
Abstract
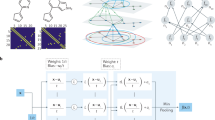
The marriage of density functional theory (DFT) and deep-learning methods has the potential to revolutionize modern computational materials science. Here we develop a deep neural network approach to represent the DFT Hamiltonian (DeepH) of crystalline materials, aiming to bypass the computationally demanding self-consistent field iterations of DFT and substantially improve the efficiency of ab initio electronic-structure calculations. A general framework is proposed to deal with the large dimensionality and gauge (or rotation) covariance of the DFT Hamiltonian matrix by virtue of locality, and this is realized by a message-passing neural network for deep learning. High accuracy, high efficiency and good transferability of the DeepH method are generally demonstrated for various kinds of material system and physical property. The method provides a solution to the accuracy–efficiency dilemma of DFT and opens opportunities to explore large-scale material systems, as evidenced by a promising application in the study of twisted van der Waals materials.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$99.00 per year
only $8.25 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
Source data are provided with this Paper. The dataset used to train the deep-learning model is available at Zenodo61.
Code availability
The code used in the current study is available at GitHub (https://github.com/mzjb/DeepH-pack) and Zenodo62.
References
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965).
Jones, R. O. Density functional theory: its origins, rise to prominence and future. Rev. Mod. Phys. 87, 897–923 (2015).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Jordan, M. I. & Mitchell, T. M. Machine learning: trends, perspectives and prospects. Science 349, 255–260 (2015).
Carleo, G. et al. Machine learning and the physical sciences. Rev. Mod. Phys. 91, 045002 (2019).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A. & Müller, K.-R. Schnet—a deep learning architecture for molecules and materials. J. Chem. Phys. 148, 241722 (2018).
Zhang, L., Han, J., Wang, H., Car, R. & E, W. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Gasteiger, J., Groß, J. & Günnemann, S. Directional message passing for molecular graphs. Proc. International Conference on Learning Representations (ICLR, 2020); https://openreview.net/forum?id=B1eWbxStPH
Unke, O. T. et al. SpookyNet: learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 12, 7273 (2021).
Brockherde, F. et al. Bypassing the Kohn-Sham equations with machine learning. Nat. Commun. 8, 872 (2017).
Grisafi, A. et al. Transferable machine-learning model of the electron density. ACS Cent. Sci. 5, 57–64 (2019).
Chandrasekaran, A. et al. Solving the electronic structure problem with machine learning. npj Comput. Mater. 5, 22 (2019).
Tsubaki, M. & Mizoguchi, T. Quantum deep field: data-driven wave function, electron density generation, and atomization energy prediction and extrapolation with machine learning. Phys. Rev. Lett. 125, 206401 (2020).
Grisafi, A., Wilkins, D. M., Csányi, G. & Ceriotti, M. Symmetry-adapted machine learning for tensorial properties of atomistic systems. Phys. Rev. Lett. 120, 036002 (2018).
Gu, Q., Zhang, L. & Feng, J. Neural network representation of electronic structure from ab initio molecular dynamics. Sci. Bull. 67, 29–37 (2022).
Schütt, K. T., Gastegger, M., Tkatchenko, A., Müller, K.-R. & Maurer, R. J. Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat. Commun. 10, 5024 (2019).
Unke, O. T. et al. SE(3)-equivariant prediction of molecular wavefunctions and electronic densities. In Proc. Advances in Neural Information Processing Systems (eds. Beygelzimer, A., Dauphin, Y., Liang, P. & Vaughan, J. W.) 14434–14447 (Curran Associates, 2021); https://openreview.net/forum?id=auGY2UQfhSu
Nagai, R., Akashi, R. & Sugino, O. Completing density functional theory by machine learning hidden messages from molecules. npj Comput. Mater. 6, 43 (2020).
Dick, S. & Fernandez-Serra, M. Machine learning accurate exchange and correlation functionals of the electronic density. Nat. Commun. 11, 3509 (2020).
Kirkpatrick, J. et al. Pushing the frontiers of density functionals by solving the fractional electron problem. Science 374, 1385–1389 (2021).
Mills, K. et al. Extensive deep neural networks for transferring small scale learning to large scale systems. Chem. Sci. 10, 4129–4140 (2019).
Zubatiuk, T. & Isayev, O. Development of multimodal machine learning potentials: toward a physics-aware artificial intelligence. Acc. Chem. Res. 54, 1575–1585 (2021).
