values of [MoI(NHx)(PCP)] were 33.8, 52.7 and 41.2 kcal mol−1 for the imide (x = 1), amide (x = 2) and ammine (x = 3) complexes, respectively. All the calculated BDFE values were comparable with or higher than that of the O–H bond (34 kcal mol−1) of water bound to a Sm(II) centre in THF41; thus, the successive addition of hydrogen atoms to 2a employing H2O in the presence of SmI2 was thermodynamically reasonable53. The smallest BDFE(N–H) value (that of the imide complex Ia) indicated that the addition of the first hydrogen atom (protonation and reduction) was the most energetically unfavourable process in the transformation of the nitride N atom of 2a into ammonia.
Next, we investigated the influence of the introduction of substituents to the benzimidazole skeleton of the PCP ligand on the BDFEs of [MoI(NHx)(PCPR1,R2)] (x = 1–3), where R1 and R2 are the substituents at positions 5 and 6 in the benzimidazole skeleton. To obtain the trends in a wide range of the Hammett substituent constants (σp) (ref. 54), we examined one electron-donating methyl group (σp = –0.17) and three electron-withdrawing groups, namely Cl, F and CF3 with σp values of 0.06, 0.23 and 0.54, respectively. For the CF3 group, its 5-substituted (R1 = CF3, R2 = H) and 5,6-substituted (R1 = R2 = CF3) PCP ligands were considered. Table 1 summarizes the BDFE(N–H) values of [MoI(NHx)(PCPR1,R2)] (x = 1–3). The BDFE(N–H) values of all the substituted complexes exhibited the same trend as those of the unsubstituted ones, in the following order: imide (x = 1) < ammine (x = 3) < amide (x = 2). Therefore, the following discussion focuses on the addition of hydrogen atoms to the nitride complexes to yield the corresponding imide complexes, as this step was the most energetically unfavourable.
Based on the BDFE(N–H) results, we examined how the substituted PCP ligands modified the electronic structure of [MoI(N)(PCP)] 2a. Figure 3a plots the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of 2a and 5,6-substituted [MoI(N)(PCPR1,R2)] (2b–2d, 2f) versus the σp values. Both molecular orbital energy levels correlated with the Hammett substituent constant, and a strong electron-withdrawing substituent, such as –CF3, lowered (or stabilized) the LUMO energy of the nitride complex more effectively than it did the HOMO energy. The lowering of the LUMO energy of 2a is expected to improve the PCET reaction to yield the corresponding imide complex, as it will enhance the electron affinity of 2a.

a, Energy plots of the LUMO (red) and HOMO (blue) energies of 2a and the 5,6-substituted nitride complexes (2b–2d and 2f) versus the Hammett substituent constant σp. b, Spatial distribution of the HOMO and LUMO of 2a. c, Mayer bond orders of the Mo–C(carbene) of Ia and the 5,6-substituted imide complexes (Ib–Id and If) versus Hammett substituent constant σp. d, Spatial distribution and schematic drawings of frontier orbitals of Ia that contribute to the σ donation from the pincer ligand to Mo (α-HOMO-10) and the π backdonation from Mo to the pincer ligand (α-HOMO). MO, molecular orbital.
NHCs coordinated to a transition-metal centre are known to serve not only as a strong σ-donor but also as a π acceptor29,55,56. As shown in Fig. 3b, the spatial distribution of the LUMO of 2a delocalizes over the Mo≡N moiety as well as the benzimidazole moiety of the PCP ligand. These moieties are connected through an overlap between the dzx orbital of Mo and the pz orbital of the carbene C atom of the PCP ligand. Therefore, the electron-donating and withdrawing substituents can affect the strength of the Mo–C(carbene) bond of the substituted imide complexes [MoI(NH)(PCPR1,R2)] generated by the PCET to give [MoI(N)(PCPR1,R2)]. Figure 3c plots the Mayer bond orders57 of the imide complexes Ia–Id and If versus the σp values. The strength of the Mo–C(carbene) bond is in good correlation with the σp values, and a strong electron-withdrawing substituent, such as –CF3, can strengthen the connection between the Mo centre and the PCP ligand. As depicted in Fig. 3d, the α-HOMO of 1a in the doublet spin state, which corresponds to the LUMO of 2a, is responsible for the π backdonation from the singly occupied dzx orbital of Mo to the vacant pz orbital of the carbene C atom. We previously reported that this unique π-accepting ability of the NHC-based PCP ligand contributed to the high thermodynamic stability of [{Mo(0)(N2)2(PCP)}2(μ-N2)] as an N2-fixing catalyst29,58, Therefore, we can expect that the use of the substituted PCP ligands improves the catalytic activity of the present Mo–PCP system for nitrogen fixation.
