
Abstract
Viruses and bacteria commonly exhibit spatial repetition of the surface molecules that directly interface with the host immune system. However, the complex interaction of patterned surfaces with immune molecules containing multiple binding domains is poorly understood. We developed a pipeline for constructing mechanistic models of antibody interactions with patterned antigen substrates. Our framework relies on immobilized DNA origami nanostructures decorated with precisely placed antigens. The results revealed that antigen spacing is a spatial control parameter that can be tuned to influence the antibody residence time and migration speed. The model predicts that gradients in antigen spacing can drive persistent, directed antibody migration in the direction of more stable spacing. These results depict antibody–antigen interactions as a computational system where antigen geometry constrains and potentially directs the antibody movement. We propose that this form of molecular programmability could be exploited during the co-evolution of pathogens and immune systems or in the design of molecular machines.
Similar content being viewed by others


Main
Due to their multiple binding domains, immunoglobulin molecules like the bivalent immunoglobulin G (IgG) antibody exhibit complex interactions with multivalent antigens, that is, clusters of multiple copies of molecules or molecular domains occurring at separation distances of the order of 1–30 nm. Multivalent interactions enhance the stability of binding interactions by enabling the simultaneous attachment of multiple ligands, increasing the magnitude of the apparent affinity or Gibbs free energy of multivalent binding, also called functional affinity in favor of the term ‘avidity’, and extending the residence times of the bound antibodies1,2,3.
Many pathogenic surfaces exhibit spatial repetition at length scales relevant to antibody multivalence. Viral capsid proteins undergo self-assembly into periodic patterns4, and some neutralizing antibodies achieve their high affinity and neutralization capability through bivalence5,6. Self-assembling crystalline arrays of surface-layer (S-layer) proteins—the outermost structure on many bacteria and archaea—are a major contact point between the pathogen and host7 and are implicated as the mediators of innate8 and adaptive immunity9,10. Their repetitive organization may be integral to their immunological role, as their removal from bacterial surfaces was seen to reduce the immune response11. Multivalence is also probably an important factor during the affinity maturation of antibodies and thus in vaccine design12,13,14.
Antibody interaction with patterned surfaces presents a challenge for both experimental control and mathematical modeling as it is a many-bodied problem occurring on the timescales of seconds to minutes. Such systems are too computationally expensive for full-atom molecular simulation. Models treating antibodies and antigens as abstract binding and non-binding units have been the most successful at capturing the relevant dynamics, and have historically treated multivalence as a function of ligand coating density where multivalence emerges statistically as the average nearest-neighbor distance between the ligands decreases15. More recently, coarse-grained molecular simulations have been fruitfully used to quantify the effects of the cooperative binding of the antibody subunits on binding affinity16,17. Nevertheless, a challenge of precisely calibrating such models remains due to the absence of experimental tools to independently assess monovalent and multivalent binding dynamics as well as a pipeline to connect such data to the models.
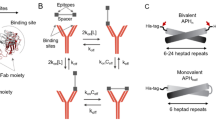
The patterned surface plasmon resonance (PSPR) technique enables the measurement of binding kinetics on precise, monodisperse patterns of ligands, achieving a robust control of geometry through the use of DNA origami nanostructures18 (Fig. 1a). Here we demonstrate a pipeline for the automated conversion of PSPR data into a flexible, experimentally parameterized model of antibody interaction with arbitrarily complex multivalent surfaces. The model is based on a coarse-grained simplification of bivalent antibody binding to antigens as a discrete Markov process with distinct states: empty antigen, monovalent antibody–antigen complexes and bivalent antibody–antigen complexes with transitions between these states governed by elementary rates (Fig. 1b). From this basis, the dynamics of more complex patterns of multiple antigens can be reduced (Fig. 1c) to combinations of these elementary states. A causal linkage between the pattern geometry and antibody dynamics could be potentially exploited as a form of spatial programmability by either immunity or pathogens during their adversarial co-evolution. We investigate this possibility and the role of spatial tolerance, that is, the range and impact of antigen separation distances on bivalent binding kinetics (Fig. 1d), in determining the effective binding affinity, walking speed of antibody migration on patterned surfaces and direction of antibody migration. Such control mechanisms might inform the development of vaccines for greater control over the affinity maturation process.

a, Illustration of patterning concept, where small-molecule antigens (haptens) are arranged using short, flexible tethers at well-defined locations on DNA origami nanostructures. This enables the multivalent interaction of antibodies with antigen patterns. b, Markov model of antibody binding where only the basic binding/unbinding and bivalent interconversion processes are used to couple discrete monovalent and bivalent binding states. c, Model extension to more complex pattern geometries is accomplished by separating the system into elementary transitions between the states comprising different combinations of empty and monovalently or bivalently occupied antigens. d, Pairs of antigens separated by different lengths elicit differing antibody-binding kinetics due to the separation-distance-dependent impact of the antibody structure on the chance of bivalent interconversion.
Results
Spatial tolerance model
We developed a model parameterization pipeline based on a progressive fitting of the transient surface plasmon resonance (SPR) profiles first for monovalent and then for bivalent binding processes to reduce the degrees of freedom at each stage of fitting. In the first stage, we used either rabbit anti-digoxygenin (DIG) IgG or mouse-derived anti-DIG IgG1-targeting DIG-decorated DNA origamis with a single-cycle kinetics program where progressively higher concentrations of antibody were exposed to the immobilized antigen substrate (Fig. 2a,b). This program was performed with a one-antigen configuration (Fig. 2c,e) to parameterize our Markov model (‘Markov model of arbitrary antigen pattern geometries’ section) by relating the SPR signal to the average occupancy Φ defined as the number of antibodies per structure averaged according to the prevalence of each possible state (‘Conversion from SPR signal RAb to bound antibody nAb’ section). This yields the respective association and dissociation rates k1 = 1.93 ± 0.05 × 107 M−1 s−1 and k−1 = 5.28 ± 0.07 × 10−4 s−1 as well as a monovalent dissociation constant KD1 = 2.7 ± 0.11 × 10−11 M defined as the ratio of the dissociation to association rates. We parameterized the interconversion between the monovalent and bivalent states by fixing the previously determined monovalent parameters and fitting the model to experiments involving multiple adjacent antigens (‘Fitting continuous-time Markov models to PSPR data using autocorrelation of residuals’ section). We fitted the model by adjusting KD2 or the interconversion constant defined by the ratio of the reverse and forward interconversion rates. For structures configured with two antigens separated by 14.3 ± 1.2 nm, we find KD2 = 8 ± 6 × 10−3 (Fig. 2d,f).

a,b, Concentration versus time plot of antibody solution exposed to patterned antigen substrates in a single-cycle kinetics PSPR experiment. c,d, Experimental binding kinetics data (black line) of one-antigen (c) and two-antigen configurations (d), superimposed over the occupancy calculated from the parameterized model. Model occupancy is divided and colored according to the state, with the height corresponding to the state’s contribution to the total antibody occupancy per structure. e,f, For the one-antigen (e) and two-antigen (f) configurations, the transient-state probabilities stratified according to the model prediction, colored and stacked to satisfy the normalization condition where all the probabilities add to 1. The legends list either all or the five most prevalent states σi along with their corresponding occupancy ϕi and probabilities pi at the end of their respective run. g, Interconversion constants (red points) plotted versus two-antigen configuration separation distance x and the fitted spatial tolerance model (blue line). The blue error bars indicate the model fits due to one standard error of the mean away from a mean input one-antigen run, propagated to KD2 values (vertical black error bars). The horizontal black error bars denote spatial uncertainty (defined elsewhere44). The vertical black error bars denote uncertainty due to one standard error of the mean variation in the input data. h, Goodness-of-fit characterization of the spatial tolerance model. The red points show an apparent random dispersion of KD2 points minus a moving average, whereas the blue points show the dispersion of model values subtracted from KD2 at each point. i, Sensitivity of the model’s minimum to shifted values of one-antigen input data by the number of standard error of mean (SEM) away from a mean input run.
