
Abstract
Dissolved primary production released into seawater by marine phytoplankton is a major source of carbon fueling heterotrophic bacterial production in the ocean. The composition of the organic compounds released by healthy phytoplankton is poorly known and difficult to assess with existing chemical methods. Here, expression of transporter and catabolic genes by three model marine bacteria (Ruegeria pomeroyi DSS-3, Stenotrophomonas sp. SKA14, and Polaribacter dokdonensis MED152) was used as a biological sensor of metabolites released from the picoeukaryote Micromonas commoda RCC299. Bacterial expression responses indicated that the three species together recognized 38 picoeukaryote metabolites. This was consistent with the Micromonas expression of genes for starch metabolism and synthesis of peptidoglycan-like intermediates. A comparison of the hypothesized Micromonas exometabolite pool with that of the diatom Thalassiosira pseudonana CCMP1335, analyzed previously with the same biological sensor method, indicated that both phytoplankton released organic acids, nucleosides, and amino acids, but differed in polysaccharide and organic nitrogen release. Future ocean conditions are expected to favor picoeukaryotic phytoplankton over larger-celled microphytoplankton. Results from this study suggest that such a shift could alter the substrate pool available to heterotrophic bacterioplankton.
Similar content being viewed by others


Introduction
Phytoplankton are a main source of the bioavailable dissolved organic carbon (DOC) that supports heterotrophic marine bacteria. DOC can be released through phytoplankton exudation to maintain physiological balance, or through senescence, grazing, or viral lysis [1, 2]. Heterotrophic bacteria recycle this pool of labile dissolved compounds on time frames of hours to days [3,4,5], maintaining standing concentrations of individual metabolites in the nanomolar range or lower [5,6,7,8,9]. This presents substantial analytical challenges for studying phytoplankton metabolite utilization by heterotrophic bacteria. Untargeted chemical analyses have uncovered hundreds of thousands of organic features in surface ocean DOC [10], but very few have been identified as bacterioplankton substrates [11,12,13].
One important marine phytoplankton group contributing metabolites to the labile DOC pool of the surface ocean is the picophytoplankton. These small autotrophs have cell diameters ranging between 0.6 and 3 μm [14], and currently account for 10% to 40% of marine planktonic primary production [15, 16]. The most abundant of the eukaryotic picophytoplankton is the class Prasinophyceae (Chlorophyta) [14], and the three dominant genera in this group, Micromonas, Ostreococcus, and Bathycoccus, together represent more than 90% of picoeukaryote 18S rRNA sequences in ocean surveys [17]. Micromonas commoda, the focus of this study, is a cosmopolitan member of the Prasinophyceae [18] and is abundant in temperate coastal ecosystems [19,20,21]. Previous studies using chemical approaches have characterized the major carotenoids and lipids in this picophytoplankter [22] and the endometabolites of close relative Micromonas pusilla [23].
Our interest in characterizing M. commoda metabolites assimilated by heterotrophic bacteria stems from forecasted shifts in picoplankton community composition as the planet warms [24,25,26,27]. In fact, picophytoplankton are predicted to be favored in warmed seawater [24, 25, 27,28,29] due to the inverse relationship observed between temperature and phytoplankton cell size [25] and expectations that smaller cells are favored under low nutrient availability [16]. Experiments addressing effects of climate-relevant factors on picophytoplankton have shown significant positive growth responses to both increased temperature and seawater acidification [21, 30].
In this study we use model bacterial species as biological sensors of labile compounds released by the eukaryotic picophytoplankter M. commoda RCC299. Our approach identified cellular machinery for organic carbon acquisition that was transcriptionally activated by the bacteria when M. commoda served as the source of growth substrates. We focused mainly on changes in expression of bacterial transporters and catabolic genes, using gene annotations to generate hypotheses about the identity of picophytoplankton-derived metabolites. The experiments involved pairwise co-cultures in which M. commoda was the sole source of organic matter for each of three bacterial species selected to represent taxa commonly found associated with natural marine phytoplankton: Ruegeria pomeroyi DSS-3 (Rhodobacteraceae; isolated from Southeastern US coastal seawater), Stenotrophomonas sp. SKA14 (Gammaproteobacteria; isolated from Swedish coastal seawater), and Polaribacter dokdonensis MED152 (Flavobacteriales; isolated from the Mediterranean Sea). These same species were used in a previous study of Thalassiosira pseudonana CCMP1335 exometabolites (35), allowing a comparison of hypothesized organic matter release by the two phytoplankton.
Methods
Co-cultures
Axenic cultures of Micromonas commoda RCC299 (National Center for Marine Algae, NMCA) were grown in 1 L of organic-carbon free defined medium L1-Si [31] as modified by NCMA (https://ncma.bigelow.org/) at a salinity of 35 in 1900 mL vented polystyrene tissue culture flasks. Flasks were maintained at 18 oC under 16 h light at 160 μmol photons m−2s−1 and 8 h dark. Pre-cultures of Micromonas were sequentially upscaled (50 ml, 200 ml, 1 L) with transfers occurring during the exponential growth phase. After growing for 7 d (early stationary growth phase; ~2.7 × 106 cells ml−1), bacteria pre-grown in YTSS medium [32] were washed 5 times in sterile L1 medium at 6000 RCF and inoculated into the axenic cultures at ~106 cells ml−1. Replicate co-cultures were established for each bacterial strain and also for an axenic phytoplankton control. Bacterial contamination of the axenic cultures was ruled out based on lack of colony formation from culture aliquots spread onto YTSS plates and absence of bacterial-size particles in flow cytometry scattergrams (see below). After incubation in the light for 8 h to allow for transcriptional changes in response to available substrates, cells were collected on 0.2 μm pore-size 47 mm Supor filters (500 ml per filter). Filters were immediately flash frozen in liquid N2 and stored at −80 oC until processing. Three additional treatments were established with bacterial strains introduced individually into L1 medium with 400 μM C glucose as the sole carbon source (which supports all 3) at the same initial cell concentration as the co-cultures. As this treatment contained a single, known metabolite, it served as a control for co-culture transcriptome analysis.
