
Abstract
Molecular modeling has been considered indispensable in studying the energy storage of supercapacitors at the atomistic level. The constant potential method (CPM) allows the electric potential to be kept uniform in the electrode, which is essential for a realistic description of the charge repartition and dynamics process in supercapacitors. However, previous CPM studies have been limited to the potentiostatic mode. Although widely adopted in experiments, the galvanostatic mode has rarely been investigated in CPM simulations because of a lack of effective methods. Here we develop a modeling approach to simulating the galvanostatic charge–discharge process of supercapacitors under constant potential. We show that, for nanoporous electrodes, this modeling approach can capture experimentally consistent dynamics in supercapacitors. It can also delineate, at the molecular scale, the hysteresis in ion adsorption–desorption dynamics during charging and discharging. This approach thus enables the further accurate modeling of the physics and electrochemistry in supercapacitor dynamics.
Similar content being viewed by others
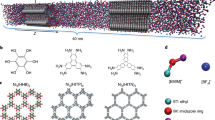
Main
Supercapacitors have been attracting much attention because of their high power densities and superior cycle times1,2. Researchers often resort to molecular modeling to investigate the thermodynamic and kinetic behavior of supercapacitors, particularly those with nanoporous electrodes, because molecular simulations can provide a precise microscopic picture of electric double layers (EDLs) and their formation in supercapacitors3,4,5. In the molecular modeling of supercapacitors, how to describe electrode polarization is a critical issue. The most straightforward way is to distribute charges uniformly on the electrode atoms in the constant charge method (CCM)4,5,6, whereas the constant potential method (CPM), an approach that is computationally expensive but can more precisely model realistic conditions, maintains electrode atoms at constant potentials by self-consistently adjusting the electrode charges based on the electrode potential and ionic environment7,8,9,10,11,12. To explore the equilibrium performance of supercapacitors, CCM can be used to approximate systems with open electrodes (where the electrode surface is in contact with the bulk electrolyte13, for example, a planar14, cylindrical6 or spherical15 surface), but it is not accurate for nanoporous electrodes10,11,12,16,17. To model the charge–discharge process of nanoporous supercapacitors, CPM is preferred because its self-consistent adjustment of electrode charges, absent in CCM, can produce the correct charging dynamics and heat generation17,18.
When studying the charging and discharging processes of supercapacitors, early CPM simulations often adopted the potential control mode, in which step-like7,8,9,10,11,12, linear19 or climbing-type20 potential differences are applied between the positive and negative electrodes. Molecular simulations in the potential control mode help to understand the fundamentals of charging and discharging supercapacitors. However, they cannot offer insights into the charging kinetics in the galvanostatic mode (that is, when the electrode is applied with a constant electric current21), which has been widely used in practical applications22 and fundamental electrochemical studies (for example, galvanostatic charge–discharge, GCD)23,24. To simulate the charging and discharging process of supercapacitors that are under more realistic conditions (for example, the current control with porous electrodes), it is desirable to have advanced modeling approaches that are able to regulate the electric current (or galvanostatic) and maintain a constant potential across each atom within the electrode. Most recently, an approach combining finite electric displacement and a constant potential technique was developed for this purpose25. This approach has been applied to open-electrode systems, but has yet to be demonstrated for nanoporous systems. As such, existing GCD simulations have largely been based on CCM, with the equivalent charge set on each electrode atom and varied linearly with time (in the following, this method is termed GCD-CCM)26,27. However, due to the lack of self-consistent adjustment of electrode charges, the accuracy of GCD-CCM, especially for nanoporous systems, remains to be delineated.
Based on previous CPM studies7,8,12, we developed a method to model the GCD of supercapacitors under constant potential, rigorously enforcing the equipotential state within an electrode at each time step (named GCD-CPM; Methods). Both GCD-CPM and GCD-CCM were applied to molecular dynamics (MD) simulations of two typical supercapacitor systems: open electrode (Fig. 1a) and nanoporous electrode (Fig. 1b). The ionic liquid ethyl-3-methyl-imidazolium tetrafluoroborate ([EMIM][BF4]) was used as the electrolyte in both systems (Fig. 1c). After equilibrating the simulation under the non-charged state (potential of zero surface charge), we applied a square-wave current with amplitude I0 and period P (Fig. 1d and Methods). We then analyzed the time evolution of the electrode potential and EDL formation in both systems. We further demonstrated the effectiveness of GCD-CPM via experimental validation.

a,b, Snapshots of supercapacitors featuring open electrodes (a) and nanoporous electrodes (b). c, Coarse-grained models of the ionic liquid [EMIM][BF4] used in the supercapacitors. d, Applied electric current with time (t) during GCD.
Results
GCD-CPM and GCD-CCM for open-electrode systems
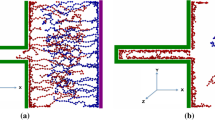
We first compared GCD-CPM and CGD-CCM in open-electrode systems. Like the GCD test in the experiments23,24, MD simulation systems were charged and discharged with a square-wave current, becoming stable after a few cycles (details are provided in Supplementary Section 1). Figure 2 shows the modeling-obtained GCD of a stable cycle with a period of 100 ps. In molecular modeling with GCD-CPM, the charges distributed on the electrode atoms are not uniform at any instant (Fig. 2a) due to the thermal motion of the electrolyte molecules5,28, as in previous works using CPM with step-like potentials4,5,28. Such a non-uniform distribution can be quantified by the probability distribution of the charges on the electrode atoms5,28. These charge probability distributions were analyzed in GCD-CPM simulations for each time step during the charge–discharge process (see Methods for the detailed probability distribution calculation). They have a volcano-like shape, and their width (or variance) changes little with time (Fig. 2b shows results for the positive electrode and Supplementary Fig. 2a for the negative electrode) in response to the applied current (Fig. 1d). On the electrolyte side, cations and anions, driven by the electrode polarization, form dynamic EDLs, which are delineated by the ion number density as a function of time and distance from the electrode surface (Fig. 2c and Supplementary Figs. 3 and 4). It is found that EDLs have typical layering structures, consistent with previous findings4,29,30. During the charging process (0–50 ps), the first cation layer decreases with time and moves slightly away from the electrode surface, and this trend is reversed during discharging (50–100 ps).