By applying progressive fitting to PSPR runs with structures patterned with two adjacent antigens of varied separation distances, we found the internal conversion process to vary accordingly. Small and large separation distances correspond to reduced bivalence, that is, larger KD2. We constructed a phenomenological equation (‘Mathematical description of spatial tolerance’ section) modeling the interconversion constant (Fig. 2g). The model is composed of a logistic tension term representing the reduced bivalence at large separation distances and an exponential compression term representing the penalty to bivalence observed at extremely close separation distances, a characteristic that has been substantiated by recent biosensing applications19. In our model, the interconversion constant is, thus, a function of adjacent antigen separation distance with the form
where ℓt and ℓc are the characteristic lengths defining the scale of the tensile and compressive terms, respectively; αt is the sharpness of the tensile penalty; αc is the decay parameter of compressive penalty to bivalence; and \({K}_{\mathrm{D}2}^{\mathrm{max}}\) is the value of KD2 at which the contributions of bivalence to binding dynamics are vanishingly small. We found that when appropriately fitted (Fig. 2h), this model predicts a theoretical min(KD2) located at ~10.6 nm separation distance with an approximately 1.0 nm uncertainty due to an expected random shift in the one-antigen input data (Fig. 2i), whereas the experimental datapoint with the lowest KD2 is located at ~15.0 nm. This result is in agreement with another study20 in which the optimal epitope separation distance is estimated using DNA origami and atomic force microscopy, although we note here that the curve is likely to differ between isotype, species and possibly even clones due to angular variation in the epitope–paratope bond.
Steady-state and transient analyses
To determine the dependence of system bivalence on solution-phase concentration, we used the parameterized model to obtain the steady-state probability distributions for a range of solution-phase concentrations (Fig. 3a). This revealed concentration regimes of differing dominant states: empty, bivalent and saturated monovalent at low, medium and high solution-phase concentrations, respectively. The entropic maxima occur at transitions between these domains (Fig. 3b), and the transitions in bivalent and monovalent contributions to chemical potential occur in accordance with the transition from bivalent to saturated monovalent regimes (Fig. 3c; ‘Determination of thermodynamic properties’ section).

a, Stationary distributions colored by state of a two-antigen system (14 nm separation) for a range of solution-phase antibody concentrations, demonstrating clear regions of predominantly empty, bivalent single-antibody occupancy and monovalent two-antibody occupancy regimes connected by smooth transition regions. b, Distribution of state entropic contributions to free energy for a range of solution-phase antibody concentrations. c, Chemical potential contributions at equilibrium from each state for a range of concentrations (legend shows the five most abundant states). d, Result of a blind test with SPR signal in RU predicted for a trimeric 7.2 × 14.3 × 16.0 nm antigen configuration and known-concentration intervals overlaid with the raw experimental (red) run. e, Occupancy of the predicted run stratified by state. f, Transient-state probability distribution for the trimeric antigen configuration. g, Transient evolution of monovalent (blue) and bivalent (red) contributions of antibodies to the average occupancy or number of antibodies per structure in the trimeric system. The cross between the two lines demonstrates the transition between regimes by changing the concentration. The legends list all or the five most prevalent states σi along with their corresponding occupancy ϕi and probabilities pi at the end of their respective run or contributions to entropy in the case of e.
In addition to simulating de novo patterns’ steady-state properties, the model enables us to simulate the transient dynamics of hypothetical systems with arbitrary geometries and arbitrary timing in the introduction of different solution-phase concentrations. To validate the pipeline, we used the model parameterized with one- and two-antigen data (Fig. 2g) to create a blind a priori prediction of the evolution of a higher-order system with three antigens arrayed in a 7.2 × 14.3 × 16.0 nm right triangle and then check its correspondence with an experimental trajectory (Fig. 3d); additional validation is shown in Supplementary Fig. 7. We found that the experimental trajectories closely conformed to the predictions. The model provides access to the individual contributions of states to the signal through their occupancy (Fig. 3e) and relative proportions (Fig. 3f), enabling us to construct a narrative explanation for the observed dynamics. We see, for example, in the final stage when the concentration was set to zero, as the total occupancy decreased and occupancy contributions from monovalently bound antibodies decreased, bivalent-state contributions counterintuitively increased. This indicates that higher concentrations in the penultimate stage inverted the system to favor monovalent-dominated saturation states that subsequently transitioned into unsaturated bivalent states as the sites became available (Fig. 3g).
Experiments on repetitive antigen patterns
To explore the potential for pattern-based control and programmability of antibody dynamics, we modeled the dynamics of larger systems with greater relevance to periodic pathogenic surfaces. For larger systems, a complete enumeration of states scales poorly with increasing numbers of adjacent antigens. We developed a Markov chain Monte Carlo (MCMC) implementation of the model (‘MCMC version of the model’ section) to sample the trajectories that converge to state probabilities with large sample numbers. Rather than enumerating all the system states (that is, combinations of antibodies and binding modes on a structure and possible transitions), the system performs a random walk through the large state space, computing its rate of escape into neighboring states at any point in time. We then examined the collections of individual trajectories for such systems to understand their average behavior. Specifically, we examined the role of repetitive antigen spacing in simple one-dimensional (1D) arrays.
Antigens arranged according to a spacing gradient in the range of 10–22 nm separation distances (that is, the interval of the steepest slope in equation (1)) elicit asymmetric accumulation towards the narrow-spaced end of the array (Fig. 4a). This system also exhibited individual walking trajectories that tend towards the narrow-spaced end (Fig. 4b), asymmetric velocity (Fig. 4c) and asymmetric net displacement (Fig. 4d) according to the direction of the gradient. The mechanism for this locomotion is that of a biased random walk, where at any point in time, a bivalently bound antibody has a random chance to dislodge one of its paratopes and then reassociate either with the same epitope or an adjacent one. Differential spacing between the adjacent epitopes leads to a statistical preference for more stable spacings with a lower interconversion ratio KD2.

a, Cumulative antibody residence times as a function of antibody location on 1D antigen gradients oriented with increasing spacing (top) and decreasing spacing (bottom). b, Random-walk trajectories of antibodies tracked from their initial landing locations on 1D antigen gradients with increasing (top) and decreasing (bottom) spacing gradients. c, Histograms of antibody velocity on 1D antigen gradients of increasing (left) and decreasing (right) spacing distance. d, Net displacement of antibodies tracked from their initial binding location on 1D antigen gradients of increasing (left) and decreasing (right) spacing gradients (sample size for c and d is 200 simulations of 10,000 s each). e, Histogram of antibody residence times on uniform 1D antigen array with a wide 22 nm spacing. f, Histogram of antibody residence times on uniform 1D antigen array with narrow 10 nm spacing. g, Histogram of antibody displacements tracked from their initial binding locations for uniform 1D antigen arrays with wide (magenta) and narrow (blue) spacings (sample size for e, f and g is 100 simulations of 10,000 s each).
Antibodies binding to 1D arrays with uniform spacing exhibited divergent residence times, with antibodies spending less cumulative time on 22-nm-spaced arrays (Fig. 4e) than those of narrow 10-nm-spaced arrays (Fig. 4f). The migration speeds of antibodies on high-strain-inducing arrays are greater than those of low-strain-inducing arrays, and a comparison of the net displacement shows that antibodies moved further from their initial binding location on widely spaced arrays relative to the narrowly spaced ones (Fig. 4g).
Discussion
Repeating epitope patterns are present in many viruses as coat proteins21,22,23,24 and in bacteria as S-layer proteins25,26,27, often with a high degree of symmetry or geometric periodicity. Such repetitive, quasi-crystalline patterns have been recognized as a marker of foreignness corresponding to major enhancements of IgG response compared with unorganized substrates28. Investigators have observed both monovalent and bivalent antibody binding to such periodic viral surfaces24,29, and binding enhancement due to bivalence is a recognized factor in determining both immune pathogen recognition and neutralization capability30,31. A high-speed atomic force microscopy study32 captured the real-time bipedal locomotion of antibodies on reconstituted pathogenic surfaces with periodic patterns of epitopes.
The authors of this study proposed that antibody locomotion is enabled by the strain induced during bivalent binding as antibodies accommodate the geometry of their target antigens, weakening the bond and triggering a bipedal step. Our results agree and indicate that precisely tuned spacing on repetitive antigen patterns would have a major impact on the strength of bivalent bonds; furthermore, differences in adjacent antigen spacings statistically drive migration, as antibodies randomly move until becoming immobilized in states with minimal strain.
One limitation of our model is that torsional flexibility of the hinge region6 is not considered. This is due to the design of the DNA origami nanostructure substrates in which hapten antigens are tethered by short, flexible spacers with rotational freedom. Future studies could explore rotational spatial tolerance as well as degrees of freedom in the Z direction by including additional terms in equation (1) calibrated with the PSPR data that systematically modulate relative epitope orientations or employ structures that incorporate pathogenic protein antigens33,34,35,36 for more realistic structural complexity and physiological relevance.