Bacterial cell numbers were determined by flow cytometry. Samples were fixed at a final concentration of 1% glutaraldehyde, incubated at 4 °C for 20 min, and stored at −80 °C. Just prior to analysis, an internal standard of 5-μm fluorescent particles (ACFP-50-5; Spherotech, Lake Forest, IL, USA) was added, followed by staining for 15 min with SYBR Green I (final concentration 0.75X; Life Technologies, Waltham, MA, USA). Samples were analyzed on an Agilent Quanteon flow cytometer (Acea, Biosciences Inc, San Diego CA) with a 405 nm laser using a 530/30 bandpass filter for SYBR Green (bacteria) and a 695/40 bandpass filter for chlorophyll a (phytoplankton).
RNAseq analysis
RNA was extracted as in Ferrer-González et al. [33]. Briefly, filters were cut into pieces under sterile conditions and incubated at room temperature for 1 h in TE buffer, SDS (0.6% final concentration), and proteinase K (120 ng μl –1 final concentration), and extracted in equal volumes of acid phenol:chloroform:isoamyl alcohol (25:24:1) and then chloroform:isoamyl alcohol (24:1). Following centrifugation, the resulting supernatant was mixed with 1 volume of isopropanol and sheared by passage through a 21 G syringe needle. Samples were incubated overnight at −20 °C and then centrifuged. The pellet was resuspended in RNase-free water. Potential traces of DNA were removed using the Turbo DNA-free kit (Invitrogen, Waltham, MA, USA), and samples were tested for residual DNA by a 35-cycle PCR targeting the 16S rRNA gene of the strains (27F, 1492R primer set, temperature program: 30 s at 98 °C, 35 cycles of 30 s at 95 °C, 30 s at 50 °C, and 60 s at 72 °C, followed by 15 min at 72 °C). The DNA was processed for sequencing with the NEBNext rRNA Depletion Kit (E7860; New England Bio Labs, Ipswich, MA), modified to remove Micromonas and bacterial rRNA using a custom pool of 160 oligonucleotide probes targeting the 18S, 5.8S, and 28S rRNA genes and ITS regions (developed using the NEBNext Custom RNA Depletion Design Tool; https://depletion-design.neb.com) (Table S1) synthesized by Integrated DNA Technologies (Coralville, Iowa, USA) and incorporated into the depletion kit protocol according to manufacturer’s recommendations. Library preparation was carried out using the NEBNext Ultra II Directional Kit (E7765), and libraries were sequenced at the Georgia Genomics and Bioinformatics Core (Athens, GA, USA) on the NextSeq 2000 platform (SE100; Illumina, San Diego, CA, USA). Raw data were deposited in the NCBI SRA BioProject database under accession PRJNA787291, and data product files are available on Zenodo (https://doi.org/10.5281/zenodo.6812122).
Quality control of sequences was performed using the FASTX toolkit, imposing a minimum quality score of 20 over 80% of read length. Reads aligning to an in-house rRNA database (https://doi.org/10.5281/zenodo.6812122) were removed (SortMeRNA 2.1-GCCcore-8.3.0). Remaining reads were mapped to the R. pomeroyi DSS-3, Stenotrophomonas sp. SKA14, or P. dokdonensis MED152 genome (Bowtie 2) and counted (HTSeq) [34, 35] (NCBI RefSeq accession numbers ASM1196v2, ASM15857v1, and ASM15294v2, respectively), with 51% mapping. Genes with differential expression compared to the glucose controls were determined using DESeq2 [36], with significance after adjusting for multiple comparisons (padj) set at ≤0.01, with n = 2, 3, or 4. Annotations of the bacterial genomes were updated by comparison to closely related genomes in JGI Integrated Microbial Genomes and Microbiomes (IMG/M) [37], NCBI RefSeq [38], and Price et al. [39]. The dbCAN web resource was used for identification of carbohydrate-active enzyme annotations [40], considering results of HMMs, peptide pattern recognitions, and protein alignments (Tables S2–S4). For the M. commoda RCC299 genome, the gene model sets and functional annotations were obtained from the most recent assembly [41] through the JGI PhycoCosm genome browser [42], referenced against Worden et al. [43] and Guo et al. [44], and verified by BLAST alignments utilizing the JGI IMG database [37] (Tables S5–S7).
Results and discussion
Ecologically relevant metabolites released extracellularly by the picophytoplankter M. commoda RCC299 were postulated based on gene expression responses from three individually co-cultured marine heterotrophic bacteria. The Micromonas exometabolites accumulating during 7 d of axenic growth prior to inoculation were the sole carbon source for the bacterial strains. The bacteria represent three taxa commonly found associated with natural marine phytoplankton: Rhodobacteraceae (represented by R. pomeroyi), Gammaproteobacteria (Stenotrophomonas sp.), and Flavobacteriales (P. dokdonensis). These same species were used as biological sensors of Thalassiosira pseudonana CCMP1335 exometabolites in a previous study [33]. Bacterial gene expression was determined by sampling 8 h after inoculation and comparing expression when co-growing with the phytoplankter to that on a single metabolite control (400 μM C as glucose). Full information on gene expression is given in Tables S2–S7, but here we focus analysis on transporters and initial steps in catabolic pathways [45, 46], indicative of bacterial detection and utilization of organic compounds in the exometabolite pool, and on carbohydrate-active enzymes (CAZymes) [47], indicative of sugar and glycoconjugate catabolism. Thus, we interpreted bacterial gene expression changes as biological sensors that predict the composition of exometabolites being released from living Micromonas cells.