In summary, DFT calculations predicted that the introduction of strong electron-withdrawing substituents, such as –CF3, to the PCP ligand would effectively lower the LUMO energy level. This increases the electron affinity of 2a, which is essential to accelerate the rate-determining N–H bond formation reactions via PCET. In addition, the introduction of the electron-withdrawing substituents enhances the π-accepting ability of the PCP ligand, which leads to a solid connection between the Mo centre and the PCP ligand through π backdonation to give an improvement in the catalytic activity for nitrogen fixation.
Properties of the molybdenum complexes that bear PCP ligands
Based on the DFT results, we designed PCP-type pincer ligands that bear different substituents at positions 5 and/or 6 of the benzimidazole ring (3a–3f), as well as their corresponding molybdenum trichloride complexes [MoCl3(R–PCP)] (1a–1f) (Fig. 4a,b, respectively). The electronic properties of these ligands and molybdenum complexes were estimated via NMR spectroscopy of the corresponding selenocarbene compounds of the PCP-type pincer ligands (4a–4f), the infrared spectroscopy of the corresponding molybdenum(0) tricarbonyl complexes that bear the same PCP-type pincer ligands, [Mo(CO)3(R–PCP)] (6a–6f)59,60 and the cyclic voltammetry of the isolated molybdenum(IV)–nitride complexes [Mo(N)I(R-PCP)] (2a and 2e). The chemical shift of the 77Se NMR spectra of the selenocarbene compounds generally reveals the π-backdonation ability of the corresponding carbene ligands54,61; the CO-stretching frequency of the carbonyl complexes measured via infrared spectroscopy reveals all the electronic properties of the pincer ligands, which includes those of carbene and two phosphine moieties. The redox potentials measured by cyclic voltammetry of the molybdenum(IV)–nitride complexes provide direct information on the electronic properties with regard to reactive intermediates.

a, Structures of the PCP-type pincer ligands that bear different substituents (3a–3f) and the corresponding selenocarbene compounds (4a–4f). b, Preparation of the molybdenum tricarbonyl and trichloro complexes that bear the PCP-type pincer ligands. aReported data29.
First, we prepared the selenocarbene compounds (4a–4f) via the reactions of the corresponding carbene ligands with elemental selenium powder (see Supplementary Section 2 for the details) and performed 77Se NMR spectroscopy of the corresponding selenocarbene compounds in THF-d8 (Fig. 4a). The chemical shifts of the selenocarbene compounds, which were identified as selenium atoms that were bonded to carbon atoms, are listed in Table 2. The 77Se NMR spectroscopy of the selenocarbene compounds revealed that the enhanced π backdonation contributes to the paramagnetic shielding term with a substantial downfield 77Se-NMR signal54,61. The shift of the selenocarbene compounds that introduced two methyl groups as the electron-donating groups (4b, 135.2 ppm) was more upfield than that for the non-substituted selenocarbene compound (4a, 147.1 ppm). Contrarily, the introductions of the fluoro- (4c, 170.2 ppm), chloro- (4d, 177.3 ppm) and trifluoromethyl (4e and 4f with one and two CF3 groups, respectively, 174.3 and 196.6 ppm) groups as the electron-withdrawing groups facilitated downfield shifts. These results demonstrated that the π-backdonation ability of the carbene moiety was effectively tuned via the introduction of substituent groups in positions 5 and/or 6 of the benzimidazole ring, as predicted by DFT.