a–c, GCD-CPM modeling: a snapshot of the positive electrode at 50 ps in the MD simulation, with atoms colored by their instantaneous charges (a); the probability distributions of electrode charges varying with time (gray contour) and their average (dashed line) (b); evolution of ion number density with time (t) and distance (z) from the electrode (c). d–f, GCD-CCM modeling: a snapshot of the positive electrode at 50 ps, with atoms colored by their instantaneous potential (d); the probability distributions of the electrode potentials (gray contour) and their average (red dashed line) (e); evolution of ion number density (f). For comparison, the solid cyan line in e is the electrode potential obtained by GCD-CPM. The error bar for the electrode potential is close to the line width and is thus not shown.
The GCD of an open-electrode system was also explored using GCD-CCM. Instead of adjusting the charges distributed on electrode atoms at each time step to keep the electrode in an equipotential state in GCD-CPM modeling, in GCD-CCM modeling, the charge on each electrode atom is preset with the same value. The electrode potential is computed accordingly, showing a distinctly non-uniform distribution (Fig. 2d). However, the averaged potential over electrode atoms at each time closely matches the results obtained from the GCD-CPM calculation (Fig. 2e and Supplementary Fig. 2b). The evolution of cation density at the positive electrode obtained by GCD-CCM calculations was also found to be nearly the same as that obtained by GCD-CPM (Fig. 2c,f and Supplementary Fig. 3), and a comparison of more dynamic EDL structures reaches the same agreement (Supplementary Fig. 4). Besides the number density, ion orientation is also an important descriptor to characterize the EDL. On further examining the ion orientation, the angular distributions of the cations adsorbed on the negative electrode, obtained by these two methods, were almost the same (Supplementary Fig. 5). With additional electric currents with different amplitudes and periods, the results obtained using these two methods are also consistent (Extended Data Fig. 1).
Importance of GCD-CPM for nanoporous-electrode systems
We next investigated the GCD of nanoporous-electrode systems. In GCD-CPM modeling, the probability distributions of electrode charges are more non-uniform and can vary more significantly with time (Fig. 3a and Supplementary Fig. 6a), compared with those in open-electrode systems (Fig. 2b and Supplementary Fig. 2a). This can be ascribed to the non-uniform distribution of electrode charges along the pore axis (Extended Data Fig. 2a). In particular, the variation of surface charges at the pore entrance is more extensive than in the central portion of the pore. Driven by electrode polarization, the in-pore charge density ρe, stemming from the electrolyte, exhibits a trend similar to the variation in electrode charges; that is, the change in charge density at the pore entrance is greater than at the center of the pore (Fig. 3b versus Extended Data Fig. 2a).

a, The gray contour shows the probability distributions of atom charges on the positive electrode, obtained from GCD-CPM simulations, and the dashed line is the average of such charges. b, Evolution of in-pore charge density from the electrolyte along the pore (where z = 0 is the pore entrance), obtained from GCD-CPM simulations. c, The gray contour indicates the probability distributions of potentials on the positive electrode, obtained from GCD-CCM simulations. The red dashed line is the average of the potentials and the solid green line is the potential obtained from GCD-CPM simulations. d, Evolution of the in-pore charge density from the electrolyte, obtained from GCD-CCM simulations. e, Effective diffusivity of the in-pore electrolyte under positive polarization. f, Heat generation of a GCD cycle. The pore width is 0.67 nm. Error bars in b and d indicate 1 s.d. of four independent simulations. The box plots in e and f represent the maximal, minimal, median, 25% and 75% of the data distribution.
The potential distributions on the nanoporous electrodes were examined using GCD-CCM (Fig. 3c and Supplementary Fig. 6b) and compared to GCD-CPM. The potential was found to be more non-uniform than that distributed on the open electrode, and its average differs from the GCD-CPM results. Remarkably, there is a parabolic-like distribution of the electrode potential from the entrance to the central portion of a pore (Extended Data Fig. 2b), which conflicts with the fact that the electrode should be equipotential in reality, suggesting that GCD-CCM could result in an unphysical phenomenon. For GCD-CCM, the in-pore charges, driven by the uniformly distributed electrode charges, vary less than those obtained using GCD-CPM (Fig. 3d versus Fig. 3b).
Ion transport under nanoconfinement has been found to have a strong correlation with the charging dynamics and power density of nanoporous supercapacitors1,3,11. The in-pore ion transport during the charge–discharge process can be evaluated by means of the effective diffusivity, Deff, which is described as11,31
where ρe is the charge density of the in-pore electrolyte, t is time and z is the location along the pore axis. Using equation (1) and the evolution of the in-pore charge density (Fig. 3 and Supplementary Fig. 7), the effective diffusivity of the electrolyte in positive nanoporous electrodes was calculated and is shown in Fig. 3e (calculation details are provided in the Methods section). Using GCD-CPM modeling, for different periods P, the effective diffusivity in the pore of the positive electrode relative to the ion self-diffusion in the bulk electrolyte, Deff/Dbulk, is 52 (P = 200 ps), 21 (P = 1,000 ps) and 13 (P = 3,000 ps), close to the results predicted by theory11,31 and molecular simulations with step-like CPM11. For GCD-CCM modeling, Deff/Dbulk is 16,566 (P = 200 ps), 2,434 (P = 1,000 ps) and 231 (P = 3,000 ps), considerably larger than the results from GCD-CPM. A similar trend is found for negative nanoporous electrodes (Supplementary Figs. 8 and 9).