An additional limitation of our model is the potential for over-extrapolation in increasingly complex systems, with any inaccuracies in spatial tolerance model or parameterization quality subject to propagation. We predicted that long-range gradients of differential spacings could be used to establish the persistent directed migration of antibodies on a surface, and we propose that PSPR and the progressive fitting pipeline presented here should be used in future studies to experimentally test the predictions. First, such designs should be possible using DNA origami; second, the stratification of states obtained by model fitting to convoluted binding data might enable one to measure the spatial distribution of antibodies on gradient structures.
Repetitive antigen arrays have been important in vaccine design37,38. The evolution of protective antibodies against malaria was shown to be dependent on a repetitive motif39, and bacteria are known to interfere with antibody binding such as Fc targeting to prevent opsonization40. The apparent importance of spatial organization in immunological signaling suggests a role for non-equilibrium spatial phenomena such as those studied here, and we might expect antigen organization itself to be under selective pressure during host–pathogen co-evolution. We suggest that the mechanism of stochastic walking predicted here might explain some of the pressures guiding the pathogen epitope organization, and such a mechanism might be exploited in the rational design of vaccines.
An inversion of this energy landscape phenomenon pertains to laterally mobile antigens such as the spike proteins in viral lipid envelopes. Mobile antigens would be expected to accommodate bivalent binding via lateral diffusion to achieve the minimum interconversion ratio KD2. Another study derived a theoretical affinity optimum for mobile spike proteins that depends on their surface density, arguing that intermediate densities invoke the greatest immune response and that the low-spike-density characteristic of human immunodeficiency virus is the key to its immune evasion41. In this respect, a spatial tolerance model and experimental parameterization pipeline could aid vaccine development by informing design choices meant to elicit a precise immune response, for example, immunostimulatory virus-like particles35,42. Our pipeline could also be used to dissect the complex state spaces of bi-, tri- or tetra-specific antibodies that are recently being developed for therapeutic and biosensing applications43. We expect these molecules to exhibit complex binding behaviors, especially as many are engineered with non-Fc-based tethering regions of various flexibilities and lengths.
The capacity for emergent dynamics and programmable behavior makes antibody–antigen interactions a subject of greater potential complexity than previously thought. Experimentally parameterized modeling provides a reality-grounded sandbox for discovery, and we anticipate that future modeling pipelines coupled to other experimental technologies will bear fruit as this subject continues to be explored.
Methods
Statistics and reproducibility
Error assessment (Fig. 2g) was performed using a boostrapping method in which the mean and mean ± one standard error of the mean input data were used to form the central, upper and lower inputs propagated to obtain the individual output points shown in the figure, with the upper and lower vertical error bars corresponding to the outputs produced by the upper and lower inputs, respectively. Goodness of fit for the phenomenological spatial tolerance function was characterized using an adapted chi-squared metric (‘Mathematical description of spatial tolerance’ section). The assessment of model robustness and predictive potential (Fig. 3 and Supplementary Figs. 7 and 8) was performed using a blinded test in which I.T.H. performed the model parameterization and prediction of experimental SPR curves for an untested three-antigen triangular structure for three independent replicates with different corresponding bound structure amounts, whereas the experimental test data obtained by A.S. and I.S. were withheld until the predictions were submitted for comparison.
Overview of computational methods
Some methods and explanations may be found elsewhere18. However, in the following, we emphasize the original developments of this work including the following: a minimal parameterization pipeline for Markov models sensitive to arbitrary antigen spacings and its experimental validation; phenomenological model with analytical equation describing spatial tolerance as a continuous function; application of the model and fitting pipeline to two different antibodies: one from rabbit and the other, mouse; a systematic approach to determining the conversion factor between the SPR response units (RUs) to that of the number of bound antibodies per structure; steady-state analysis and determination of thermodynamic quantities from equilibrium-state probability distributions; a random-walk MCMC variant of the model that can be used to simulate larger systems with too many connected antigens to be feasible for enumerative approaches.
Briefly, the pipeline is executed in three parts. The first part requires the empirical estimation of the maximum SPR response due to the saturation of antibody-binding sites, based on the measured signal due to origami structures binding to the surface and a standard curve constructed to relate structure binding to the maximum SPR response. This information is used as a conversion factor to relate the SPR signal for a given experiment to an average quantity of antibodies bound per structure. The second aspect of the pipeline is the fitting of a continuous-time Markov chain model to the SPR binding data of both one-antigen and two-antigen structures over a range of separation distances. This enables the construction of a parameterized analytical spatial tolerance equation that is used to predict the binding kinetics for arbitrary separation distances and antigen geometry. The third part of the pipeline entails the deployment of this fitted model for predictive purposes. The steady-state properties of a given system geometry are simulated by determining the distribution of states when the net flux between the states is zero. Large systems are simulated using an MCMC simulation that generates many individual trajectories of single antibodies walking on a user-specified pattern geometry. Mathematical explanation can be found in the following sections where this approach is described in more detail.
Model assumptions and constraints
We assume a coarse-grained model of binding states that equates all the physical states in which antibodies are bound by one arm as monovalent and which equates all the physical states in which antibodies bound by two arms as bivalent.
We assume a fixed amount of bound structures that does not change with time t, that is,
where Rstruct is the SPR signal due to the structures and
where nstruct is the molar amount of structures.
The system has an IgG reservoir that is large compared with the available binding surface and thus has an effectively fixed concentration, that is,
where cAb is the concentration of the solution-phase antibody.
Conversion from SPR signal R Ab to bound antibody n Ab
Consider first the simple 1–1 interaction of an antibody analyte that binds and unbinds to a structure containing a single antigen ligand.
where σ_ is the state corresponding to an unoccupied structure; σAb is the state corresponding to a structure with a bound antibody; and k1 and k−1 are the association and dissociation rates, respectively.
We may work in terms of molar quantities rather than concentrations or surface densities, as the dimensions of the system do not change, that is,
or we may work in terms of the molar amount of structures, both occupied and unoccupied (units of mol), where nAb is the number of bound antibody-structure complexes and n_ is the number of unoccupied structures; nAb and n_ are functions of t.
Therefore, the state probabilities are
and
corresponding to the unoccupied and one-antibody-occupied states, respectively.
We also define occupancy as the number of antibodies that are bound to a single structure for a given state. For simple one-antigen structures, this value is zero for the empty state and 1 for the bound state, namely,
and
respectively. The average occupancy is a macroscopic description of the state of the system comprising N states, or the average fraction of bound antibodies per structure.
For the case of the one-antigen structure, this becomes
In a 1–1 binding model, a change in the SPR signal is proportional to the amount of bound material or, in other words, a change in the molar amount of structures with occupied binding sites nAb.
where ξAb is a conversion factor corresponding to the expected change in the SPR response signal per mole of bound antibodies.
The rate of change of the occupied sites is equal to the rate of conversion of unoccupied sites via binding events minus the rate of the conversion of occupied sites via unbinding events.
The SPR signal after the structure-binding step is proportional to the molar amount of bound structures, that is,
where ξstruct is a conversion factor corresponding to the expected change in the SPR response signal per mole of structures.
Substituting RU-based expressions of molar amounts into equation (6), we get
Substituting RU-based expressions of molar amounts into equation (14) yields
which simplifies to
Since we have gathered both conversion constants into one term in equation (18), we now define the occupancy signal factor as
or the dimensionless ratio of molar conversion factors: bound antibody relative to the structure.
Note, by rearrangement, the relationship to average occupancy—that is, the occupancy signal factor—is the ratio of occupancy (in terms of SPR signal) to the molar quantities.
Substituting ξ*, we then arrive at the expression for the rate of change in the SPR signal with respect to time as a function of the structure-binding signal and antibody-binding signal:
In the case of a monovalent structure (one antigen available for binding) at the point of maximum saturation when the average occupancy is unitary (Φ = 1), the molar quantities of the bound antibody and structures are equal:
where \({n}_{\mathrm{Ab}}^{\mathrm{max}}\) is the maximum number of moles of antibody that can bind to the system.
Under the maximum saturation conditions, the monovalent occupancy signal factor then reduces to
where \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) is the maximum SPR response signal due to the saturation of antibody-binding sites.