Next, the molybdenum tricarbonyl complexes that bear the PCP-type pincer ligands, [Mo(CO)3(R-PCP)] (6a–6f) were synthesized (Fig. 4b). The treatment of the pincer ligands (3a–3f), which were generated in situ via the reactions of the precursors of the PCP-pincer ligands (5a–5f) with 1.4 equiv. potassium hexamethyldisilazide (KN(SiMe3)2) in toluene for one hour at room temperature and 0.9 equiv. [Mo(CO)3(η6–C6H5CH3)] in toluene for 18 hours at 80 °C yielded the corresponding molybdenum tricarbonyl complexes that bear the PCP-type pincer ligands, [Mo(CO)3(R-PCP)] (6a–6f), in yields of 8–26%. X-ray analysis confirmed that the molecular structures of these complexes were almost the same (Supplementary Figs. 9–14). We measured the CO-stretching frequencies of 6a–6f in a THF solution. Two signals, which corresponded to the CO-stretching frequency, were observed in all the complexes. A slightly smaller wavenumber of the CO-stretching frequency of 6b, which introduced two methyl groups (1,831 and 1,935 cm−1) as the electron-donating groups, than that of the non-substituted complex (6a, 1,833 and 1,936 cm−1) was obtained. Conversely, the complexes that contained the fluoro-, chloro- and trifluoromethyl groups as electron-withdrawing groups exhibited higher wavenumbers (6c, 1,836 and 1,939 cm−1; 6d, 1,838 and 1,940 cm−1; 6e, 1,837 and 1,940 cm−1 and 6f, 1,842 and 1,942 cm−1). These infrared results indicated that the tendency of the effect of the substituent groups was consistent with that observed by NMR spectroscopy for the selenocarbene compounds (4), although we observed only a small difference in the CO-stretching frequencies of the molybdenum(0) tricarbonyl complexes (6) by the introduction of the substituent groups to positions 5 and/or 6 of the benzimidazole ring.
To obtain more detailed and direct information on the electronic property with regard to reactive intermediates, we measured the cyclic voltammetry of the molybdenum(IV)–nitride complex [Mo(N)I(PCP)] (2a). In this case, an irreversible reduction wave was observed at −2.85 V versus FeCp2+/0, which was estimated by differential pulse voltammetry measurements. However, in the case of the molybdenum(IV)–nitride complex that bear a CF3-substituted PCP-type pincer ligand, [Mo(N)I(CF3-PCP)] (2e), the comparable reduction potential shifted to −2.63 V versus FeCp2+/0 (Supplementary Fig. 1). These experimental results indicated that the introduction of electron-withdrawing groups, such as a CF3 group at the 5-position in the PCP-type pincer ligand, substantially contributed to the electron-deficient molybdenum centre at the molybdenum(IV)–nitride complex. Thus, the electronic properties of the substituent groups that were introduced to positions 5 and/or 6 of the benzimidazole ring strongly affected the overall electronic properties of the pincer ligands.
We prepared molybdenum trichloro complexes that bear the PCP-type pincer ligands [MoCl3(R-PCP)] (1a–1f) (Fig. 4b). The treatments of the pincer ligands 3c–3f, which were generated in situ from the reactions of the precursors of the PCP-type pincer ligands 5c–5f, respectively, with 0.9 equiv. [MoCl3(THF)3] in toluene for 18 hours at 80 °C, afforded the corresponding molybdenum trichloro complexes that bear the PCP-type pincer ligands [MoCl3(R-PCP)] (1c–1f) in 13–60% yields. The molecular structures of 1c–1f were confirmed by X-ray analysis (Supplementary Figs. 2–5). These complexes (1c–1f) exhibited solution magnetic moments of 3.8, 3.5, 3.8 and 3.9 μB, respectively, which indicates that these complexes were characterized by the S = 3/2 spin-state (3.87 μB) (see Supplementary Section 2 for the details).