Previous work reported that CCM modeling gives a large value for heat generation during the charging process, contradicting the Joule effect17. We investigated the heat generated during the charge–discharge process in nanoporous supercapacitors, calculating it as the energy removed from the system by a thermal bath (details of the heat calculation are provided in the Methods section). Figure 3f and Supplementary Fig. 10 show the total heat generation of a charge–discharge cycle and heat generation varying with time, respectively, demonstrating that the value of heat generation computed by GCD-CCM is much larger than that obtained from GCD-CPM calculations. Accordingly, GCD-CCM cannot be used for modeling the dynamics of nanoporous supercapacitors, at least for the nanoporous electrodes studied here.
Experimental validation of GCD-CPM modeling
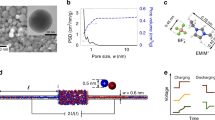
We next validated the molecular modeling with GCD-CPM through experimental galvanostatic measurements (experimental details are provided in the Methods section). The supercapacitor uses the same electrolyte as modeled in our simulation. The nanoporous carbon electrode has an average pore size very close to that in the molecular models. In the experiment, the GCD curve includes an ohmic drop caused by the electrode resistance (Supplementary Fig. 12)23,32. Because the electrode resistance is neglected in molecular modeling, the ohmic drop was removed from the experimental GCD curves, as described in the Methods section. This treatment aids a comparison of the experiment and modeling, but does not change the charge–discharge process of the nanoporous supercapacitors (Fig. 4a and Supplementary Fig. 12a).

a, GCD curve in the experiment with a period of 20 s and its fitting by the dimensionless diffusion model. b, GCD curve of GCD-CPM with a period of 200 ps and its fitting by the dimensionless diffusion model.
The charging dynamics of the nanoporous electrodes can be explained by the diffusion model, which is rooted in mean-field theory11,31. To compare macroscale experiments and nanoscale modeling, we derive the dimensionless form of the diffusion model for the charging and discharging process of the nanoporous supercapacitors. The dimensionless diffusion model shows that the GCD curve, described by the dimensionless potential \({{{\mathrm{{\Delta}}}\bar {\varPhi}}}\) varying with dimensionless time \({{{\bar{T}}}}\), is only determined by the dimensionless period α (defined as the ratio of the electric current period to the system charging time constant). Details of the dimensionless diffusion model are provided in the Methods section. As shown in Fig. 4a, the diffusion model provides a good fit for the experimental GCD data, with a dimensionless period of 0.42. It was then used to fit the GCD curve, also with period α = 0.42, obtained by GCD-CPM modeling of nanoporous systems (Fig. 4b), and this also shows good agreement. Because the GCD curves in the experiment and modeling have the same dimensionless period, their fitted curves are the same, with actual periods in the experiment and modeling of 20 s and 200 ps, respectively. Therefore, with regulation of the electric current period, the GCD-CPM modeling is consistent with the experiment (Supplementary Fig. 13). However, the fitting reveals an apparent discrepancy between the experiment and GCD-CCM modeling (Supplementary Fig. 14). Accordingly, only GCD-CPM could predict the correct dynamic process for the charge–discharge of the nanoporous electrodes. Furthermore, the capacitances extracted from the GCD curves obtained from molecular simulations with GCD-CPM are compatible with those from our experimental measurements (details are provided in the Methods section and Supplementary Fig. 15), and both are consistent with previous experiments with the similar porous carbon electrodes and the same electrolyte33,34.
Hysteresis in the charge–discharge process
Figure 5a shows the hysteresis of the ion adsorption–desorption during the charge–discharge process of nanoporous supercapacitors, demonstrated by the time evolution of the in-pore electrolyte density. Specifically, in the positive porous electrode, driven by the applied current, counterions (that is, anions) are attracted into pores during charging and rejected out of the pores during discharging, while a reverse trend exists for co-ions (that is, cations). The desorption of the counterions during discharging does not backtrack with the adsorption during charging (that is, at the same surface charge, σ, the number of counterions during charging is different from that during discharging), and the change of co-ions in the desorption process is also different from that in the adsorption process. These findings indicate that there is a hysteresis in the ion response during the charge–discharge process, which is similar to the hysteresis loop experimentally observed in porous electrode supercapacitors probed by an electrochemical quartz crystal microbalance (note that the areal densities shown in our work are based on the specific surface area of the porous electrode rather than the area of quartz-crystal surface used in experiments)35,36.

a, The mass density of the in-pore electrolyte (magenta line) and the number densities of the in-pore cations and anions (red and blue lines, respectively). The solid lines show the discharging process and the dashed lines the charging process. b, The charge density coming from the in-pore electrolyte (blue line) and the charge density on the electrode (red line). The shown results are for the positive electrode.
The hysteresis of the ion adsorption–desorption can be understood by comparing the charges from the in-pore electrolyte and on the electrode (Fig. 5b). The change in the charge from the in-pore electrolyte exhibits a trend (increase or decrease with time) that is essentially the same as that on the electrode, but the rate of change on the electrolyte side is slower than that on the electrode side. This indicates that the ion transport in the electrolyte, especially under nanoconfinement, is sluggish compared to the charge transport through the electrode. As shown in Fig. 5b, at the beginning of a stable cycle at the positive electrode (at 0 ps), the total charge on the electrode is zero (shown in red) and that from the electrolyte is still negative (shown in blue). At the time when charging and discharging switch (at 100 ps), the charge from the in-pore electrolyte is less than that from the electrode. Moreover, the change in the charge from the in-pore electrolyte in the initial stage is slower than in the rest, because both cations and anions change at a lower rate in the initial stage of the charging and discharging process (Fig. 5a). The hysteresis in the negative electrode exhibits the same trend (Supplementary Fig. 16).