This relationship is then used to produce a standard curve from monovalent structure-binding data to obtain the following linear relationship:
where an empirically determined ξ* enables one to estimate the SPR signal corresponding to an occupancy of one antibody per structure from the Rstruct signal. This is useful for structures with valence greater than 1 and whose binding kinetics do not obey simple 1–1 equations. Since \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) on a multivalent structure will not resemble that of the monovalent 1–1 system, we refer to this conversion factor obtained from monovalent \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) as \({R}_{\mathrm{Ab}^\mathrm{mono}}\), that is, an SPR signal to antibody number conversion factor:
where Vstruct is the valence.
In such cases, we obtain the average occupancy using the estimated \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) from linear regression.
Empirical estimation of \({R}_{\mathrm{Ab}}^{\mathrm{max}}\)
Thus, we obtain a standard curve used to convert the SPR signals for arbitrary structure configurations by empirically determining the correlation between the structure-binding signals and the maximum signals corresponding to saturated monovalent (one-antigen) structures, enabling conversion from SPR signal to occupancy in the absence of a well-understood binding model, provided the structure-binding signal is known.
The structure-bound signal (Supplementary Fig. 1a) is taken to be the difference between the signals before and after the structures are flowed over the chip and allowed to bind. Estimates of the parameters k1, k−1 and ξ* are supplied to a numerical minimization of the autocorrelation of residuals between the experimental and theoretical curves for the fourth-order Runge–Kutta approximation of equation (22), that is, the function \(\frac{\mathrm{d}{R}_{\mathrm{Ab}}}{\mathrm{d}t}=f({R}_{\mathrm{Ab}})\) recursively approximated according to the formula
where h is a small timestep and the constituent terms have the form
For each monovalent run (Supplementary Fig. 1b,c for rabbit and mouse antibodies, respectively) with a unique value of Rstruct, a projected value of \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) is computed using equation (25). By fitting the monovalent models to 1–1 kinetics, we obtain the rate constants that allow the computational prediction of \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) (Supplementary Fig. 1d,e for rabbit and mouse antibodies, respectively) in the absence of experimental saturation conditions. This enables us to make a standard curve to adjust \({R}_{\mathrm{Ab}}^{\mathrm{mono}}\) according to Rstruct in the absence of a 1–1 \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) (Supplementary Fig. 1f,g for rabbit and mouse antibodies, respectively). We use this value as a conversion factor, enabling us to convert the SPR RUs into the number of antibodies per structure (Supplementary Fig. 1h,i for rabbit and mouse antibodies, respectively). By knowing Rstruct, we can estimate this conversion factor for non-trivial antigen configurations where the multivalence influences the ease of reaching a saturation value corresponding to \({R}_{\mathrm{Ab}}^{\mathrm{max}}\).
Equilibrium characterization with dissociation constants
The equilibrium dissociation constant concisely describes the relationship between analyte and ligand, and provides a good basis for comparison between the systems across experimental conditions in which the dynamic behavior can vary substantially. Given a model of the process, we can derive a formula for the equilibrium dissociation constant by solving the system of equations. For a 1–1 process, we have the following:
at the steady state,
Rearranging equation (34) yields
and
For a 1–1 monovalent model, the dissociation constant is
Thus, at equilibrium, the SPR signal is
An empirical measurement of the dissociation constant is obtained by determining the equilibrium binding signals at multiple concentrations and fitting the linearized form of equation (38) or
where \({R}_{\mathrm{Ab}^{\mathrm{eq}}}\) is the steady-state SPR signal due to the bound antibody.
The equilibrium dissociation constant is a good descriptive parameter that concisely captures the essential dynamics.
From the dissociation constant, we know the occupancy as
where Φeq is the expected occupancy at the steady state.
Such a concise description is desirable for complex structures as well. However, the difficulty arises in the case of multivalent structures that no longer exhibit simple 1–1 dynamics. One approach is to simply approximate the dynamics with a 1–1 model and obtain an apparent dissociation constant.
For the only modestly more complicated bivalent system, we can derive the relationship between an apparent dissociation constant and a complete model with two dissociation constants to describe the multiple processes taking place.
In the case of the two-antigen structure, there are N = 5 total states: one empty structure (σ__), two states with one monovalently occupied antigen each (σAb_ and σ_Ab), one state with both antigens bivalently occupied by one antibody (σ.Ab.) and a state with both antigens monovalently occupied by antibodies (σAbAb).
First, the reaction system can be represented according to the diagram in Fig. 1 or the set of reactions below:
We have the two dissociation constants for the processes of monovalent (KD1) binding and bivalent (KD2) interconversion as follows.
The system can be represented with a system of differential equations as follows:
where \({p}_{{\sigma }_{{{{\_}}}{{{\_}}}}}\), \({p}_{{\sigma }_{\mathrm{Ab}\_}}\,\), \({p}_{{\sigma }_{\_\mathrm{Ab}}}\,\), \({p}_{{\sigma }_{.\mathrm{Ab}.}}\,\) and \({p}_{{\sigma }_{\mathrm{Ab}\mathrm{Ab}}}\) are the probabilities of each of the five states in the bivalent systems, subject to the following normalization condition:
Given the knowledge of the constituent equilibrium constants, we can—in the simple case of the bivalent system—solve for the apparent dissociation constant as a function of the microconstants. This is, in effect, specifying a certain equilibrium value predicted on the basis of the complete bivalent model, and assuming instead that it is the result of the 1–1 kinetics. However, for multiple concentrations, the equilibrium will not shift proportionately; therefore, the apparent binding constant is a function of the concentration from which the equilibrium value is derived, making its value dependent on the conditions rather than serving as a concise description of the system as a whole.
The bivalent system has—at equilibrium—the condition that the rate of change of each of its states is zero, that is,
This condition, in addition to the normalization conditions, allows us to solve for the equilibrium concentrations of each of the species in terms of rate constants and the fixed solution concentration of the analyte antibody, as follows:
We can combine the states according to their corresponding occupancy—that is, the number of antibodies that the state contributes to the overall signal due to the bound antibody, where
The probabilistic definition of occupancy is the expectation value of state occupancy. Each state has a corresponding integer occupancy associated with the number of antibodies bound to the structure in that state as well as a respective probability of that state at any point in time. The equilibrium occupancy is, thus, the average occupancy of all the states weighted by their equilibrium probabilities:
Substituting equations (53) through (57), we arrive at
Apparent dissociation constant
Taking the equilibrium occupancy of the two-antigen system from equation (60) and applying it to the equilibrium occupancy in terms of the 1–1 dissociation constant, equation (40) can be used to solve for an apparent equilibrium dissociation constant of the form
This constant is a value that would be obtained from a 1–1 fit to an equilibrium SPR value that arose from the two-antigen kinetics. Rearranging and substituting equations (44) and (45) into equation (61), the formula simplifies to
which is a function of concentration and has a root at the critical value when \({c}_{\mathrm{Ab}}{K}_{\mathrm{D}2}^{2}={K}_{\mathrm{D}1}^{2}\), that is, the point at which the average equilibrium occupancy greater than 1 is expected in the two-antigen system, and rendering any 1–1 kinetics description as impossible.
The rearrangement of equation (62) enables us to determine the interconversion constant from an apparent dissociation constant provided that we know the monovalent binding constant.
At concentrations where cAb ≪ KD1, the relationship between \({K}_{\mathrm{D}2}^{2}\) and \({K}_{\mathrm{Dapp}}^{2}\) is relatively constant (Supplementary Fig. 2). Note that this is only valid for PSPR data with a two-antigen topology of a single separation distance.
Mathematical description of spatial tolerance
Spatial tolerance refers to the favourability of bivalent antibody binding according to the spatial distribution of the two adjoining antigens. Some antibodies stretch and compress more than others, leading to a greater chance of entering and remaining in a bivalent state. In our model, we propose that the monovalent binding step occurs separately from the bivalent binding step, and that it is purely dependent on the solution-phase concentration and the epitope–paratope binding affinity. Spatial tolerance, therefore, is a property of the interconversion step from the monovalent to bivalent states and the reverse process (from bivalent back to monovalent). For antigens separated by very small distances, electrostatic repulsion in response to compression and steric hindrance within the IgG molecule occurs, penalizing the conversion to bivalent binding and/or favouring unbinding back to the monovalent states. Conversely, at larger separation distances, the molecule must stretch to accommodate the gap, again penalizing the conversion to bivalence and/or favouring conversion back to monovalence.