Ruegeria pomeroyi DSS-3 transcriptional responses
We found 513 of the 4332 R. pomeroyi protein-encoding genes significantly enriched in the Micromonas co-culture relative to the glucose control (2- to 216-fold; DeSeq2, padj ≤0.01), representing 12% of coding sequences in the bacterium’s genome (Table S2). Expression of transporters, catabolic genes diagnostic of initial degradation of known compounds, and CAZymes accounted for 103 of the significantly enriched R. pomeroyi genes (2.4% of all genes; Table 1). Twenty-seven transporter systems annotated for uptake of five chemical classes of organic compounds were among the differentially expressed genes. The classes included essential amino acids (leucine, isoleucine, valine, and alanine); organic sulfur compounds [dimethylsulfoniopropionate (DMSP), isethionate, taurine, and N-acetyltaurine]; organic acids (glycerol, lactate, and malate); carbohydrates (glycosides, galactosides; and glycerol-3-phosphate), and a nitrogen-containing osmolyte [trimethylamine N-oxide (TMAO)] (Fig. 1). Other predicted extracellular metabolites available to R. pomeroyi included polyamines, oligopeptides, nucleosides, and urea (Table 1). Seven of these compounds were found previously by direct chemical analysis in the endometabolome of close relative Micromonas pusilla: leucine, isoleucine, DMSP, malate, glycerol-3-phosphates, nucleosides, and polyamines [23]. Six were recently confirmed experimentally by analysis of R. pomeroyi transporter mutants [48] (Table 1).

Circles specify genes which enable transport (green) or catabolism (orange). Shaded ovals identify chemical classes of metabolites. Cobalamin (vitamin B12) was added to the growth medium (light gray font).
In some cases, enriched genes in diagnostic catabolic pathways supported the signals from transporters; they included genes for the catabolism of TMAO (dmmDABC), alanine (ald), DMSP (dmdACD, megL), taurine (tpa, xsc, pta), isethionate (iseJ), glycerol-3-phosphate (glpD) and urea (ureABD), many of which have been shown to be assimilated by R. pomeroyi [49,50,51,52,53]. In one case, the catabolic gene N-acetyltaurine amidohydrolase (naaS), specific for the catabolism of N-acetyltaurine, added a new compound to the suite of exometabolites hypothesized from transporter expression (Table 1). CAZyme genes enriched in the transcriptome indicated catabolism or modifications of peptidoglycan and its components (CAZyme annotations CE11, GH23, GH25, GT51; Table 1). The full biosynthesis pathway for peptidoglycan synthesis is present in the closely-related species M. pusilla CCMP1545 (although peptidoglycan itself has not been detected), while the M. commoda pathway appears incomplete [54]. Nonetheless, several genes present in the M. commoda genome could generate peptidoglycan intermediates (MURE, MRAY, MURG, and PBP). Finally, one R. pomeroyi CAZyme with higher relative expression was annotated for the oxidation of the osmolyte choline (AA3), which R. pomeroyi can use as a substrate [55].
In the glucose control, 219 R. pomeroyi genes were significantly enriched (2- to 75-fold; DeSeq2, padj ≤0.01, Table S2). As expected, enriched genes included those encoding glucose transport and metabolism to pyruvate via glycolysis and the Entner-Doudoroff pathway.
Stenotrophomonas sp. SKA14 transcriptional responses
We found 337 of the 4469 Stenotrophomonas sp. protein-encoding genes with significantly higher relative expression in the Micromonas culture relative to the glucose control (2- to 33-fold; DeSeq2, padj ≤0.01), representing 7.5% of coding sequences in the bacterium’s genome (Table S3). Expression of transporters, diagnostic catabolic genes, and CAZymes accounted for 20 of the significantly enriched genes (0.5% of all genes; Table 2). The Stenotrophomonas sp. genome contains 9 enriched organic compound transporters that do not yet have a definitive functional assignment. One is an enriched TonB-dependent transporter (TBDT), a component of a polysaccharide utilization locus-like (PUL-like) structure with a putative annotation for the degradation of α-glucans (Table 2). Others included a transporter that potentially mediates the uptake of nucleosides (nupC), and a transporter with a putative annotation for uptake of nucleobases xanthine and uracil. Putative osmolyte transporters with differential expression included a betaine-choline-carnitine transporter (BCCT) gene predicted for choline uptake (betT), and a major facilitator superfamily (MFS) transporter predicted for proline uptake (proP). Finally, two TBDT transporters with higher relative expression are annotated as potential cobalamin (vitamin B12) receptors. The Stenotrophomonas sp. genome does not contain genes for de novo B12 biosynthesis, and therefore the signal from these transporters may represent uptake from the culture medium. Like R. pomeroyi, enriched CAZymes in the Stenotrophomonas sp. transcriptome are indicative of modification or catabolism of peptidoglycan-like substrates (CBM50 + GH23, GH103, GH103, GH23, CBM50, GT2). Phenotypic studies of Stentrophomonas sp. SKA14 are not available, but other members of the genus, including beneficial plant-associated strains [56], share the ability to grow on peptidoglycan monomer N-acetylglucosamine and glucose [57].
In the glucose control, 358 genes were significantly enriched (2- to 19-fold; DeSeq2, padj ≤0.01; Table S3) compared to Stenotrophomonas in co-culture. Glucose transporter genes are not annotated in the Stenotrophomonas sp. genome, and catabolism of glucose through glycolysis and the pentose phosphate pathway did not stand out in the transcriptional data. We suspect that uptake and metabolism of α-1,4-glucan monomers released by Micromonas led to similar expression levels of the glucose transporter and glycolysis genes in both the co-culture and control transcriptomes.