There is also a hysteresis in ion adsorption–desorption during charging and discharging in the open-electrode system (Supplementary Fig. 17), though weaker than in the nanoporous-electrode system. This hysteresis originates from the fact that the electrolyte ions do not respond to the electrode polarization in as timely a manner as the electrode charges (Supplementary Fig. 18a). This hysteresis could account for the negative potentials occurring at the initial stage of the charging and at the end of the discharging at the positive electrode (Fig. 2e and Extended Data Fig. 1). Details of the hysteresis and its contribution to the negative potential are provided in Supplementary Section 8 and Supplementary Figs. 17 and 18. Similarly, the negative value of potentials on the positive electrode of nanoporous supercapacitors can also be understood by the hysteresis of ion adsorption–desorption resulting in the negative charge from the electrolyte at the beginning of charging or the end of discharging (Fig. 5b).
Discussion
The GCD-CPM method developed here can not only model the charging and discharging process of supercapacitors on applying electric currents of any form, but can also be combined with the previous CPM in potentiostatic mode to investigate complex charging modes at the molecular level, thereby helping to gain deep insights into the dynamics mechanisms and optimal charge–discharge modes. The dependence of capacitance on pore size has been intensively investigated by molecular simulations in the potentiostatic mode3,37,38,39. Such a dependence of charging dynamics, especially in the galvanostatic mode, is of significance and can be investigated by GCD-CPM. This method could also be used for other applications involving the dynamic formation and release of double layers, such as capacitive deionization40, batteries41, superlubricity42 and electrolyte gating43. Moreover, our work provides guidance for selecting molecular simulation methods for given simulation systems.
It is worthwhile noting that the position of the electrode is set as fixed in our modeling, which works for electrode systems with negligible electrode deformation during charging and discharging. Future directions in the modeling development will include the consideration of electrode deformation, which is essential for electrode materials experiencing large deformations, such as MXenes44,45.
Methods
GCD-CPM for modeling galvanostatic charge–discharge
We considered a classical electrochemical system containing N immobile electrode atoms (including Npos positive electrode atoms and Nneg negative electrode atoms) and M mobile electrolyte atoms. The CPM considers Gaussian charge distributions for electrode atoms and point charges for electrolyte atoms7,8. The fluctuating charges on electrodes, with magnitude q, are extra degrees of freedom to keep the electrodes equipotential. The electrostatic energy of the system, Uele, is a function of atom positions X and electrode charge magnitude q, written as8,14,46
The first and second terms in the right-hand side of equation (2) represent the electrode–electrode and electrode–electrolyte interactions, respectively, and the last term C(X) describes the electrolyte–electrolyte interaction. Matrix A with size N × N, depending on the position of electrode atoms, is defined as8,14,46
where α and η are the parameters of the Gaussian distribution, k is a reciprocal lattice vector, Xij is the vector from electrode atom i to j, V is the volume of the simulation box, and erfc and δij are the complementary error function and Dirac delta function, respectively. Vector B in length N is defined as
where S(k) is the structure factor of the electrolyte, Xij is the vector from electrode atom i to electrolyte atom j, and qj is the charge of electrolyte atom j. The potential on each electrode atom is \(\frac{{\partial U_{\rm{ele}}}}{{\partial q_i}}\) (i for the electrode atom index).
To model the galvanostatic charge–discharge, the total electrode charge is constrained as \(Q\left( t \right) = \mathop {\sum}\nolimits_{i = 1}^{N_{\rm{pos}}} {q_i}\) and \(\mathop {\sum}\nolimits_{i = N_{\rm{pos}} + 1}^{N_{\rm{pos}} + N_{\rm{neg}}} {q_i} = - Q(t)\), where qi is the charge of electrode atom i. In this work, Q(t) is a triangular wave for GCD due to the applied square-wave current (Fig. 1d). To keep the electrode equipotential at any time step and meet the constraints of the total charge of the electrode, we need to minimize the energy
The vector \({\bf{\upphi}} ^{{{\intercal}}} = [\phi _{\rm{pos}}{{{\mathbf{I}}}}_{{{{\rm{pos}}}}},\phi _{\rm{neg}}{{{\mathbf{I}}}}_{{{{\rm{neg}}}}}]\) is for the potentials on all electrode atoms, where \(\phi _{\rm{pos}}\,(\phi _{\rm{neg}})\) is the potential on the positive (negative) electrode. \({{{\mathbf{I}}}}_{{{{\rm{pos}}}}}\,\,({{{\mathbf{I}}}}_{{{{\rm{neg}}}}})\) is a vector of size 1 × Npos (1 × Nneg), the elements of which are all one. Similar to the previous CPM simulations with step-like potentials, we solve a set of linear equations to minimize Ut at each time step, then obtain ϕpos, ϕneg and electrode atom charges q. Mathematically, we have
The size of matrix \({{{{M}}}} = \left[ {\begin{array}{*{20}{c}} {{{{A}}}} & {{{{E}}}} \\ {{{{D}}}} & 0 \end{array}} \right]\) is (N + 2) × (N + 2), where \({{{{D}}}} = \left[ {\begin{array}{*{20}{c}} {{{{\mathbf{I}}}}_{{{{\rm{pos}}}}}} & 0 \\ 0 & {{{{\mathbf{I}}}}_{{{{\rm{neg}}}}}} \end{array}} \right]\) and \({{{{E}}}} = \left[ {\begin{array}{*{20}{c}} { - {{{\mathbf{I}}}}_{{{{\rm{pos}}}}}^{{{\intercal}}}} & 0 \\ 0 & { - {{{\mathbf{I}}}}_{{{{\rm{neg}}}}}^{{{\intercal}}}} \end{array}} \right]\). Unknowns of the linear equations are \({{{\mathbf{x}}}}^{{{\intercal}}} = \left[ {{{{\mathbf{q}}}}^{{{\intercal}}},\phi _{\rm{pos}},\phi _{\rm{neg}}} \right]\), and \({{{\mathbf{n}}}}^{{{\intercal}}} = \left[ { - {{{\mathbf{B}}}}^{{{\intercal}}},Q\left( t \right), - Q(t)} \right]\).