Spatial tolerance is a description of the landscape of this tradeoff—the breadth of the favorable region in between extremes that is conducive to bivalent binding, sharpness and degree of symmetry of the transitions to monovalent preference at close and far separations. Progressive fitting allows us to obtain KD2 for a single two-antigen system provided that we have determined KD1 for a one-antigen system, for which we take a mean run of multiple one-antigen runs (n = 6) with ±one and two standard errors of the mean away from the mean run as uncertainty intervals (Supplementary Fig. 3a,i for rabbit and mouse antibodies, respectively). Determining KD2 for different antigen separation distances gives us an empirical basis for spatial tolerance. We can phenomenologically model spatial tolerance with an equation for determining the interconversion constant KD2 as a function of the separation distance between two antigens x:
where KD2-compression (Supplementary Fig. 3b,j for rabbit and mouse antibodies, respectively) and KD2-tensile (Supplementary Fig. 3c,k for rabbit and mouse antibodies, respectively) are the exponential and logistic terms, respectively. These separately model the decrease in interconversion due to the tensile stretch of the molecule at increasing distances and that due to the onset of excluded volume, electrostatic repulsion or steric hindrance caused by compression of the molecule to bridge close distances.
The tensile term is built from a logistic function and has the following form:
where \({K}_{\mathrm{D}2}^{\mathrm{max}}\) is an upper limit of the value of KD2, αt is the logistic growth rate or steepness with which the tensile penalty grows at increasing separation distances and has units of inverse length, and ℓt is the value of the midpoint of the sigmoidal curve that can be thought of as a characteristic length that defines the scale below which favorable interconversion occurs and above which the function approaches minimal interconversion.
The exponential compressive term has the form:
where αc is the exponential decay rate that has units of inverse length and ℓc is another characteristic length parameter with units of length. The model is subject to the constraint ℓc < ℓt.
The combined expression yields equation (1), which predicts the interconversion constant as a function of separation distance (Supplementary Fig. 3d,e for rabbit antibodies and Supplementary Fig. 3l,m for mouse antibodies). Uncertainty represented with vertical error bars is due to variation in the one-antigen input data that has been propagated to obtain different KD2 values fitted with correspondingly different KD1 values as constraints. The horizontal error bars represent uncertainty in the separation distance of protruding sites on DNA origami nanostructures, estimated according to the method employed elsewhere44.
This can be converted into an effective or apparent dissociation constant as a 1–1 model on the basis of the bivalent model’s prediction of equilibrium occupancy (Supplementary Fig. 3f,n for rabbit and mouse antibodies, respectively, and the ‘Apparent dissociation constant’ section). The propagation of one-antigen input data uncertainties yields slightly different parameterizations of the model due to the shifted KD2 values, and thus, we see a corresponding shift in the minimum of the function where bivalent binding is the strongest (Supplementary Fig. 3g,o for rabbit and mouse antibodies, respectively).
To assess the goodness of fit, we employ a chi-squared metric where the expected error is computed by projecting KD2 onto a straight axis by subtracting a three-point moving average. Random noise in the data should be approximately Gaussian distributed about the moving average, and we can thus compute a standard error of the mean E(x) from this straightened profile of the data. Supplementary Fig. 3h,p for rabbit and mouse antibodies, respectively, compare the distribution of KD2 minus the moving average (red) and model prediction (blue), a dispersion that should be Gaussian/random, if appropriately fitted. The chi-squared metric is
where KD2-obs(x) is the observed interconversion value at a distance x and KD2-pred(x) is that predicted by the model. A good chi-squared metric should be neither much less than 1.000 (indicating overfitting) nor much greater than 1.000 (indicating poor fit).
Markov model of arbitrary antigen pattern geometries
For the binding kinetics of multi-antigen patterns of systems of sizes on the order of 2–8 adjacent antigens, we employ a fully enumerative Markov chain model based on a complete transfer matrix, that is, all the possible states and transitions of the system. The antigen pattern itself is modeled as a discrete network of antigen sites with a Euclidean distance matrix as follows:
This matrix can be simplified by applying a cutoff dcrit above which the antigens are considered too far apart to be neighbors. This reduces the number of possible states, eliminating those that are so unfavorable that they can be considered negligible.
A single state σi of the system is defined as a set of antigens, their status (empty, monovalently occupied or bivalently occupied) and a pointer indicating which bivalent-status antigens are linked to each other. The state space of a system is the set of all the states that a structure in the system can assume, namely, \({\mathbb{S}}:=\{{\sigma }_{0},{\sigma }_{1},\ldots {\sigma }_{N}\}\). The set of states are, thus, all the possible configurations of empty, monovalently bound and bivalently bound antibodies given the constraints of pattern geometry (Supplementary Fig. 4).
Each state is linked to adjacent states by elementary transitions, that is, the change in status of individual antibodies comprising the state. Those transitions are either the concentration-dependent addition or the subtraction of a single antibody to the system via monovalent binding or unbinding:
or a bivalent interconversion event where a monovalently bound antibody binds to an adjacent antigen site, changing its status to bivalently bound and vice versa:
Not all the states are necessarily connected. An adjacency matrix describes which states are connected by transitions.
The system parameters are the set of zero-order transition rates {λ1 = cAbk1, λ−1 = k−1, λ2 = k2, λ−2 = k−2}. A multi-antigen–antibody system is, thus, fully described by the continuous-time Markov model \(({\mathbb{S}},{{{\bf{\Lambda }}}})\) defined as the set of states and its corresponding transition rate matrix of the form:
The automated enumeration of states and their connections in systems of arbitrary antigen pattern geometry are accomplished using an implementation of the breadth-first search algorithm. The algorithm searches for edges between adjacent states and assigns the appropriate elementary rate process. A queue of neighboring states is made on the visitation of any state. One by one, the algorithm visits each state in the queue, populating it with additional states when they are discovered, and skipping the addition of states that have already been visited. The algorithm, thus, is characterized by an initial expansion phase of the queue followed by a systematic reduction of the queue until all the states have been visited, and the queue becomes empty. This exhaustive enumeration is deterministic and enables us to assemble a complete transition matrix regardless of the antigen geometry. However, as the number of adjacent antigens grows, the number of combinations increases dramatically; thus, for larger systems, a sampling-based approach must be used instead.
Supplementary Algorithm 1 describes the process by which the states and transitions are discovered starting from a single starting state. Here states are distinguished by the status of each of the sites ζk in the pattern, being either empty, monovalently occupied or bivalently occupied, as well as connected to another adjacent site ζs. The colored text is used to separate the different classes of transition.
The time complexity of the breadth-first search can be expressed as O(∣V∣ + ∣E∣), where ∣V∣ and ∣E∣ are the numbers of vertices and edges, respectively. In the case of antigen patterns, the former correspond to the number of antigens in the pattern. The latter correspond to the number of adjacent pairwise connections that are possible between two antigens. This is determined by the bivalent flexibility of the antibody in question; as a rule of thumb, we could say that antigens further than 25 nm apart are not close enough for bivalent bonds to form.
Transient (non-equilibrium) dynamics of enumerative PSPR models
The continuous-time Markov model enables us to compute the transient evolution of a system. The probability distribution
is a vector whose elements pi(t) are the probabilities of the respective system states σ0, σ1,…, σN at time t. A uniform probability distribution would, for example, represent equal probabilities of finding a structure in any one of the possible states. Another example is at the start of a single-cycle kinetics PSPR run, when the initial condition p(t0) is that of a distribution where p_(t0) = 1 for the state σ_ corresponding to an empty structure and pi(t0) = 0 for all the other states.
The transient evolution of state probabilities is computed from an initial condition using the linear system of Chapman–Kolmogorov differential equations:
which uses an infinitessimal generator matrix Q obtained from the rate matrix and used to determine the relative rates at which state the probabilities change with incremental time.
The infinitessimal generator is then used to compute the change in state probability distribution going from one timepoint to the next by the matrix exponential formula:
where η is the computation’s depth of recursion—the higher it is, the more accurate the result is; Δt is an incremental advancement in time. Due to the numerical instability of this solution, we employ the uniformized discrete-time Markov chain method of Fox and Glynn to stably compute equation (76) (ref. 45). The continuous-time Markov model \(({\mathbb{S}},{{{\bf{\Lambda }}}})\) is approximated by a discrete model \(({\mathbb{S}},{{{\bf{U}}}})\) by renormalizing the generator matrix with respect to the fastest outgoing rate or the uniformization rate q:
where I is the identity matrix.