Goedecker, S. Linear scaling electronic structure methods. Rev. Mod. Phys. 71, 1085–1123 (1999).
Hegde, G. & Bowen, R. C. Machine-learned approximations to density functional theory Hamiltonians. Sci. Rep. 7, 42669 (2017).
Thomas, N. et al. Tensor field networks: rotation- and translation-equivariant neural networks for 3D point clouds. Preprint at https://arxiv.org/abs/arXiv:1802.08219 (2018).
Anderson, B., Hy, T. S. & Kondor, R. Cormorant: covariant molecular neural networks. In Proc. Advances in Neural Information Processing Systems Vol. 32 (eds. Wallach, H. et al.) 14537–14546 (Curran Associates, 2019); https://proceedings.neurips.cc/paper/2019/file/03573b32b2746e6e8ca98b9123f2249b-Paper.pdf
Fuchs, F. B., Worrall, D. E., Fischer, V. & Welling, M. SE(3)-transformers: 3D roto-translation equivariant attention networks. In Proc. Advances in Neural Information Processing Systems Vol. 33 (eds. Larochelle, H., Ranzato M., Hadsell, R., Balcan, M. F. & Lin, H.) 1970–1981 (Curran Associates, 2020); https://proceedings.neurips.cc/paper/2020/file/15231a7ce4ba789d13b722cc5c955834-Paper.pdf
Martin, R. M. Electronic Structure: Basic Theory and Practical Methods (Cambridge Univ. Press, 2004); https://doi.org/10.1017/CBO9780511805769
Kohn, W. Density functional and density matrix method scaling linearly with the number of atoms. Phys. Rev. Lett. 76, 3168–3171 (1996).
Prodan, E. & Kohn, W. Nearsightedness of electronic matter. Proc. Natl. Acad. Sci. USA 102, 11635 (2005).
Wang, C. et al. First-principles calculation of optical responses based on nonorthogonal localized orbitals. New J. Phys. 21, 093001 (2019).
Marzari, N., Mostofi, A. A., Yates, J. R., Souza, I. & Vanderbilt, D. Maximally localized wannier functions: theory and applications. Rev. Mod. Phys. 84, 1419–1475 (2012).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In Proc. 34th International Conference on Machine Learning (ICML) PMLR 70 (eds. Precup, D. & Teh, Y. W.) 1263–1272 (2017); http://proceedings.mlr.press/v70/gilmer17a.html
Schütt, K. T., Arbabzadah, F., Chmiela, S., Müller, K.-R. & Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 8, 13890 (2017).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Wang, Z. et al. Symmetry-adapted graph neural networks for constructing molecular dynamics force fields. Sci. China Phys. Mech. Astron. 64, 117211 (2021).
Ba, J. L., Kiros, J. R. & Hinton, G. E. Layer normalization. Preprint at https://arxiv.org/abs/arXiv:1607.06450 (2016).
Morimoto, T. & Nagaosa, N. Topological nature of nonlinear optical effects in solids. Sci. Adv. 2, e1501524 (2016).
Wang, C. et al. First-principles calculation of nonlinear optical responses by Wannier interpolation. Phys. Rev. B 96, 115147 (2017).
Bistritzer, R. & MacDonald, A. H. Moiré bands in twisted double-layer graphene. Proc. Natl. Acad. Sci. USA 108, 12233 (2011).
Cao, Y. et al. Correlated insulator behaviour at half-filling in magic-angle graphene superlattices. Nature 556, 80–84 (2018).
Cao, Y. et al. Unconventional superconductivity in magic-angle graphene superlattices. Nature 556, 43–50 (2018).
Yankowitz, M. et al. Tuning superconductivity in twisted bilayer graphene. Science 363, 1059–1064 (2019).
Xie, Y. et al. Fractional Chern insulators in magic-angle twisted bilayer graphene. Nature 600, 439–443 (2021).
Carr, S., Fang, S. & Kaxiras, E. Electronic-structure methods for twisted moiré layers. Nat. Rev. Mater. 5, 748–763 (2020).
Jeong, W., Yoo, D., Lee, K., Jung, J. & Han, S. Efficient atomic-resolution uncertainty estimation for neural network potentials using a replica ensemble. J. Phys. Chem. Lett. 11, 6090–6096 (2020).