Molecular dynamics simulations
For the supercapacitor with open electrodes, each electrode is modeled by three layers of graphene sheets (Fig. 1a). The simulation box has a volume of 4.254 × 4.176 × 48 nm3, with a distance of 10 nm between the innermost layers of the positive and negative electrodes. For the supercapacitor with nanoporous electrodes, each electrode is modeled as a slit-shaped pore with walls composed of three layers of graphene sheets (Fig. 1b). The pore size of 0.67 nm is defined as the accessible gap between the two innermost slit sheets in contact with the electrolyte, and the pore length is 8 nm. The dimensions of the simulation box are 4.1 nm, 3 nm and 32 nm. The simulation box of all systems is sufficiently large to ensure that the central region of the electrolyte reservoir is in a bulk-like state. The Lennard–Jones model of the carbon atom47 is taken for the electrode, and a coarse-grained model (Fig. 1b), which provides accurate thermodynamic and dynamic properties, is adopted for the ionic liquid, [EMIM][BF4] (ref. 48).
All simulations are performed in the NVT ensemble using customized MD software, GROMACS49 (see the ‘Code availability’ section for more information), with a time step of 2 fs and a Nosé–Hoover thermostat50 at a temperature of 400 K. The effects of the thermostat settings were explored (Supplementary Section 9 and Supplementary Figs. 19 and 20). The heat generation during GCD was calculated as the energy removed from the system to maintain its temperature50,51. To avoid energy drift and attain an accurate heat generation, our simulations are carried out in double precision49. A cutoff length of 1.2 nm is chosen to calculate non-electrostatic interactions and electrostatic interactions in real space. The electrostatic interactions in reciprocal space are computed using the particle mesh Ewald method52 with a fast Fourier transform grid spacing of 0.1 nm. Because the open-electrode system has a slab geometry, the three-dimensional Ewald summation with the correction term for the slab geometry is adopted in electrostatic interactions5,53.
For each simulation, the system is first run for 40 ns to reach equilibrium at the potential of zero surface charge. A square-wave current of 30 cycles is then applied on the supercapacitors (Fig. 1b). We average the data of the last 10 cycles to obtain results for a stable cycle, so the time mentioned above refers to the time relative to the beginning of the stable cycle. For the open-electrode system, the charge density of the positive electrode grows linearly from 0 to 9 μC cm−2 in the first half of each cycle and then decreases linearly to 0 in the second half cycle. The period of applied electric current is set as 50, 100 and 500 ps. For the nanoporous-electrode system, the surface charge density on the electrode is set to change linearly between ~0 and 4.96 μC cm−2, with periods of 200, 1,000 and 3,000 ps. For GCD-CPM, the charge on electrode atoms is obtained using the method mentioned above. For GCD-CCM, the potential at each electrode atom is computed as the sum of the potentials in the real space and reciprocal space. To ensure the accuracy of the simulation results, four to five independent simulations are run with different initial configurations for each system.
Calculation of the probability distribution of the electrode atom charge and potential
For GCD-CPM, the probability distribution of electrode atom charges is f(q, t), defined as the ratio of N(q, t) to the total atoms of the electrode. N(q, t) refers to the number of electrode atoms with charges in the range of \(q - {\rm{d}}q/2\) and \(q + {\rm{d}}q/2\) at time t. We set dq as \(\frac{{Q_{\rm{max}} - Q_{\rm{min}}}}{{N_{\rm{bin}}}}\), where Qmax and Qmin are the maximum and minimum charge values of all atoms at the electrode during the charging process, and Nbin is set as 300 here. Similarly, the probability distributions of electrode atom potentials in GCD-CCM can be obtained.
Calculation of effective diffusion
The spatial and temporal distribution of charge densities inside the electrode pore, ρe, is described by the charge diffusion equation (equation (1)). Deff can be calculated by minimizing the difference in the in-pore charge density evolution between the molecular simulation and the diffusion equation. The initial and boundary conditions used to solve the minimization problem are taken directly from molecular simulations. Supplementary Figs. 7 and 8 show the evolution of in-pore charge density inside the electrode pore, obtained by GCD-CPM and GCD-CCM simulations and their fitting based on the minimization problem. Figure 3e and Supplementary Fig. 9 show the effective diffusion inside the positive and negative electrode pores obtained by solving the minimization problem.
Dimensionless diffusion model for GCD
As already mentioned, the charge–discharge process of ionic liquids in a nanopore can be described by the charge diffusion equation (equation (1)) derived from mean-field theory11,31. For GCD, the initial and boundary conditions are set as
where l is the pore length and S is a square-wave function with period and amplitude of one. Equation (7a) indicates that the pore is not charged at t = 0. The boundary condition in equation (7b) is the zero normal flux at z = l (corresponds to the middle position of the open pore in our simulations), and the boundary in equation (7c) indicates an applied square-wave current with period P and amplitude I0, as shown in Fig. 1d.