Polaribacter dokdonensis MED152 transcriptional responses
We found 185 of the 2614 P. dokdonensis protein-encoding genes significantly enriched in the Micromonas co-culture relative to the glucose control (2- to 38-fold; DeSeq2, padj ≤0.01), representing 7% of coding sequences in the bacterium’s genome (Table S4). Expression of transporters, diagnostic catabolic genes, and CAZymes accounted for 11 of the significantly enriched genes (0.4% of all genes; Table 3). There were no transporter systems with annotations for uptake of organic compounds among the genes with significantly higher relative expression. There were five CAZymes enriched, however, including two in PUL3 (as designated in the CAZy database): a trehalase (GH37) for hydrolysis of the α-glucosidic linkages of α,α-trehalose, and an amylo-1,4 → 1,6-transglucosidase (GH13_18) that transfers a segment of a α-1,4-glucan chain. These two enzymes are suggestive of the presence of a α-1,4-glucan in the exometabolome of Micromonas. Members of the green algal lineage, like their true plant relatives, carry genes for carbon storage as starch, consisting of α-1,4-glucan units as amylose and amylopectin [58, 59]. In PUL5, enriched genes included an N-acetylglucosamine kinase. Two other CAZymes in this PUL, β-N-acetylhexosaminidase (GH20) and glucosamine-6-phosphate deaminase, had annotations for N-acetyl-D-glucosamine catabolism, although these were significant at a less stringent cutoff of padj ≤0.05 (Table 3). These results suggest that P. dokdonensis had access to N-acetylglucosamine, consistent with evidence from the other two bacteria that components of a peptidoglycan-like polymer were available in the Micromonas exometabolome. Other enriched CAZymes not located within PULs included a glycosyl transferase (GT2) and an endo-β-N-acetylglucosaminidase (GH163). The transcriptional patterns found here are consistent with previous findings of very low numbers of amino acid and sugar transporters in the P. dokdonensis genome, suggesting specialization for growth instead on polymeric substrates (polysaccharides and proteins) [60].
In the P. dokdonensis glucose control, 236 genes were significantly enriched (2- to 30-fold; DeSeq2, padj ≤0.01). These included the genes that encode glucose transport and metabolism via glycolysis (Table S4).
Micromonas commoda RCC299 transcriptional responses
Of the 10,240 Micromonas protein-encoding genes, 456 were significantly enriched in the presence of R. pomeroyi relative to the axenic control (DeSeq2, padj ≤0.01), with 78 unique to this co-culture. In the Stenotrophomonas sp. co-culture, 807 genes were significantly enriched, with 191 unique. In the P. dokdonensis co-culture, 873 genes were significantly enriched, with 501 unique (Fig. 2a; Tables S5, 6, 7). We identified five categories of enriched genes with potential relevance to the metabolite interactions inferred from the bacterial transcription: starch metabolism, synthesis of peptidoglycan-like intermediates, organic compound transport, thiamine synthesis, and nitrogen acquisition (Fig. 2b; Table S8). Seven enriched Micromonas genes function in starch metabolism. Two of these responded to the presence of all three bacteria: the debranching enzyme isoamylase (ISA1) that removes wrongly positioned branches during starch biosynthesis, and the α-amylase AMYA2 that metabolizes branched starch into linear glucans during breakdown [58] (Fig. 2). Other enriched starch-related enzymes were the glucan water dikinase (GWD2) which phosphorylates and catalyzes degradation of starch [58, 61, 62], two other α-amylases (AMYA3 and 6), a starch synthase (SSIIB) [58], and a 4-α-glucanotransferase (DPE) that breaks down linear glucans to maltose [58] (Table S8). These changes in relative gene expression suggest that Micromonas activated starch metabolism components such as amylopectin, and they complement the enriched bacterial catabolic genes for α-1,4-glucan degradation.

a Transcription was enriched for 1393 genes in Micromonas across all treatments. Boxed values indicate pair-wise comparisons of the genes enriched in both species as a percent of the genes enriched in only one or the other. Complete gene information and fold-change values are given in Tables S5, S6, and S7. Significance was determined based on padj values ≤0.01. b Transcription was enriched for 19 potential substrate-related genes in Micromonas, 10 of which were found for more than one co-cultured bacterium. Complete gene information and fold-change values are given in Table S8.
In all three bacterial co-cultures compared to the axenic culture, relative increases in Micromonas expression was observed for UDP-N-acetylmuramoylalanyl-D-glutamate-2,6-diaminopimelate ligase (murE), an enzyme in the green alga’s incomplete peptidoglycan-like biosynthesis pathway (Fig. 2b; Table S8). This is consistent with the enriched signals of peptidoglycan catabolism or modification in all three bacterial transcriptomes, but whether the substrates inducing the bacterial enzymes came from bacterial or phytoplankton metabolite pools could not be distinguished. A number of enriched Micromonas transporter proteins were annotated for targeting organic substrates. Their direction of transport is not known, however, and they could either import or export compounds. Predicted substrates of the transporters are purines, betaine/carnitine/choline, cholic acid, and urea (Table S8). All have been reported previously as components of marine phytoplankton endometabolomes [63,64,65], and all except cholic acid complement transcription signals by one or more of the co-cultured bacteria.