Catalytic activities of the molybdenum PCP complexes
We examined the catalytic activities of 1a–1f in the catalytic reduction of dinitrogen into ammonia by investigating the degree of progress of the catalytic reaction. The amount of ammonia produced from the reaction of dinitrogen with 28,800 equiv. SmI2 as the reductant and 28,800 equiv. H2O as the proton source in the presence of a catalytic amount (25 nmol) each of 1a–1f in THF at 25 °C was measured in the reaction time range between five minutes and four hours (Fig. 5a). The time profiles of the amounts of ammonia in the catalytic reaction are shown in Fig. 5b,c. The amount of ammonia decreased significantly when a complex that bear two methyl groups, 1b, was employed as the electron-donating group. Conversely, the utilization of complexes that bear the electron-withdrawing groups (1c–1f), as well as 1a, accelerated the reaction in the early stage. The turnover number (TON) values, as calculated by the amount of ammonia based on the Mo atom in the reaction after 60 minutes, are presented in Table 2. Notably, the TON tended to increase as the electron-withdrawing strength of the substituents at positions 5 and/or 6 of the benzimidazole ring increased; maximum values were obtained in the presence of 1e and 1f as the catalysts. However, the TOFs, as calculated by the amount of ammonia based on the Mo atom in the reaction within five minutes (Table 2), slightly decreased when the complex (1f), which exhibited a stronger electron-withdrawing property than that of 1e was utilized. Conversely, the amount of hydrogen gas produced as a by-product in this catalytic reaction system exhibited an inverse trend against the produced amount of ammonia when employing the catalysts (Fig. 5d). Thus, the amount of hydrogen gas increased significantly when the complex that bear two methyl groups was employed as the electron-donating groups (1b). The experimental results (Fig. 5) indicated that the DFT-based predictions of the designed molybdenum complexes as to the LUMO energies of the corresponding molybdenum–nitride complexes availed an efficient method to develop very effective molybdenum complexes as catalysts for the production of ammonia under ambient reaction conditions. In the present reaction system, the rate of ammonia formation decreased over time as reactants, such as SmI2 and H2O, were consumed after the start of the reaction. As a result, we consider that the reason for the decrease in the rate of ammonia formation over time is due to the consumption of reactants such as SmI2 and H2O. Thus, we do not believe that the reason is due to the effect of the produced ammonia and dihydrogen or the deactivation of the catalyst.

a, Reaction conditions of the catalytic production of ammonia for the comparison of the catalytic activities of 1a–1f. b, Time profiles of the amounts of ammonia produced from the catalytic production of ammonia catalysed by 1a–1f. c, The data of panel b for the amounts of the ammonia (equiv. based on the Mo atom of the catalyst) that were produced by 1a–1f. Data are means of multiple individual experiments (two to four times) with error bars based on s.d. d, Time profiles of the amounts of dihydrogen produced during the catalytic production of ammonia catalysed by 1a–1f as a by-product. e, Catalytic reaction of dinitrogen (1 atm) with high amounts of SmI2 and H2O as the reductant and proton source, respectively.
Finally, we conducted the catalytic reaction in the presence of 1e, which exhibited the highest TON and TOF values among the prepared catalysts under the present reaction conditions higher amounts of the reductant and a proton source were employed (Fig. 5e). The reaction of dinitrogen (1 atm) with 230,000 equiv. each of SmI2 and H2O as the reductant and proton source, respectively, in the presence of a catalytic amount (25 nmol) of 1e in THF for 72 hours at 25 °C was performed by adding the reductant and proton source in three portions. Therefore, 60,000 equiv. ammonia was obtained based on the Mo atom of the catalyst (with a yield of 78% based on SmI2). This very high value is ~14 times larger than our previously reported one (4,350 equiv. ammonia)30, and the highest achieved among the catalytic activities of reported reactions that employed transition metal complexes as the catalysts.
Conclusion
In summary, we confirmed that molybdenum trichloride complexes bearing a trifluoromethyl-substituted PCP-type pincer ligand functioned as the most effective catalyst for the catalytic production of ammonia from dinitrogen under ambient reaction conditions based on the mechanistic insight afforded by the experimental results, as well as on the prediction of the DFT calculations on the reactive intermediates. In this novel reaction system, up to 60,000 equiv. ammonia were produced based on the Mo atom of the catalyst, demonstrating a TOF of up to 800 equiv. Mo min−1. The amount of the ammonia produced in this study, as well as the production rate, were approximately one order of magnitude higher than those observed under previous reaction conditions30.