Supplementary Algorithm 2 shows how this matrix is generated in practice.
Equation (76) becomes the approximation
The matrix exponential is then approximated with the following Taylor series expansion.
Using equation (79), we can stably compute the transient evolution of a system from an initial condition.
The system entropy can by computed as
where kB is the Boltzmann constant.
Supplementary Algorithm 3 shows how the probability distributions at different timepoints are computed from an initial condition.
Fitting continuous-time Markov models to PSPR data using autocorrelation of residuals
By using equation (79) to compute the transient probability distribution of the system, we are able to also compute the occupancy at each timepoint using the definition from equation (11). The system occupancy is, thus, a function of time of the form
The continuous-time Markov model is fitted to the experimental data by comparing occupancies computed on the basis of equations (79) and (81) with that of the occupancy computed from the normalizing PSPR data via equation (27); evidently, the theoretical curve either correctly or incorrectly fits the experimental data depending on the parameterization (Supplementary Figs. 5a and 6a). The residuals (Supplementary Figs. 5b and 6b) are computed by
Although fitting by minimizing the sum of the squared residuals can be used to obtain acceptable model parameterizations, we used residual analysis with autocorrelation to improve the robustness of fitting and reduce the systematic mis-parameterization by making fits more sensitive to divergence in curve shapes. We compute an absolute, average autocorrelation over a fixed interval kΔt with k = 50 as
where e(t, k) = [e(t), e(t + Δt),…, e(t + kΔt)], \(\,\overrightarrow{v}=[0,1,\ldots k]\) and \(| \overrightarrow{v}|\) is the conjugate of \(\overrightarrow{v}\). The objective function \(\mathrm{min}({{{\mathcal{E}}}})\) numerically minimized to obtain fits to the experimental PSPR data is then the sum-squared residual vector weighted by its autocorrelation vector.
This provides an error function sensitive to sustained divergence of the model and experimental data (Supplementary Fig. 6c) even if the two curves cross paths, and like summing the residuals provides a low value when the alignment is good (Supplementary Fig. 5c).
We performed a cross-validation of the Markov model fitting by parameterizing based on the experimental data from various antigen patterns (all with fixed nearest-neighbor separation distances between the antigens to remove the complication of separation-distance dependence of the binding kinetics). The rate parameters derived from these training data were then fixed and the model was applied to other patterns as a limited test of the extrapolation of a parameterized model to different antigen pattern geometries. The absolute sums of residuals (Supplementary Fig. 7a) and absolute sums of residuals weighted by autocorrelation (Supplementary Fig. 7b) show that the best-performing models were those that are the most complex and exhibit bivalence such as hexagonal and pentagonal configurations in the last two rows. This suggests that downward extrapolation in pattern complexity is more viable than upward.
To validate the spatial tolerance model, we conducted a blinded test in which an a priori prediction was made using the spatial tolerance parameters (Fig. 2f). This was done with the rabbit anti-DIG IgG antibody. The data used for prediction consisted entirely of the averaged one-antigen data and the series of two-antigen varied-separation-distance data used to parameterize the model and the structure-binding data to determine the conversion factor from RU to occupancy, and vice versa. Thus, no data with structures configured with more than two antigens was used for parameterization. To perform the test, we chose to predict the evolution of a trimeric 7.2 × 14.3 × 16.0 nm configuration with the same single-cycle kinetics protocol of timed known-concentration injections used for the other runs in this study (Supplementary Fig. 7c). Predictions were made first by computing the expected occupancy values on a per-structure basis. These were then converted into SPR RUs by multiplying them with the \({R}_{\mathrm{Ab}}^{\mathrm{max}}\) value determined from the structure-binding curve and standard curve (Supplementary Fig. 1). The experimental results were withheld until predictions were made, and were then revealed and compared with the theoretical curves each done in triplicate with the respective structure-binding data used for each one (Supplementary Fig. 7d).
An important question to consider is how sensitive the stratified-state probability distribution predictions are to error in the antibody concentration. As shown earlier, the distribution is dependent on concentration and timing; however, the answer is probably fairly complex. This is because different phases (Fig. 3a–c) are structured in a complex manner in terms of their concentration intervals and shape of transitions between phases. To get only a very rough idea of the sensitivity though, what we have done for the triangle structure is to change each of the concentrations by factors of 0.5, 0.9, 1.1 and 2.0 (Supplementary Fig. 8a) to see how this affects both the resulting transient probability distribution (Supplementary Fig. 8b) and the relative correspondence of the predicted curve to that of the experimental data (Supplementary Fig. 8c). Evidently, the probability distribution is fairly robust, with the five most represented states remaining unchanged in all the five conditions, albeit their relative ranking changes slightly, for example, going from 1.1x to 2.0x.
Determination of thermodynamic properties
We can obtain the equilibrium probabilities from uniformized continuous-time Markov chain first by simulation out to long timescales at a fixed solution concentration until the probabilities cease to change.
We can determine the steady-state probability distribution more expediently on the basis of the infinitessimal generator matrix, that is, equation (75), numerically solving for the probability distribution which—when multiplied with the generator matrix—produces a vector of zeros, meaning that there is zero change from one moment to the next, subject to the normalization condition in which all the probabilities must sum to 1. That is, the steady-state probability distribution is the solution to the matrix equation
The multi-antigen structure in the context of a PSPR experiment is an open system, freely allowed to exchange particles with the large external reservoir connected to it. With (T, V, cAb) held constant, the system (an antigen-patterned structure) will approach a minimum free energy at the steady state by exchanging antibodies with the bath, obeying the Boltzmann distribution law
where \({\mathbb{Z}}\) is a grand canonical partition function that predicts equilibrium at a grand potential free-energy minimum dΦ(T, V, μAb) = 0 with chemical potential µAb = –kBTln[cAb], and Eipi = μAb + μmononmono + μbivnbiv are the state energies determined by the environmental potential due to solution-phase antibody concentration and individual potentials of antibody monovalent and bivalent bonds populating the state; further, nmono denotes the monovalent bonds and nbiv denotes the bivalent bonds with chemical potentials μmono and μbiv, respectively. The value of \({\mathbb{Z}}\) and the state energies are numerically solved for a given steady-state probability distribution, making it possible for us to determine the thermodynamic quantities.
We can obtain the thermodynamic quantities such as the solution-concentration-dependent equilibrium grand potential free energy via
The equilibrium probabilities enable us to calculate the relative potential differences as
This makes it possible to compute the chemical potentials of monovalent and bivalent bonds, for example, from the basic two-antigen system of a fixed separation, by comparing the equilibrium probabilities in states that differ by exactly one bond of a given type. This is equivalent to obtaining the change in free energy via the dissociation constant for that process.
where nempty, nmono and nbiv are the degeneracies of empty, monovalently one-occupied and bivalently one-occupied states, respectively. For the two-antigen system, these values are 1, 2 and 1, respectively. For the rabbit anti-DIG IgG model and a separation distance of 15 nm, the chemical potentials are μ0−1 = 1.805 × 10−20 J per particle and μ1−2 = 8.889 × 10−21 J per particle.
We can also obtain a stand-alone bivalent-binding chemical potential such that tallying the number of monovalent and bivalent molecules in a state would yield the potential of that state.
This gives us μ0−2 = 2.693 × 10−20 J per particle for 15 nm separation. By this method, the distance-dependent KD2 model of equation (1) can be converted into a chemical potential curve.
MCMC version of the model
Systems with larger numbers of bivalent connections have many states and transitions, and the fully enumerative continuous-time Markov chain does not scale well. For these systems, we use the MCMC sampling approach in which only the local states are computed throughout the trajectory of a single system. Multiple trajectories are sampled to approach and approximate the probability distributions that would otherwise be computed to a higher precision for the enumerative method. Instead of computing the flux of state probabilities over fixed intervals of time, we instead computed Poisson intervals (dwell times) of states according to the rates of processes that each state is subject to here. For any particular state σi, there is a set of adjacent states σj ∀ j such that Ai,j ≠ 0.
The exit rate of that state is then the summation of all the outgoing rates:
and the corresponding dwell time in that state comes from the exponential cumulative distribution function
where p is the probability that a transition to a neighboring state occurs within the time interval and dwell time τ is a random variable that we may sample by choosing random values of p from the interval [0, 1].
The choice of state given that a transition occurs is then a matter of the relative rates λi,j for the different states σj, with each state having a probability \({p}_{j}=\frac{{\lambda }_{i,j}}{{\lambda }_{\mathrm{exit}}}\) of becoming the destination state.