A subset of the Micromonas transporter genes enriched in the presence of bacteria were annotated for mediating the uptake or assimilation of exogenous compounds, potentially of bacterial origin (Table S8). A gene that encodes a TPP riboswitch regulating thiamine biosynthesis (UNK1) was enriched in the presence of Stenotrophomonas sp. and P. dokdonensis. Previous studies in M. commoda have shown this gene to be activated in response to thiamine depletion [66], and could indicate depletion in the co-culture. Correspondingly, an N-myristoyltransferase 1 (NMT1) [43] associated with thiamine biosynthesis was enriched in the presence of P. dokdonensis. Another key transcriptional signal consisted of 6 enriched genes related to nitrogen acquisition. The co-culture medium contained nitrate as the only nitrogen source, which Micromonas can use. Nonetheless, in the presence of two bacterial species (Stenotrophomonas sp. and P. dokdonensis) the picoeukaryote’s transcriptome was enriched for ammonium transporters, glutamate synthase GSN1 and glutamine synthetase GSIII (key components of the glutamine synthetase–glutamate synthase cycle or GS-GOGAT system for ammonium incorporation) [67, 68], and the aspartate aminotransferase ASP1 (which transaminates glutamate and oxaloacetate to 2-oxoglutarate and aspartate reversibly, depending on nitrogen availability) [67, 69]. The activation by M. commoda of these uptake and metabolism genes suggests that the bacteria induced a shift in nitrogen acquisition strategies that did not occur under axenic conditions.
Bacterial sensing of Micromonas versus Thalassiosira exometabolites
A previous study of metabolite availability with these same three bacteria in co-culture with Thalassiosira pseudonana CCMP1335 [33] allows for a comparison of the biologically-sensed substrates between a representative picoeukaryote and diatom (Tables S9, S10, S11). Transporters or catabolic genes that were induced in one or more bacterial species when growing on the exometabolome of each phytoplankter indicated overlap in composition for the metabolites glycerol, sn-glycerol-3-phosphate, lactate, nucleosides, urea, and proline. Three organic sulfur compounds were also predicted to be shared between the phytoplankton exometabolomes; these are DMSP, N-acetyltaurine, and an unidentified sulfonate.
Bacterial transcription signals also suggested that several metabolites were unique to the Micromonas metabolome. These included the organic sulfur compound isethionate, organic nitrogen compounds TMAO, choline, carnitine, and polyamines, and organic sulfur and nitrogen compound taurine. Expression signals unique to the Thalassiosira exometabolome were the organic sulfur compound 2,3-dihydroxypropane-1-sulfonate (DHPS) and the organic nitrogen compounds glutamine, N-acetyl-galactosamine, and peptides. Other unique Thalassiosira compounds suggested by bacterial expression were benzoate, citrate, β-1,3-glucans, and an alginate-like oligosaccharide.
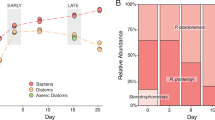
A comparison of bacterial cell numbers at the beginning and end of the experiment indicated that all three bacteria increased in abundance during the 8 h co-cultures with phytoplankton (T-test; p < 0.01) (Fig. 3). R. pomeroyi grew at a faster rate than the other bacteria in Micromonas exometabolites (μ = 0.10 h−1), while P. dokdonensis grew faster rate than the others in Thalassiosira exometabolites (μ = 0.32 h−1), emphasizing that exometabolite composition differentially affected bacterial species’ fitness relative to one another.

Replicates (n = 4) are plotted individually, and standard errors of the means are indicated. Specific growth rates (μ, in units of h−1) are estimated from the T0 and T8 cell numbers, assuming exponential growth. Pink symbols, Ruegeria pomeroyi DSS-3; blue symbols, Stenotrophomonas sp. SKA14; orange symbols, Polaribacter dokdonensis MED152. Initial phytoplankton exometabolite pools consisted of organic compounds released during 7 d of axenic culture (exponential through early stationary phase).
Conclusions
Identifying the specific compounds responsible for microbial carbon flux in surface ocean environments is challenging from a chemical analysis perspective because of their low concentrations, rapid uptake, and partitioning with sea salts in extraction protocols [70]. Here we used transcriptional signals of bacteria representative of phytoplankton-associated taxa to postulate molecules mediating microbial carbon flux. This approach assumes that the expression of bacterial transport or catabolism genes indicate the presence of their target metabolite, which is generally the case [33, 71]. Increased transporter expression in the absence of substrates can be energetically unfavorable for bacteria [72], and marine bacterial communities have been shown experimentally to respond to the availability of new substrates by increasing transporter expression [73,74,75]. However, caveats to this approach include posttranscriptional or constitutive gene regulation, and incorrect bacterial gene annotation (Table S1, S2, and S3) [76].
The Micromonas exometabolome is hypothesized to include at least 38 metabolites based on transcriptional regulation by the bacteria, many of which contained the heteroatoms nitrogen (55%), sulfur (13%), or phosphorus (8%). There was evidence of resource partitioning among the three heterotrophic bacteria, as was also found earlier for the Thalassiosira exometabolome [33]. For example, enrichment of transcripts for uptake of low molecular weight organic sulfur compounds was unique to R. pomeroyi, agreeing with previous findings that utilization of phytoplankton-derived sulfur metabolites is characteristic of marine Rhodobacteraceae [6, 51, 77]. In contrast, both Stenotrophomonas and P. dokdonensis had higher proportions of significant genes annotated for catabolism of α-glucans compared to R. pomeroyi.
Comparative analysis of hypothesized metabolite release by M. commoda and T. pseudonana based on gene expression patterns of the co-cultured bacteria indicated that Micromonas exometabolites overlapped with the diatom’s at the level of broad chemical categories (such as fatty acids, nucleosides, amino acids, and organic sulfur compounds), but differed in the specific compounds within those categories. For example, bacterial gene expression predicts that Micromonas is a more important source of the organic sulfur compounds taurine and isethionate and of the methylamines TMAO and choline, but a less important source of DHPS, glutamine, and oligosaccharides. Several of these metabolites are hypothesized to be used selectively by marine bacterial taxa; for instance, DHPS catabolism genes are concentrated in specific groups of marine Alphaproteobacteria that also have complete biosynthesis pathways for B12 [71], a vitamin which is required yet not synthesized by M. commonda based genome analysis and experimental data from close relative M. pusilla [78]. On the other hand, degradation pathways for diverse oligosaccharides are common in Flavobacteriia and Gammaproteobacteria [79, 80]. Continued increases in surface ocean temperature coupled to decreases in nutrient supply [16, 25] are predicted to favor smaller phytoplankton (e.g., picoeukaryotes such as Micromonas) over larger phytoplankton (i.e., microphytoplankton such as diatoms) [21, 30]. Our results suggest that phytoplankton community shifts will cascade to changes in the pool of microbial-derived metabolites, potentially selecting for different communities of associated heterotrophic bacteria. Given that the movement of carbon from phytoplankton to bacteria via the labile DOC pool is one of the largest annual organic carbon transfers in the ocean [70], future modifications to this flux may have important consequences to global biogeochemical cycles.