As shown in Supplementary Fig. 21, the ions are distributed in the nanopore, and the electrode is equipotential. Taking the areal capacitance, C, along the nanopore as a constant, we divide the pore into numerous microcapacitors in parallel, with each having capacitance Cdz. We then get
where ϕ1(z, t) is the potential in the electrolyte and ϕ0(t) the potential on the electrode. Using the notation for the potential drop from the electrolyte to electrode \(\phi \left( {z,t} \right) = \phi _1(z,t) - \phi _0(t)\) and substituting equation (8) into equations (1) and (7), we respectively obtain the diffusion equation for the potential
and initial and boundary conditions
It is worth noting that equations (9) and (10) are in the same form as the transmission line model54,55, because the charging process predicted by the transmission model also exhibits diffusive behavior11.
We define the dimensionless potential as \({{{\bar{{\varPhi}}}}} = \frac{{CD_{\rm{eff}}}}{{I_0l}}\phi\), dimensionless position as Z = z/l, dimensionless time as \({{{\bar{T}}}} = t/\tau\) (\(\tau = l^2/D_{\rm{eff}}\) is the charging time constant) and the dimensionless period as α = P/τ. Hence, equation (9) can be written in dimensionless form as
The initial and boundary conditions become
Accordingly, the potential difference between the positive and negative electrodes, \({{{\mathrm{{\Delta}}}\bar {\varPhi}}}({{{\bar{T}}}})\), could be obtained during the charge–discharge process, given by the GCD curve, as
where
and θ is the Heaviside step function. Therefore, the relation between \({{{{{\Delta}}}\bar {\varPhi}}}\) and \({{{\bar{T}}}}\), described by equation (13), reveals that the dimensionless GCD curve is only determined by the dimensionless period α (that is, the ratio of the electric current period to the charging time constant of the system).
Materials for the experiment
Commercial activated-carbon cloth (ACC-5092-15) was purchased from Kynol, fabricated into the free-standing and binder-free electrode. Before use, it was dried at 120 °C under vacuum for 12 h. The ionic liquid [EMIM][BF4] (Iolitec Technologies) was purified at 85 °C by the Schlenk line for 24 h.
Material characterization
Nitrogen sorption experiments were performed with an Autosorb iQ system (Quantachrome) at 77 K. The activated-carbon cloth was degassed at 10−2 Pa at a temperature of 200 °C for 10 h. The sample was found to exhibit a gravimetric Brunauer–Emmett–Teller surface area of 1,534 m2 g−1. The pore size analysis was calculated (Supplementary Fig. 11) using quenched-solid density functional theory, assuming a slit-shaped pore model. The average pore size of the micropores was calculated as ~0.72 nm on the basis of volume-weighted pore size d50.
Assembly and instrumentation
Before fabrication, two 10-mm-diameter activated-carbon-cloth electrodes were soaked in a 10-ml glass vial filled with [EMIM][BF4] in an argon-filled glove box containing less than 0.01 ppm H2O and O2. A two-electrode electrochemical cell was then fabricated in the same glove box. After adding the electrolyte, the electrochemical cells were hermetically sealed and transferred out of the glove box. Electrochemical measurements were performed on a CS-350H workstation (Wuhan Corrtest Instrument Co.).
Electrochemical measurements
For comparison with modeling, GCD tests were carried out for our cell. The setting times for charging and discharging were equal, which is not the regular setting with voltage limitation. A series of electric currents with different charge–discharge periods (~12–400 s) were used to find the same dimensionless period as for the simulation. Supplementary Fig. 12 shows the GCD curves obtained in the experiment, and their dimensionless periods are the same as those from the GCD-CPM simulations (their actual periods are 20, 260 and 360 s). To enable comparison with modeling, ohmic drops, mainly caused by the electrode resistance23,32 and not reflected in the simulations, were removed from the experimental GCD curves. The treated experimental curves were then compared with the GCD-CPM results using the dimensionless diffusion model, as shown in Supplementary Figs. 13 and 14. Areal capacitances extracted from GCD curves were obtained from24
where C is the areal capacitance of a single electrode, I is the constant current, Δt is the discharge time, A is the surface area of the electrode and ΔV is the voltage change during discharge (the voltage change removes the ohmic drop from the experiments). The areal capacitances obtained by simulations and experiments are shown in Supplementary Fig. 15.
Data availability
The simulation setting data for the galvanostatic charge–discharge of supercapacitors using the CPM and CCM approaches are available from Code Ocean56. The experimental data for the galvanostatic charge–discharge tests were generated on the laboratory electrochemical workstation with the settings described in the Methods section. Source data are provided with this paper.
Code availability
The code for modeling the galvanostatic charge–discharge process in the molecular dynamics simulation using CPM and CCM approaches is available from Code Ocean56.
References
Wang, X. et al. Electrode material–ionic liquid coupling for electrochemical energy storage. Nat. Rev. Mater. 5, 787–808 (2020).
Simon, P. & Gogotsi, Y. Perspectives for electrochemical capacitors and related devices. Nat. Mater. 19, 1151–1163 (2020).
Salanne, M. et al. Efficient storage mechanisms for building better supercapacitors. Nat. Energy 1, 16070 (2017).
Bedrov, D. et al. Molecular dynamics simulations of ionic liquids and electrolytes using polarizable force fields. Chem. Rev. 119, 7940–7995 (2019).