A single iteration, thus, starts with an initial state, followed by the enumeration of each of the adjacent states, and random sampling to determine both dwell time and destination after the transition. The simulation involves performing multiple random walks and keeping track of the occupancy and state distribution over a discretized timeline.
Supplementary Algorithm 4 shows how this random-walk MCMC approach is used to simulate the antibody dynamics by computing the dwell times and local state-to-state transitions.
Experimental methods
Some experimental data used in this work were presented previously18 and were not analysed further than a basic fitting to a standard 1–1 model. In the current work, we have used the data to develop a more accurate mechanistic model, a pipeline for constructing such models from a minimal dataset, an in-depth physical characterization framework and a de novo simulation that goes beyond the previous work.
Reagents
Oligonucleotides (unmodified and DIG modified) in 96-well plates were purchased from IDT. Chemicals (NaCl, KCl, MgCl2, Tris-HCl, EDTA, PEG800, NaOH, KOAc, KOH and NaOAc) for buffer preparation were purchased from Sigma-Aldrich. Rabbit anti-DIG IgG (no. 9H27L19) was purchased from Thermo Scientific. Streptavidin was purchased from Sigma-Aldrich and mouse anti-DIG IgG1 (no. ab420) was purchased from Abcam. Phosphate-buffered saline (1 M stock solution) was purchased from Sigma-Aldrich. BIAcore consumables (CM3 sensor chip and HBS-EP (HEPES 10 mM, NaCl 150 mM, EDTA 3 mM, 0.005% Tween 20) running buffer) were purchased from GE Healthcare. Amicon centrifugal filters with 100 kDa molecular weight cutoff were purchased from Merck Millipore.
Origami design and production
The 18-helix bundle DNA origami nanotube was designed with caDNAno46 using the honeycomb lattice. This structure has been characterized earlier18,33,34,47. In short, the p7560 scaffold was extracted from the M13 phage, and the 18-helix bundle DNA nanotube was folded under the following conditions: 20 nM scaffold, 100 nM each staple oligonucleotide, 13 mM MgCl2, 5 mM Tris at pH 8.5, 1 mM EDTA. The mixture was subjected to heat denaturation at 80 °C for 5 min followed by a slow cooling ramp from 80 to 60 °C over 20 min and 60 to 24 °C over 14 h. The excess staples were removed by ultrafiltration with Amicon 100 kDa molecular weight cutoff filters. The wash buffer used was 1× phosphate-buffered saline supplemented with 10 mM MgCl2.
PSPR protocol
A BIAcore t200 instrument (GE Healthcare) was used for the SPR experiments. The running buffer used in all the experiments is HBS-EP supplemented with 10 mM MgCl2. The flow rate used for all the kinetics experiments is 30 μl min–1. Streptavidin was diluted to a final concentration of 10 μg ml–1 in 10 mM NaOAc at pH 4.5 and chemically attached to the CM3 sensor chip with N-hydroxysuccinimide and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide coupling (using the standard protocol from GE Healthcare). Anchor oligonucleotides containing a 3'-biotin modification were diluted to 200 nM in 1× HBS-EP running buffer and injected over the surface for 20 min followed by the washing of non-specifically bound oligos by injecting 50 mM NaOH for 5 min. The DNA nanostructures were diluted to 5 nM and injected over the surface for 20 mins followed by washing with the running buffer for 10 min. The antibodies were diluted to various concentrations in the running buffer, ranging from 0.025 to 0.500 nM. The single-cycle kinetics injection method was used to sequentially inject the antibody solution over the surface, starting from the lowest concentration; the contact time for each concentration is 5 min. After the final antibody injection, the dissociation curve was recorded for 15 min. The immobilized DNA nanostructures and bound antibodies were removed from the surface by injecting 50 mM NaOH for 5 min; then, the surface is ready for the next round of experiments. The t200 evaluation software was initially used to fit the acquired data; for this, we used a 1:1 Langmiur binding model to fit the data and estimate the values of ka, kd, the association and dissociation rates, respectively, and dissociation constant KD and antibody-binding capacity (Supplementary Section 6 details the apparent dissociation constant).
Run design
The parameterization pipeline first requires a dosing scheme for the single-cycle kinetics program, that is, the timing and concentrations of the staged antibody injections into the system. Since different antibody–antigen systems are going to exhibit different kinetic profiles, we believe the following considerations could help inform the initial choice of dosing scheme.
First, an approximate knowledge of KD1 is probably the best starting point. Some antibody suppliers report an in-house measured KD (KD1 by our terminology) or else report those values published by researchers who used their product, and this value can also be determined with a single ELISA experiment48. Knowing this, it is possible to choose a dosing scheme that elicits both monovalent (KD1-dominated) kinetics and bivalent (KD2-dominated) kinetics. KD1-dominated kinetics occur when the magnitude of KD1/cAb is smaller than KD2, that is, at higher relative concentrations. However, KD2-dominated effects are the most apparent at lower concentrations or during the dissociation phase when the concentration is set to zero. Although KD2 is molecule specific, this value is probably subject to less variation among the commonly used isotypes. Therefore, even if KD2 is unknown at first, it may be a reasonable starting estimate to assume that it is similar to the values found in our study, that is, of the order of 10−2 around the optimal separation distances. Thus, framing a dosing series based on a supplier’s reported KD1 value and concentrations that span a range where relative KD1/cAb to KD2 goes through an inversion is likely to capture a useful range of kinetics well suited to parameterizing the model. The following concentrations and corresponding injection timings were used for most experiments in our study: timepoints (s): 0, 84, 384, 475, 775, 866, 1,166, 1,257, 1,557, 1,656 and 1,956; concentrations (nM): 0, 0.025, 0, 0.050, 0, 0.100, 0, 0.250, 0, 0.500 and 0.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw experimental data used to produce the results of this study can be found at https://github.com/Intertangler/spatial_tolerance/tree/master/data_repository and Zenodo49 under the subfolder ‘Data repository’. Data are licensed under the GNU General Public License. Source data for Figs. 2–4 are provided with this paper. In addition to the raw data, the GitHub/Zenodo repository contains the Jupyter Notebooks detailing the generation of our results including intermediate data and figures and plots are posted in ready-to-run form for reproduction.
Code availability
All the codes used to produce the results of this study including installation, demonstration and result reproduction instructions are available on GitHub (https://github.com/Intertangler/spatial_tolerance) and Zenodo49. The code is licensed under the GNU General Public License.
References
Martinez-Veracoechea, F. J. & Leunissen, M. E. The entropic impact of tethering, multivalency and dynamic recruitment in systems with specific binding groups. Soft Matter 9, 3213–3219 (2013).
Macken, C. A. & Perelson, A. S. Aggregation of cell surface receptors by multivalent ligands. J. Math. Biol. 14, 365–370 (1982).
Rheinnecker, M. et al. Multivalent antibody fragments with high functional affinity for a tumor-associated carbohydrate antigen. J. Immunol.157, 2989–2997 (1996).
Uetrecht, C., Barbu, I. M., Shoemaker, G. K., Van Duijn, E. & Heck, A. J. R. Interrogating viral capsid assembly with ion mobility–mass spectrometry. Nat. Chem. 3, 126–132 (2011).
Emini, E. A., Ostapchuk, P. P. & Wimmer, E. Bivalent attachment of antibody onto poliovirus leads to conformational alteration and neutralization. J. Virol. 48, 547–550 (1983).
Thouvenin, E. et al. Bivalent binding of a neutralising antibody to a calicivirus involves the torsional flexibility of the antibody hinge. J. Mol. Biol. 270, 238–246 (1997).
Boot, H. J. & Pouwels, P. H. Expression, secretion and antigenic variation of bacterial S-layer proteins. Mol. Microbiol. 21, 1117–1123 (1996).
Taverniti, V. et al. S-layer protein mediates the stimulatory effect of Lactobacillus helveticus MIMLh5 on innate immunity. Appl. Environ. Microbiol. 79, 1221–1231 (2013).
Konstantinov, S. R. et al. S layer protein A of Lactobacillus acidophilus NCFM regulates immature dendritic cell and T cell functions. Proc. Natl Acad. Sci. USA 105, 19474–19479 (2008).
Kajikawa, A. et al. Mucosal immunogenicity of genetically modified Lactobacillus acidophilus expressing an HIV-1 epitope within the surface layer protein. PLoS ONE 10, e0141713 (2015).