Data availability
Raw data are deposited in the NCBI SRA BioProject database under accession PRJNA787291, and data product files are available on Zenodo (https://doi.org/10.5281/zenodo.6812122).
References
Moran MA, Ferrer‐González FX, Fu H, Nowinski B, Olofsson M, Powers MA, et al. The ocean’s labile DOC supply chain. Limnol Oceanogr. 2022;67:1007–21.
Thornton DC. Dissolved organic matter (DOM) release by phytoplankton in the contemporary and future ocean. Eur J Phycol. 2014;49:20–46.
Simon M, Rosenstock B. Different coupling of dissolved amino acid, protein, and carbohydrate turnover to heterotrophic picoplankton production in the Southern Ocean in austral summer and fall. Limnol Oceanogr. 2007;52:85–95.
Suttle CA, Chan AM, Fuhrman JA. Dissolved free amino acids in the Sargasso Sea: uptake and respiration rates, turnover times, and concentrations. Mar Ecol Prog. 1991;70:189–99.
Zubkov MV, Tarran GA, Mary I, Fuchs BM. Differential microbial uptake of dissolved amino acids and amino sugars in surface waters of the Atlantic Ocean. J Plankton Res. 2008;30:211–20.
Durham BP, Boysen AK, Carlson LT, Groussman RD, Heal KR, Cain KR, et al. Sulfonate-based networks between eukaryotic phytoplankton and heterotrophic bacteria in the surface ocean. Nat Microbiol. 2019;4:1706–15.
Button D, Robertson BR, Lepp PW, Schmidt TM. A small, dilute-cytoplasm, high-affinity, novel bacterium isolated by extinction culture and having kinetic constants compatible with growth at ambient concentrations of dissolved nutrients in seawater. Appl Environ Microbiol. 1998;64:4467–76.
Lee C, Bada JL. Amino acids in equatorial Pacific Ocean water. EPSL. 1975;26:61–8.
Yamashita Y, Tanoue E. Chemical characteristics of amino acid-containing dissolved organic matter in seawater. Org Geochem. 2004;35:679–92.
Hertkorn N, Frommberger M, Witt M, Koch BP, Schmitt-Kopplin P, Perdue EM. Natural organic matter and the event horizon of mass spectrometry. Anal Chem. 2008;80:8908–19.
Becker JW, Berube PM, Follett CL, Waterbury JB, Chisholm SW, DeLong EF, et al. Closely related phytoplankton species produce similar suites of dissolved organic matter. Front Microbiol. 2014;5:1–14.
Bouslimani A, Sanchez LM, Garg N, Dorrestein PC. Mass spectrometry of natural products: current, emerging and future technologies. Nat Prod Rep. 2014;31:718–29.
Johnson WM, Longnecker K, Kido Soule MC, Arnold WA, Bhatia MP, Hallam SJ, et al. Metabolite composition of sinking particles differs from surface suspended particles across a latitudinal transect in the South Atlantic. Limnol Oceanogr. 2020;65:111–27.
Vaulot D, Eikrem W, Viprey M, Moreau H. The diversity of small eukaryotic phytoplankton (≤ 3 μm) in marine ecosystems. FEMS Microbiol Rev. 2008;32:795–820.
Raven J. The twelfth Tansley Lecture. Small is beautiful: the picophytoplankton. Funct Ecol. 1998;12:503–13.
Agawin NS, Duarte CM, Agustí S. Nutrient and temperature control of the contribution of picoplankton to phytoplankton biomass and production. Limnol Oceanogr. 2000;45:591–600.
Monier A, Worden AZ, Richards TA. Phylogenetic diversity and biogeography of the Mamiellophyceae lineage of eukaryotic phytoplankton across the oceans. Environ Microbiol Rep. 2016;8:461–9.
Simon N, Foulon E, Grulois D, Six C, Desdevises Y, Latimier M, et al. Revision of the genus Micromonas Manton et Parke (Chlorophyta, Mamiellophyceae), of the type species M. pusilla (Butcher) Manton & Parke and of the species M. commoda van Baren, Bachy and Worden and description of two new species based on the genetic and phenotypic characterization of cultured isolates. Protist. 2017;168:612–35.
Not F, Latasa M, Marie D, Cariou T, Vaulot D, Simon N. A single species, Micromonas pusilla (Prasinophyceae), dominates the eukaryotic picoplankton in the Western English Channel. Appl Environ Microbiol. 2004;70:4064–72.
Not F, Massana R, Latasa M, Marie D, Colson C, Eikrem W, et al. Late summer community composition and abundance of photosynthetic picoeukaryotes in Norwegian and Barents Seas. Limnol Oceanogr. 2005;50:1677–86.
Demory D, Baudoux A-C, Monier A, Simon N, Six C, Ge P, et al. Picoeukaryotes of the Micromonas genus: sentinels of a warming ocean. ISME J. 2019;13:132–46.
Marcellin-Gros R, Piganeau G, Stien D. Metabolomic insights into marine phytoplankton diversity. Mar Drugs. 2020;18:78.