As described in here, we consider that the use of the BDFE(N–H) values of key reactive intermediates, such as molybdenum–imide, molybdenum–amide and molybdenum–ammine complexes estimated by DFT calculations provides a suitable and reliable predicting method to develop more effective catalysts under ambient reaction conditions. The catalysts successfully developed will be applied not only to the catalytic nitrogen fixation driven by visible light62 and electrochemical energy, but also to the catalytic formation of nitrogen-containing organic compounds directly from nitrogen gas under mild reaction conditions63. We believe that these findings can contribute to the development of an environmentally friendly next-generation nitrogen-fixation system in the near future.
Methods
In a 50 ml Schlenk flask was placed a CH2Cl2 solution of 1e (0.05 mM, 500 μl, 25 nmol) and the solvent was removed under reduced pressure. To the flask were added SmI2(THF)2 (1.05 g, 1.92 mmol) and THF (6 ml) under N2. Then a THF solution (1 ml) that contained H2O (1.92 mmol) was added to the stirred solution in the Schlenk flask in one portion. After the addition of the water, the mixture was further stirred at 25 °C for 24 h. This procedure was then repeated twice (in total, SmI2(THF)2 (1.92 mmol × 3), H2O (1.92 mmol × 3), 24 h × 3). Aqueous potassium hydroxide solution (30 wt%, 5 ml) was added to the reaction mixture. The mixture was evaporated under reduced pressure, and the distillate trapped in a dilute H2SO4 solution (0.5 M, 10 ml). The amount of ammonia (1.42 mmol, 56,800 equiv. based on the molybdenum atom, a 74% yield based on SmI2(THF)2) present in the H2SO4 solution was determined by the indophenol method.
Data availability
Source data are provided with this paper. The crystallographic data for 1c·CH2Cl2, 1d·0.5CH2Cl2, 1e, 1f, [Mo(N)I(PCP)]OTf, 2a, 2e, 6a, 6b, 6c, 6d, 6e·C4H4O, and 6f have been deposited with the Cambridge Crystallographic Data Centre under accession numbers 2069962, 2069963, 2069964, 2069965, 2069966, 2069967, 2222890, 2069968, 2069969, 2069970, 2069971, 2069972 and 2069973, respectively. The data supporting the findings of the current study are available within the paper and its Supplementary Information.
References
Mineral Commodity Summaries 2021 (US Geological Survey, 2021).
Liu, H. Ammonia Synthesis Catalysts: Innovation and Practice (World Scientific, 2013).
Boerner, L. K. Taking the CO2 out of NH3. Chem. Eng. News 97, 18–21 (2019).
Foster, S. L. et al. Catalysts for nitrogen reduction to ammonia. Nat. Catal. 1, 490–500 (2018).
Valera-Medina, A., Xial, H., Owen-Jones, M., David, W. I. F. & Bowen, P. J. Ammonia for power. Prog. Energy Combust. Sci. 69, 63–102 (2018).
Guo, J. & Chen, P. Catalyst: NH3 as an energy carrier. Chem 3, 709–712 (2017).
Ye, L., Nayak-Luke, R., Bañares-Alcántara, R. & Tsang, E. Reaction: ‘green’ ammonia production. Chem 3, 712–714 (2017).
Service, R. F. Ammonia—a renewable fuel made from Sun, air, and water—could power the globe without carbon. Science https://doi.org/10.1126/science.aau748 (2018).
Marakatti, V. S. & Gaigneaux, E. M. Recent advances in heterogeneous catalysis for ammonia synthesis. Chem. Cat. Chem. 12, 5838–5857 (2020).
Hosono, H. & Kitano, M. Advances in materials and applications of inorganic electrides. Chem. Rev. 121, 3121–3185 (2021).
Hattori, M., Iijima, S., Nakao, T., Hosono, H. & Hara, M. Solid solution for catalytic ammonia synthesis from nitrogen and hydrogen gases at 50 °C. Nat. Commun. 11, 2001 (2020).
Ye, T.-N. et al. Vacancy-enabled N2 activation for ammonia synthesis on an Ni-loaded catalyst. Nature 583, 391–395 (2020).