Scalfi, L., Salanne, M. & Rotenberg, B. Molecular simulation of electrode–solution interfaces. Annu. Rev. Phys. Chem. 72, 189–212 (2021).
Yang, L., Fishbine, B. H., Migliori, A. & Pratt, L. R. Molecular simulation of electric double-layer capacitors based on carbon nanotube forests. J. Am. Chem. Soc. 131, 12373–12376 (2009).
Siepmann, J. I. & Sprik, M. Influence of surface topology and electrostatic potential on water/electrode systems. J. Chem. Phys. 102, 511–524 (1995).
Reed, S. K., Lanning, O. J. & Madden, P. A. Electrochemical interface between an ionic liquid and a model metallic electrode. J. Chem. Phys. 126, 084704 (2007).
Vatamanu, J., Borodin, O. & Smith, G. D. Molecular insights into the potential and temperature dependences of the differential capacitance of a room-temperature ionic liquid at graphite electrodes. J. Am. Chem. Soc. 132, 14825–14833 (2010).
Merlet, C. et al. On the molecular origin of supercapacitance in nanoporous carbon electrodes. Nat. Mater. 11, 306–310 (2012).
Kondrat, S., Wu, P., Qiao, R. & Kornyshev, A. A. Accelerating charging dynamics in subnanometre pores. Nat. Mater. 13, 387–393 (2014).
Bi, S. et al. Molecular understanding of charge storage and charging dynamics in supercapacitors with MOF electrodes and ionic liquid electrolytes. Nat. Mater. 19, 552–558 (2020).
Vatamanu, J., Borodin, O., Olguin, M., Yushin, G. & Bedrov, D. Charge storage at the nanoscale: understanding the trends from the molecular scale perspective. J. Mater. Chem. A 5, 21049–21076 (2017).
Wang, Z., Yang, Y., Olmsted, D. L., Asta, M. & Laird, B. B. Evaluation of the constant potential method in simulating electric double-layer capacitors. J. Chem. Phys. 141, 184102 (2014).
Feng, G., Jiang, D. & Cummings, P. T. Curvature effect on the capacitance of electric double layers at ionic liquid/onion-like carbon interfaces. J. Chem. Theory Comput. 8, 1058–1063 (2012).
Yang, J. Y. et al. Reliability of constant charge method for molecular dynamics simulations on EDLCs in nanometer and sub-nanometer spaces. Chemelectrochem 4, 2486–2493 (2017).
Merlet, C. et al. Simulating supercapacitors: can we model electrodes as constant charge surfaces? J. Phys. Chem. Lett. 4, 264–268 (2013).
Pean, C. et al. On the dynamics of charging in nanoporous carbon-based supercapacitors. ACS Nano 8, 1576–1583 (2014).
He, Y. D., Huang, J. S., Sumpter, B. G., Kornyshev, A. A. & Qiao, R. Dynamic charge storage in ionic liquids-filled nanopores: insight from a computational cyclic voltammetry study. J. Phys. Chem. Lett. 6, 22–30 (2015).
Breitsprecher, K. et al. How to speed up ion transport in nanopores. Nat. Commun. 11, 6085 (2020).
Kwabi, D. G., Ji, Y. & Aziz, M. J. Electrolyte lifetime in aqueous organic redox flow batteries: a critical review. Chem. Rev. 120, 6467–6489 (2020).
Li, H. et al. Cooperative CC-CV charging of supercapacitors using multicharger systems. IEEE Trans. Ind. Electron. 67, 10497–10508 (2020).
Noori, A., El-Kady, M. F., Rahmanifar, M. S., Kaner, R. B. & Mousavi, M. F. Towards establishing standard performance metrics for batteries, supercapacitors and beyond. Chem. Soc. Rev. 48, 1272–1341 (2019).
Zhang, S. & Pan, N. Supercapacitors performance evaluation. Adv. Energy Mater. 5, 1401401 (2015).
Dufils, T., Sprik, M. & Salanne, M. Computational amperometry of nanoscale capacitors in molecular simulations. J. Phys. Chem. Lett. 12, 4357–4361 (2021).
Jiang, X., Huang, J., Zhao, H., Sumpter, B. G. & Qiao, R. Dynamics of electrical double layer formation in room-temperature ionic liquids under constant-current charging conditions. J. Phys. Condens. Matter 26, 284109 (2014).
Xu, K. et al. Effects of functional groups and anion size on the charging mechanisms in layered electrode materials. Energy Storage Mater. 33, 460–469 (2020).
Limmer, D. T. et al. Charge fluctuations in nanoscale capacitors. Phys. Rev. Lett. 111, 106102 (2013).
Bazant, M. Z., Storey, B. D. & Kornyshev, A. A. Double layer in ionic liquids: overscreening versus crowding. Phys. Rev. Lett. 106, 046102 (2011).
Fedorov, M. V. & Kornyshev, A. A. Ionic liquids at electrified interfaces. Chem. Rev. 114, 2978–3036 (2014).
Kondrat, S. & Kornyshev, A. Charging dynamics and optimization of nanoporous supercapacitors. J. Phys. Chem. C 117, 12399–12406 (2013).
González, A., Goikolea, E., Barrena, J. A. & Mysyk, R. Review on supercapacitors: technologies and materials. Renew. Sustain. Energy Rev. 58, 1189–1206 (2016).
Lewandowski, A., Olejniczak, A., Galinski, M. & Stepniak, I. Performance of carbon-carbon supercapacitors based on organic, aqueous and ionic liquid electrolytes. J. Power Sources 195, 5814–5819 (2010).
Yamazaki, S., Ito, T., Yamagata, M. & Ishikawa, M. Non-aqueous electrochemical capacitor utilizing electrolytic redox reactions of bromide species in ionic liquid. Electrochim. Acta 86, 294–297 (2012).