Fiore CL, Alexander H, Soule MCK, Kujawinski EB. A phosphate starvation response gene (psr1-like) is present and expressed in Micromonas pusilla and other marine algae. Aquat Microb Ecol. 2021;86:29–46.
Hoegh-Guldberg O, Bruno JF. The impact of climate change on the world’s marine ecosystems. Science. 2010;328:1523–8.
Morán XAG, López-Urrutia Á, Calvo‐Díaz A, Li WK. Increasing importance of small phytoplankton in a warmer ocean. Glob Change Biol. 2010;16:1137–44.
Winder M, Sommer U. Phytoplankton response to a changing climate. Hydrobiologia. 2012;698:5–16.
Dutkiewicz S, Cermeno P, Jahn O, Follows MJ, Hickman AE, Taniguchi DA, et al. Dimensions of marine phytoplankton diversity. Biogeosciences. 2020;17:609–34.
Daufresne M, Lengfellner K, Sommer U. Global warming benefits the small in aquatic ecosystems. Proc Natl Acad Sci. 2009;106:12788–93.
Huertas IE, Rouco M, Lopez-Rodas V, Costas E. Warming will affect phytoplankton differently: evidence through a mechanistic approach. Proc Royal Soc B. 2011;278:3534–43.
Hoppe CJM, Flintrop CM, Rost B. The Arctic picoeukaryote Micromonas pusilla benefits synergistically from warming and ocean acidification. Biogeosciences. 2018;15:4353–65.
Berges JA, Franklin DJ, Harrison PJ. Evolution of an artificial seawater medium: improvements in enriched seawater, artificial water over the last two decades. J Phycol. 2001;37:1138–45.
González JM, Covert JS, Whitman WB, Henriksen JR, Mayer F, Scharf B, et al. Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int J Syst Evol Microbiol. 2003;53:1261–9.
Ferrer-González FX, Widner B, Holderman NR, Glushka J, Edison AS, Kujawinski EB, et al. Resource partitioning of phytoplankton metabolites that support bacterial heterotrophy. ISME J. 2021;15:762–73.
Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21.
Chen I-MA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, et al. The IMG/M data management and analysis system v. 6.0: new tools and advanced capabilities. Nucleic Acids Res. 2021;49:D751–D63.
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D45.
Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, et al. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature. 2018;557:503–9.
Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–101.
van Baren MJ, Bachy C, Reistetter EN, Purvine SO, Grimwood J, Sudek S, et al. Evidence-based green algal genomics reveals marine diversity and ancestral characteristics of land plants. BMC Genom. 2016;17:1–22.
Grigoriev IV, Hayes RD, Calhoun S, Kamel B, Wang A, Ahrendt S, et al. PhycoCosm, a comparative algal genomics resource. Nucleic Acids Res. 2021;49:D1004–D11.
Worden AZ, Lee J-H, Mock T, Rouzé P, Simmons MP, Aerts AL, et al. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science. 2009;324:268–72.
Guo J, Wilken S, Jimenez V, Choi CJ, Ansong C, Dannebaum R, et al. Specialized proteomic responses and an ancient photoprotection mechanism sustain marine green algal growth during phosphate limitation. Nat Microbiol. 2018;3:781–90.
Karp PD, Billington R, Caspi R, Fulcher CA, Latendresse M, Kothari A, et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief Bioinformatics. 2019;20:1085–93.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D5.
Schroer WF, Kepner HE, Uchimiya M, Mejia C, Rodriguez LT, Reisch CR, et al. Function and importance of marine bacterial transporters of plankton exometabolites. BioRxiv. 2023. https://doi.org/10.1101/2023.01.19.524783.
Cunliffe M. Purine catabolic pathway revealed by transcriptomics in the model marine bacterium Ruegeria pomeroyi DSS-3. FEMS Microbiol Ecol. 2016;92,:fiv150.
Denger K, Mayer J, Buhmann M, Weinitschke S, Smits THM, Cook AM. Bifurcated degradative pathway of 3-sulfolactate in Roseovarius nubinhibens ISM via sulfoacetaldehyde acetyltransferase and (S)-cysteate sulfolyase. J Bacteriol. 2009;191:5648–56.
Howard EC, Henriksen JR, Buchan A, Reisch CR, Bürgmann H, Welsh R, et al. Bacterial taxa that limit sulfur flux from the ocean. Science. 2006;314:649–52.
Lidbury I, Murrell JC, Chen Y. Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc Nat Acad Sci. 2014;111:2710–5.
Wirth JS, Wang T, Huang Q, White RH, Whitman WB. Dimethylsulfoniopropionate sulfur and methyl carbon assimilation in Ruegeria species. mBio. 2020;11:e00329–20.
Bachy C, Wittmers F, Muschiol J, Hamilton M, Henrissat B, Worden AZ. The land–sea connection: Insights into the plant lineage from a green algal perspective. Annu Rev Plant Biol. 2022;73:585–616.
Lidbury I, Kimberley G, Scanlan DJ, Murrell JC, Chen Y. Comparative genomics and mutagenesis analyses of choline metabolism in the marine Roseobacter clade. Environ Microbiol. 2015;17:5048–62.
Berg G, Egamberdieva D, Lugtenberg B, Hagemann M. Symbiotic plant–microbe interactions: stress protection, plant growth promotion, and biocontrol by Stenotrophomonas. In: Grube M, White J, Seckbach J, editors. Symbioses and stress: Springer; 2010. 445–60.
Coenye T, Vanlaere E, Falsen E, Vandamme P. Stenotrophomonas africana Drancourt et al. 1997 is a later synonym of Stenotrophomonas maltophilia (Hugh 1981) Palleroni and Bradbury 1993. Int J Syst Evol. 2004;54:1235–7.