Yandulov, D. V. & Schrock, R. R. Catalytic reduction of dinitrogen to ammonia at a single molybdenum center. Science 301, 76–78 (2003).
Nishibayashi, Y. Nitrogen Fixation (Topics in Organometallic Chemistry Vol. 60) (Springer, 2017).
Chalkley, M. J., Drover, M. W. & Peters, J. C. Catalytic N2‑to-NH3 (or -N2H4) conversion by well-defined molecular coordination complexes. Chem. Rev. 120, 5582–5636 (2020).
Masero, F., Perrin, M. A., Dey, S. & Mougel, V. Dinitrogen fixation: rationalizing strategies utilizing molecular complexes. Chem. Eur. J. 27, 3892–3928 (2021).
Tanabe, Y. & Nishibayashi, Y. Comprehensive insights into synthetic nitrogen fixation assisted by molecular catalysts under ambient or mild conditions. Chem. Soc. Rev. 50, 5201–5242 (2021).
Anderson, J. S., Rittle, J. & Peters, J. C. Catalytic conversion of nitrogen to ammonia by an iron model complex. Nature 501, 84–87 (2013).
Hill, P. J., Doyle, L. R., Crawford, A. D., Myers, W. K. & Ashley, A. E. Selective catalytic reduction of N2 to N2H4 by a simple Fe complex. J. Am. Chem. Soc. 138, 13521–13524 (2016).
Fajardo, J. Jr. & Peters, J. C. Catalytic nitrogen-to-ammonia conversion by osmium and ruthenium complexes. J. Am. Chem. Soc. 139, 16105–16108 (2017).
Doyle, L. R. et al. Catalytic dinitrogen reduction to ammonia at a triamidoamine–titanium complex. Angew. Chem. Int. Ed. 57, 6314–6418 (2018).
Arashiba, K., Miyake, Y. & Nishibayashi, Y. A molybdenum complex bearing PNP-type pincer ligands leads to the catalytic reduction of dinitrogen into ammonia. Nat. Chem. 3, 120–125 (2011).
Kuriyama, S. et al. Catalytic transformation of dinitrogen into ammonia and hydrazine by iron–dinitrogen complexes bearing pincer ligand. Nat. Commun. 7, 12181 (2016).
Kuriyama, S. et al. Direct transformation of molecular dinitrogen into ammonia catalyzed by cobalt dinitrogen complexes bearing anionic PNP pincer ligands. Angew. Chem. Int. Ed. 55, 14291–14295 (2016).
Sekiguchi, Y. et al. Catalytic reduction of molecular dinitrogen to ammonia and hydrazine using vanadium complexes. Angew. Chem. Int. Ed. 57, 9064–9068 (2018).
Meng, F. et al. Ammonia formation catalyzed by dinitrogen-bridged dirhenium complex bearing PNP-pincer ligands under mild reaction conditions. Angew. Chem. Int. Ed. 60, 13906–13912 (2021).
Ashida, Y. et al. Catalytic reduction of dinitrogen into ammonia and hydrazine using chromium complexes bearing PCP-type pincer ligand. Chem. Eur. J. 28, e202200557 (2022).
Kuriyama, S. et al. Catalytic reduction of dinitrogen to ammonia and hydrazine using iron–dinitrogen complexes bearing anionic benzene-based PCP-type pincer ligands. Bull. Chem. Soc. Jpn 95, 683–692 (2022).
Eizawa, A. et al. Remarkable catalytic activity of dinitrogen-bridged dimolybdenum complexes bearing NHC-based PCP-pincer ligands toward nitrogen fixation. Nat. Commun. 8, 14874 (2017).
Ashida, Y., Arashiba, K., Nakajima, K. & Nishibayashi, Y. Molybdenum-catalysed ammonia production with samarium diiodide and alcohols or water. Nature 568, 536–540 (2019).
Arashiba, K. et al. Catalytic nitrogen fixation via direct cleavage of nitrogen–nitrogen triple bond of molecular dinitrogen under ambient reaction conditions. Bull. Chem. Soc. Jpn 90, 1111–1118 (2017).