Lucignano, P., Alfè, D., Cataudella, V., Ninno, D. & Cantele, G. Crucial role of atomic corrugation on the flat bands and energy gaps of twisted bilayer graphene at the magic angle θ ~ 1.08°. Phys. Rev. B 99, 195419 (2019).
David, A., Rakyta, P., Kormányos, A. & Burkard, G. Induced spin-orbit coupling in twisted graphene-transition metal dichalcogenide heterobilayers: twistronics meets spintronics. Phys. Rev. B 100, 085412 (2019).
Gou, J. et al. The effect of moiré superstructures on topological edge states in twisted bismuthene homojunctions. Sci. Adv. 6, eaba2773 (2020).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Ozaki, T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B 67, 155108 (2003).
Ozaki, T. & Kino, H. Numerical atomic basis orbitals from H to Kr. Phys. Rev. B 69, 195113 (2004).
Morrison, I., Bylander, D. M. & Kleinman, L. Nonlocal Hermitian norm-conserving Vanderbilt pseudopotential. Phys. Rev. B 47, 6728–6731 (1993).
Sipe, J. E. & Shkrebtii, A. I. Second-order optical response in semiconductors. Phys. Rev. B 61, 5337–5352 (2000).
Fey, M. & Lenssen, J. E. Fast graph representation learning with PyTorch Geometric. In Proc. ICLR Workshop on Representation Learning on Graphs and Manifolds (ICLR, 2019); https://arxiv.org/abs/1903.02428
Li, H. Dataset for deep-learning density functional theory Hamiltonian for efficient ab initio electronic-structure calculation (Zenodo, 2022); https://doi.org/10.5281/zenodo.6555484
Li, H. Code for deep-learning density functional theory Hamiltonian for efficient ab initio electronic-structure calculation (Zenodo, 2022); https://doi.org/10.5281/zenodo.6555482
Acknowledgements
This work was supported by the Basic Science Center Project of NSFC (grant no. 51788104), the National Science Fund for Distinguished Young Scholars (grant no. 12025405), the National Natural Science Foundation of China (grant no. 11874035), the Ministry of Science and Technology of China (grant nos. 2018YFA0307100 and 2018YFA0305603), the Beijing Advanced Innovation Center for Future Chip (ICFC) and the Beijing Advanced Innovation Center for Materials Genome Engineering. M.Y. was supported by the Shuimu Tsinghua Scholar Program and Postdoctoral International Exchange Program. R.X. was funded by the China Postdoctoral Science Foundation (grant no. 2021TQ0187).
Author information
Authors and Affiliations
Contributions
Y.X. and W.D. proposed the project and supervised H.L., Z.W. and N.Z. in carrying out the research, with the help of M.Y., R.X. and X.G. All authors discussed the results. Y.X. and H.L. prepared the manuscript with input from the other co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Computational Science thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Kaitlin McCardle, in collaboration with the Nature Computational Science team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Details of computational methods and results, Supplementary Figs. 1–21 and Tables 1–5.
Supplementary Data 1
Atomic structures of the three distorted graphene supercells in crystallographic information file (CIF) format.
Supplementary Data 2
Atomic structures of the three distorted MoS2 supercells in CIF format.
Supplementary Data 3
The atomic structure of the distorted silicon supercell in CIF format.
Source data
Source Data Fig. 3
Source data for plot.
Source Data Fig. 4
Source data for plot.
Source Data Fig. 5
Source data for plot.
Source Data Fig. 6
Source data for plot.
Rights and permissions
About this article

Cite this article
Li, H., Wang, Z., Zou, N. et al. Deep-learning density functional theory Hamiltonian for efficient ab initio electronic-structure calculation. Nat Comput Sci 2, 367–377 (2022). https://doi.org/10.1038/s43588-022-00265-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43588-022-00265-6
This article is cited by
-
Physics-inspired transfer learning for ML-prediction of CNT band gaps from limited data
npj Computational Materials (2024)
-
Overcoming the barrier of orbital-free density functional theory for molecular systems using deep learning
Nature Computational Science (2024)
-
Equivariant neural network force fields for magnetic materials
Quantum Frontiers (2024)
-
Machine learning electronic structure methods based on the one-electron reduced density matrix
Nature Communications (2023)
-
A deep-learning method for studying magnetic superstructures
Nature Computational Science (2023)