Busi MV, Barchiesi J, Martín M, Gomez‐Casati DF. Starch metabolism in green algae. Starch‐Stärke. 2014;66:28–40.
Hedin N, Barchiesi J, Gomez-Casati DF, Iglesias AA, Ballicora MA, Busi MV. Identification and characterization of a novel starch branching enzyme from the picoalgae Ostreococcus tauri. Arch Biochem Biophys. 2017;618:52–61.
González JM, Fernández-Gómez B, Fernàndez-Guerra A, Gómez-Consarnau L, Sánchez O, Coll-Lladó M, et al. Genome analysis of the proteorhodopsin-containing marine bacterium Polaribacter sp. MED152 (Flavobacteria). Proc Nat Acad Sci. 2008;105:8724–9.
Mikkelsen R, Baunsgaard L, Blennow A. Functional characterization of alpha-glucan, water dikinase, the starch phosphorylating enzyme. Biochem J. 2004;377:525–32.
Glaring MA, Baumann MJ, Hachem MA, Nakai H, Nakai N, Santelia D, et al. Starch‐binding domains in the CBM45 family–low‐affinity domains from glucan, water dikinase and α‐amylase involved in plastidial starch metabolism. FEBS J. 2011;278:1175–85.
McLean C, Haley ST, Swarr GJ, Soule MCK, Dyhrman ST, Kujawinski EB. Harmful algal bloom-forming organism responds to nutrient stress distinctly from model phytoplankton. bioRxiv. 2021: https://doi.org/10.1101/2021.02.08.430350.
Boysen AK, Carlson LT, Durham BP, Groussman RD, Aylward FO, Ribalet F, et al. Particulate metabolites and transcripts reflect diel oscillations of microbial activity in the surface ocean. mSystems. 2021;6:1–18.
Allen AE, Dupont CL, Oborník M, Horák A, Nunes-Nesi A, McCrow JP, et al. Evolution and metabolic significance of the urea cycle in photosynthetic diatoms. Nature. 2011;473:203–7.
McRose D, Guo J, Monier A, Sudek S, Wilken S, Yan S, et al. Alternatives to vitamin B1 uptake revealed with discovery of riboswitches in multiple marine eukaryotic lineages. ISME J. 2014;8:2517–29.
McDonald SM, Plant JN, Worden AZ. The mixed lineage nature of nitrogen transport and assimilation in marine eukaryotic phytoplankton: a case study of Micromonas. Mol Biol Evol. 2010;27:2268–83.
Sanz-Luque E, Chamizo-Ampudia A, Llamas A, Galvan A, Fernandez E. Understanding nitrate assimilation and its regulation in microalgae. Front Plant Sci. 2015;6:1–17.
Gaufichon L, Rothstein SJ, Suzuki A. Asparagine metabolic pathways in Arabidopsis. Plant Cell Physiol. 2016;57:675–89.
Moran MA, Kujawinski EB, Schroer WF, Amin SA, Bates NR, Bertrand EM, et al. Microbial metabolites in the marine carbon cycle. Nat Microbiol. 2022;7:508–23.
Durham BP, Sharma S, Luo H, Smith CB, Amin SA, Bender SJ, et al. Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Nat Acad Sci. 2015;112:453–7.
Ferraris RP, Diamond JM. Specific regulation of intestinal nutrient transporters by their dietary substrates. Annu Rev Physiol. 1989;51:125–41.
McCarren J, Becker JW, Repeta DJ, Shi Y, Young CR, Malmstrom RR, et al. Microbial community transcriptomes reveal microbes and metabolic pathways associated with dissolved organic matter turnover in the sea. Proc Natl Acad Sci. 2010;107:16420–7.
Poretsky RS, Sun S, Mou X, Moran MA. Transporter genes expressed by coastal bacterioplankton in response to dissolved organic carbon. Environ Microbiol. 2010;12:616–27.
Landa M, Burns AS, Roth SJ, Moran MA. Bacterial transcriptome remodeling during sequential co-culture with a marine dinoflagellate and diatom. ISME J. 2017;11:2677–90.
Salzberg SL. Next-generation genome annotation: we still struggle to get it right. Genome Biol. 2019;20:1–3.
Moran MA, Durham BP. Sulfur metabolites in the pelagic ocean. Nat Rev Microbiol. 2019;17:665–78.
Helliwell KE, Wheeler GL, Leptos KC, Goldstein RE, Smith AG. Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Mol Biol Evol. 2011;28:2921–33.
Arnosti C, Wietz M, Brinkhoff T, Hehemann J-H, Probandt D, Zeugner L, et al. The biogeochemistry of marine polysaccharides: sources, inventories, and bacterial drivers of the carbohydrate cycle. Ann Rev Mar Sci. 2021;13:81–108.
Kirchman DL. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100.
Acknowledgements
The authors thank J. Nelson and W. Schroer for advice and assistance, and J. Pinhassi for bacterial strains. A. Worden provided the M. commoda strain and curated annotations.
Funding
This research was supported by National Science Foundation grants OCE-1948104 and OCE-2019589, and Simons Foundation grant 542391 within the Principles of Microbial Ecosystems (PriME) Collaborative to MAM. This is the NSF Center for Chemical Currencies of a Microbial Planet (C-CoMP) publication #016.
Author information
Authors and Affiliations
Contributions
FXF-G and MAM conceptualized and designed the study. FXF-G and MH generated and analyzed data, CBS prepared and analyzed samples, and JES and MO contributed to experimental design and sample collection and analysis. FXF-G and MAM wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferrer-González, F.X., Hamilton, M., Smith, C.B. et al. Bacterial transcriptional response to labile exometabolites from photosynthetic picoeukaryote Micromonas commoda. ISME COMMUN. 3, 5 (2023). https://doi.org/10.1038/s43705-023-00212-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43705-023-00212-0