Arashiba, K., Tanaka, H., Yoshizawa, K. & Nishibayashi, Y. Cycling between molybdenum–dinitrogen and –nitride complexes to support the reaction pathway for catalytic formation of ammonia from dinitrogen. Chem. Eur. J. 26, 13383–13389 (2020).
Warren, J. J., Tronic, T. A. & Mayer, J. M. Thermochemistry of proton-coupled electron transfer reagents and its implications. Chem. Rev. 110, 6961–7001 (2010).
Weinberg, D. R. et al. Proton-coupled electron transfer. Chem. Rev. 112, 4016–4093 (2012).
Miller, D. C., Tarantino, K. T. & Knowles, R. R. Proton-coupled electron transfer in organic synthesis: fundamentals, applications, and opportunities. Top. Curr. Chem. 374, 30 (2016).
Chciuk, T. V., Anderson, W. R. Jr & Flowers, R. A. II. Proton-coupled electron transfer in the reduction of carbonyls by samarium diiodide–water complexes. J. Am. Chem. Soc. 138, 8738–8741 (2016).
Chciuk, T. V., Anderson, W. R. Jr & Flowers, R. A. II. Interplay between substrate and proton donor coordination in reductions of carbonyls by SmI2–water through proton-coupled electron-transfer. J. Am. Chem. Soc. 140, 15342–15352 (2018).
Kolmar, S. S. & Mayer, J. M. SmI2(H2O)n reduction of electron rich enamines by proton-coupled electron transfer. J. Am. Chem. Soc. 139, 10687–10692 (2017).
Shaffer, D. W., Xie, Y., Szalda, D. J. & Concepcion, J. J. Manipulating the rate-limiting step in water oxidation catalysis by ruthenium bipyridine−dicarboxylate complexes. Inorg. Chem. 55, 12024–12035 (2016).
Xie, Y., Shaffer, D. W. & Concepcion, J. J. O−O radical coupling: from detailed mechanistic understanding to enhanced water oxidation catalysis. Inorg. Chem. 57, 10533–10542 (2018).
Bartulovich, C. O. & Flowers, R. A. II. Coordination-induced O–H bond weakening in Sm(II)–water complexes. Dalton Trans. 48, 16142–16147 (2019).
Kuriyama, S. et al. Catalytic formation of ammonia from molecular dinitrogen by use of dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing PNP-pincer ligands: remarkable effect of substituent at PNP-pincer ligand. J. Am. Chem. Soc. 136, 9719–1931 (2014).
Itabashi, T. et al. Effect of substituents on molybdenum triiodide complexes bearing PNP-type pincer ligands toward catalytic nitrogen fixation. Dalton Trans. 48, 3182–3186 (2019).
Becke, A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988).
Becke, A. D. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Vosko, S. H., Wilk, L. & Nusair, M. J. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 58, 1200–1211 (1980).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Phys. Chem. 132, 154104 (2010).
Wang, D., Loose, F., Chirik, P. J. & Knowles, R. R. N–H bond formation in a manganese(V) nitride yields ammonia by light-driven proton-coupled electron transfer. J. Am. Chem. Soc. 141, 4795–4799 (2019).
Loose, F. et al. Evaluation of excited state bond weakening for ammonia synthesis from a manganese nitride: stepwise proton coupled electron transfer is preferred over hydrogen atom transfer. Chem. Commun. 55, 5595–5598 (2019).
Park, Y. et al. Visible light enables catalytic formation of weak chemical bonds with molecular hydrogen. Nat. Chem. 13, 969–976 (2021).
Ashida, Y. et al. Molybdenum-catalyzed ammonia formation using simple monodentate and bidentate phosphines as auxiliary ligands. Inorg. Chem. 58, 8927–8932 (2019).
Boekell, N. G. & Flowers, R. A. II. Coordination-induced bond weakening. Chem. Rev. 122, 13447–13477 (2022).
Hansch, C., Leo, A. & Taft, R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 91, 165–195 (1991).
Verlinden, K., Buhl, H., Frank, W. & Ganter, C. Determining the ligand properties of N-heterocyclic carbenes from 77Se NMR parameters. Eur. J. Inorg. Chem. 2015, 2416–2425 (2015).
Vummaleti, S. V. C. et al. What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the N-accepting abilities of N-heterocyclic carbenes? Chem. Sci. 6, 1895–1904 (2015).