Tsai, W. Y., Taberna, P. L. & Simon, P. Electrochemical quartz crystal microbalance (EQCM) study of ion dynamics in nanoporous carbons. J. Am. Chem. Soc. 136, 8722–8728 (2014).
Levi, M. D., Salitra, G., Levy, N., Aurbach, D. & Maier, J. Application of a quartz-crystal microbalance to measure ionic fluxes in microporous carbons for energy storage. Nat. Mater. 8, 872–875 (2009).
Wu, P., Huang, J., Meunier, V., Sumpter, B. G. & Qiao, R. Complex capacitance scaling in ionic liquids-filled nanopores. ACS Nano 5, 9044–9051 (2011).
Vatamanu, J., Vatamanu, M. & Bedrov, D. Non-Faradaic energy storage by room temperature ionic liquids in nanoporous electrodes. ACS Nano 9, 5999–6017 (2015).
Mo, T. et al. Ion structure transition enhances charging dynamics in subnanometer pores. ACS Nano 14, 2395–2403 (2020).
Srimuk, P., Su, X., Yoon, J., Aurbach, D. & Presser, V. Charge-transfer materials for electrochemical water desalination, ion separation and the recovery of elements. Nat. Rev. Mater. 5, 517–538 (2020).
Swift, M. W., Swift, J. W. & Qi, Y. Modeling the electrical double layer at solid-state electrochemical interfaces. Nat. Comput. Sci. 1, 212–220 (2021).
Hod, O., Meyer, E., Zheng, Q. & Urbakh, M. Structural superlubricity and ultralow friction across the length scales. Nature 563, 485–492 (2018).
Ye, J. T. et al. Liquid-gated interface superconductivity on an atomically flat film. Nat. Mater. 9, 125–128 (2010).
Wang, X. et al. Influences from solvents on charge storage in titanium carbide MXenes. Nat. Energy 4, 241–248 (2019).
Lukatskaya, M. R. et al. Cation intercalation and high volumetric capacitance of two-dimensional titanium carbide. Science 341, 1502–1505 (2013).
Gingrich, T. R. & Wilson, M. On the Ewald summation of Gaussian charges for the simulation of metallic surfaces. Chem. Phys. Lett. 500, 178–183 (2010).
Cornell, W. D. et al. A second generation force field for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 117, 5179–5197 (1995).
Merlet, C., Salanne, M. & Rotenberg, B. New coarse-grained models of imidazolium ionic liquids for bulk and interfacial molecular simulations. J. Phys. Chem. C 116, 7687–7693 (2012).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008).
Evans, D. J. & Holian, B. L. The Nose-Hoover thermostat. J. Chem. Phys. 83, 4069–4074 (1985).
Esposito, M. & Monnai, T. Nonequilibrium thermodynamics and Nose-Hoover dynamics. J. Phys. Chem. B 115, 5144–5147 (2011).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Yeh, I.-C. & Berkowitz, M. L. Ewald summation for systems with slab geometry. J. Chem. Phys. 111, 3155–3162 (1999).
De Levie, R. On porous electrodes in electrolyte solutions. I. Capacitance effects. Electrochim. Acta 8, 751–780 (1963).
Janssen, M. Transmission line circuit and equation for an electrolyte-filled pore of finite length. Phys. Rev. Lett. 126, 136002 (2021).
Zeng L. & Feng G. Modeling galvanostatic charge-discharge of nanoporous supercapacitors. Code Ocean https://doi.org/10.24433/co.5990421.V1 (2021).
Acknowledgements
We acknowledge funding support from the National Natural Science Foundation of China (5211101181, 51876072) and the Hubei Provincial Natural Science Foundation of China (2020CFA093). The work at HUST is also supported by the Program for HUST Academic Frontier Youth Team.
Author information
Authors and Affiliations
Contributions
G.F. conceived this research and designed the simulation and experiment. L.Z. performed all simulations. T.W. carried out the experiment. L.Z. and T.Y. derived the dimensionless diffusion model. L.Z. and T.W. drafted the manuscript. All authors contributed to the discussion of results, editing and revising the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Computational Science thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Handling editor: Jie Pan, in collaboration with the Nature Computational Science team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 GCD curves with various periods for open electrode systems.
GCD curves are obtained from GCD-CPM and GCD-CCM simulations.
Extended Data Fig. 2 Distributions of charge and potential on electrodes atoms in nanoporous electrode systems.
a, Spatial distributions of electrode atom charges along pore axis for GCD-CPM simulations. b, Spatial distributions of electrode atom potentials along pore axis for GCD-CCM simulations. The data of each time counts the data of 5 ps around it. Error bars indicate one standard deviation of 4 independent simulations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–20 and Discussion.
Source data
Source Data Fig. 2
Source data for Fig. 2.
Source Data Fig. 3
Source data for Fig. 3.
Source Data Fig. 4
Source data for Fig. 4.
Source Data Fig. 5
Source data for Fig. 5.
Source Data Extended Data Fig. 1
Source data for Extended Data Fig. 1.
Source Data Extended Data Fig. 2
Source data for Extended Data Fig. 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, L., Wu, T., Ye, T. et al. Modeling galvanostatic charge–discharge of nanoporous supercapacitors. Nat Comput Sci 1, 725–731 (2021). https://doi.org/10.1038/s43588-021-00153-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43588-021-00153-5
This article is cited by
-
Molecular Understanding of Heat Transfer in Ionic-Liquid-based Electric Double Layers
Journal of Thermal Science (2023)
-
Engineering a passivating electric double layer for high performance lithium metal batteries
Nature Communications (2022)