Suzuki, S., Yokota, K., Igimi, S. & Kajikawa, A. Comparative analysis of immunological properties of S-layer proteins isolated from Lactobacillus strains. Microbiology 165, 188–196 (2019).
Veneziano, R. et al. Role of nanoscale antigen organization on B-cell activation probed using DNA origami. Nat. Nanotechnol. 15, 716–723 (2020).
LoBue, A. D. et al. Multivalent norovirus vaccines induce strong mucosal and systemic blocking antibodies against multiple strains. Vaccine 24, 5220–5234 (2006).
Baschong, W., Hasler, L., Häner, M., Kistler, J. & Aebi, U. Repetitive versus monomeric antigen presentation: direct visualization of antibody affinity and specificity. J. Struct. Biol. 143, 258–262 (2003).
Müller, K. M., Arndt, K. M. & Plückthun, A. Model and simulation of multivalent binding to fixed ligands. Anal. Biochem. 261, 149–158 (1998).
De Michele, C., De Los Rios, P., Foffi, G. & Piazza, F. Simulation and theory of antibody binding to crowded antigen-covered surfaces. PLoS Comput. Biol. 12, e1004752 (2016).
Bongini, L. et al. A dynamical study of antibody–antigen encounter reactions. Phys. Biol. 4, 172 (2007).
Shaw, A. et al. Binding to nanopatterned antigens is dominated by the spatial tolerance of antibodies. Nat. Nanotechnol. 14, 184–190 (2019).
Pfeiffer, M. et al. Single antibody detection in a DNA origami nanoantenna. iScience 24, 103072 (2021).
Zhang, P. et al. Capturing transient antibody conformations with DNA origami epitopes. Nat. Commun. 11, 3114 (2020).
Grant, R. A. et al. Structures of poliovirus complexes with anti-viral drugs: implications for viral stability and drug design. Curr. Biol. 4, 784–797 (1994).
Munoz, N., Castellsagué, X., Berrington de González, A. & Gissmann, L. HPV in the etiology of human cancer. Vaccine 24, S1–S10 (2006).
Modis, Y., Trus, B. L. & Harrison, S. C. Atomic model of the papillomavirus capsid. EMBO J. 21, 4754–4762 (2002).
Hewat, E. A., Marlovits, T. C. & Blaas, D. Structure of a neutralizing antibody bound monovalently to human rhinovirus 2. J. Virol. 72, 4396–4402 (1998).
Carl, M. and Dasch, G. A. The importance of the crystalline surface layer protein antigens of rickettsiae in T-cell immunity. in T–Cell Activation in Health and Disease 81–91 (Academic Press, 1989).
Bahl, H. et al. IV. Molecular biology of S-layers. FEMS Microbiol. Rev. 20, 47–98 (1997).
Ilk, N., Egelseer, E. M. & Sleytr, U. B. S-layer fusion proteins—construction principles and applications. Curr. Opin. Biotechnol. 22, 824–831 (2011).
Hinton, H. J., Jegerlehner, A. and Bachmann, M. F. Pattern recognition by B cells: the role of antigen repetitiveness versus toll-like receptors. Specialization and Complementation of Humoral Immune Responses to Infection 1–15 (2008).
Hewat, E. A. & Blaas, D. Structure of a neutralizing antibody bound bivalently to human rhinovirus 2. EMBO J. 15, 1515–1523 (1996).
Edeling, M. A. et al. Potent dengue virus neutralization by a therapeutic antibody with low monovalent affinity requires bivalent engagement. PLoS Pathog. 10, e1004072 (2014).
Lee, P. S. et al. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl Acad. Sci. USA 109, 17040–17045 (2012).
Preiner, J. et al. IgGs are made for walking on bacterial and viral surfaces. Nat. Commun. 5, 4394 (2014).
Shaw, A. et al. Spatial control of membrane receptor function using ligand nanocalipers. Nat. Methods 11, 841–846 (2014).
Shaw, A., Benson, E. & Högberg, B. Purification of functionalized DNA origami nanostructures. ACS Nano 9, 4968–4975 (2015).
Knappe, G. A., Wamhoff, E.-C., Read, B. J., Irvine, D. J. & Bathe, M. In situ covalent functionalization of DNA origami virus-like particles. ACS Nano 15, 14316–14322 (2021).
Hellmeier, J. et al. Strategies for the site-specific decoration of DNA origami nanostructures with functionally intact proteins. ACS Nano 15, 15057–15068 (2021).
Lee, S. Y., Choi, J. H. & Xu, Z. Microbial cell-surface display. Trends Biotechnol. 21, 45–52 (2003).
Mikawa, Y. G., Maruyama, I. N. & Brenner, S. Surface display of proteins on bacteriophage λ heads. J. Mol. Biol. 262, 21–30 (1996).
Murugan, R. et al. Evolution of protective human antibodies against Plasmodium falciparum circumsporozoite protein repeat motifs. Nat. Med. 26, 1135–1145 (2020).
KumraAhnlide, V., de Neergaard, T., Sundwall, M., Ambjörnsson, T. & Nordenfelt, P. A predictive model of antibody binding in the presence of IgG-interacting bacterial surface proteins. Front. Immunol. 12, 661 (2021).
Amitai, A., Chakraborty, A. K. & Kardar, M. The low spike density of HIV may have evolved because of the effects of T helper cell depletion on affinity maturation. PLoS Comput. Biol. 14, e1006408 (2018).
Hellmeier, J. et al. DNA origami demonstrate the unique stimulatory power of single pMHCs as T cell antigens. Proc. Natl Acad. Sci. USA 118, e2016857118 (2021).
Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today 20, 838–847 (2015).
Reuss, M. et al. Measuring true localization accuracy in super resolution microscopy with DNA-origami nanostructures. New J. Phys. 19, 025013 (2017).
Fox, B. L. & Glynn, P. W. Computing Poisson probabilities. Commun. ACM 31, 440–445 (1988).
Douglas, S. M. et al. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 37, 5001–5006 (2009).
Zhao, Y.-X. et al. DNA origami delivery system for cancer therapy with tunable release properties. ACS Nano 6, 8684–8691 (2012).
Heinrich, L., Tissot, N., Hartmann, D. J. & Cohen, R. Comparison of the results obtained by ELISA and surface plasmon resonance for the determination of antibody affinity. J. Immunol. Methods 352, 13–22 (2010).
Hoffecker, I. Intertangler/spatial_tolerance: spatial tolerance initial release. Zenodo https://doi.org/10.5281/zenodo.5961096 (2022).
Acknowledgements
We would like to acknowledge F. Fördős for helpful discussions. We would like to acknowledge support from A. W. Stiftelsen (M19-0547) to I.T.H. and the Knut and Alice Wallenberg foundation (KAW 2017.0114 and KAW 2017.0276) and from the European Research Council ERC (GA no. 724872) to B.H.
Funding
Open access funding provided by Royal Institute of Technology.
Author information
Authors and Affiliations
Contributions
I.T.H. conceived and implemented the core model and carried out the computational experiments. A.S. and I.S. obtained and preprocessed the PSPR data. I.T.H. and V.S. developed and tested the fitting pipeline and the MCMC simulation. I.T.H., A.S. and B.H. conceived the study concept. I.T.H., A.S., V.S., I.S. and B.H. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Computational Science thanks Pontus Nordenfelt, Philippe Robert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Handling editor: Ananya Rastogi, in collaboration with the Nature Computational Science team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–8 and Algorithms 1–4.
Source data
Source Data Fig. 2
Data used for results presented in Fig. 2.
Source Data Fig. 3
Data used for results presented in Fig. 3.
Source Data Fig. 4
Data used for results presented in Fig. 4.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hoffecker, I.T., Shaw, A., Sorokina, V. et al. Stochastic modeling of antibody binding predicts programmable migration on antigen patterns. Nat Comput Sci 2, 179–192 (2022). https://doi.org/10.1038/s43588-022-00218-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43588-022-00218-z
This article is cited by
-
Theoretical analysis of immunochromatographic assay and consideration of its operating parameters for efficient designing of high-sensitivity cardiac troponin I (hs-cTnI) detection
Scientific Reports (2023)
-
DNA-origami-directed virus capsid polymorphism
Nature Nanotechnology (2023)
-
Unconstrained generation of synthetic antibody–antigen structures to guide machine learning methodology for antibody specificity prediction
Nature Computational Science (2022)