Mayer, I. Charge, bond order and valence in the ab initio SCF theory. Chem. Phys. Lett. 97, 270–274 (1983).
Egi, A., Tanaka, H., Konomi, A., Nishibayashi, Y. & Yoshizawa, K. Nitrogen fixation catalyzed by dinitrogen-bridged dimolybdenum complexes bearing PCP- and PNP-type pincer ligands: a shortcut pathway deduced from free energy profiles. Eur. J. Inorg. Chem. 2020, 1490–1498 (2020).
Nelson, D. J. & Nolan, S. P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 42, 6723–6753 (2013).
Huynh, H. V. Electronic properties of N‑heterocyclic carbenes and their experimental determination. Chem. Rev. 118, 9457–9492 (2018).
Liske, A., Verlinden, K., Buhl, H., Schaper, K. & Ganter, C. Determining the π‑acceptor properties of N‑heterocyclic carbenes by measuring the 77Se NMR chemical shifts of their selenium adducts. Organometallics 32, 5269–5272 (2013).
Ashida, Y. et al. Catalytic nitrogen fixation using visible light energy. Nat. Commun. 13, 7263 (2022).
Itabashi, T. et al. Direct synthesis of cyanate anion from dinitrogen catalysed by molybdenum complexes bearing pincer-type ligand. Nat. Commun. 13, 6161 (2022).
Acknowledgements
This project was supported by CREST, JST (JPMJCR1541). We acknowledge the Grants-in-Aid for Scientific Research (nos. JP20H05671, JP20K21203 and 22K19041) from JSPS and MEXT. Y.A. is a recipient of the JSPS Research Fellowships for Young Scientists. This paper is based on results obtained from project JPNP21020 commissioned by the New Energy and Industrial Technology Development Organization (NEDO).
Author information
Authors and Affiliations
Contributions
Y.N. and K.Y. conceived and designed this project. T.M., Y.A. and K.A. conducted the experimental work, which included the X-ray analysis. H.T. and A.E. conducted the theoretical studies. All the authors discussed the results and drafted the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y.A., K.A. and Y.N. have filed patents based on the research reported here (Japanese patent application nos. 2018-158595, 2018-036967 and 2018-036967 and international patent application JP2019/007793). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Peter Seavill, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental Details, Supplementary Sections 1–8, Figs. 1–124 and Tables 1–15.
Supplementary Data 1
Crystallographic Data for 1c, CCDC 2069962.
Supplementary Data 2
Crystallographic Data for 1d, CCDC 2069963.
Supplementary Data 3
Crystallographic Data for 1e, CCDC 2069964.
Supplementary Data 4
Crystallographic Data for 1f, CCDC 2069965.
Supplementary Data 5
Crystallographic Data for 2a, CCDC 2069967.
Supplementary Data 6
Crystallographic Data for 2e, CCDC 2222890.
Supplementary Data 7
Crystallographic Data for 6a, CCDC 2069968.
Supplementary Data 8
Crystallographic Data for 6b, CCDC 2069969.
Supplementary Data 9
Crystallographic Data for 6c, CCDC 2069970.
Supplementary Data 10
Crystallographic Data for 6d, CCDC 2069971.
Supplementary Data 11
Crystallographic Data for 6e, CCDC 2069972.
Supplementary Data 12
Crystallographic Data for 6f, CCDC 2069973.
Supplementary Data 13
Crystallographic Data for [Mo(N)I(PCP)]OTf, CCDC 2069966.
Supplementary Data 14
Coordinate file.
Supplementary Data 15
Source Data for Supplementary Figure 1.
Source data
Source Data Fig. 5
Source data for Figure 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ashida, Y., Mizushima, T., Arashiba, K. et al. Catalytic production of ammonia from dinitrogen employing molybdenum complexes bearing N-heterocyclic carbene-based PCP-type pincer ligands. Nat. Synth 2, 635–644 (2023). https://doi.org/10.1038/s44160-023-00292-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44160-023-00292-9
This article is cited by
-
Mechanistic study for efficient nitrogen fixation
Nature Synthesis (2023)
