
Abstract
Exposures of the Earth’s crust-mantle transition are scarce, thus, limiting our knowledge about the formation of subcontinental underplate cumulates, and their significance for metal storage and migration. Here, we investigated chalcophile metals to track sulfide crystallization within the Contact Series, an <150-m-thick pyroxenite-gabbronorite sequence, formed by mantle-derived melts, highlighting the boundary between the Balmuccia mantle peridotite and gabbronoritic Mafic Complex of the Ivrea-Verbano Zone. Within the Contact Series, numerous sulfides crystallized in response to the differentiation of mantle-derived underplated melts. Such sulfide-controlled metal differentiation resulted in anomalous Cu contents (up to ~380 ppm), compared to reference mantle (~19 ppm) and crustal samples (~1 ppm). We propose that the assimilation of continental crust material is a critical mechanism driving sulfide segregation and sulfide-controlled metal storage. Our results evidence that sulfides are trapped in the underplated mafic-ultramafic cumulates and that their enrichment in Cu may provide essential implications for crustal metallogeny.
Similar content being viewed by others


Introduction
Exploring the Earth’s crust-mantle transition, a globally-distributed zone beneath the oceanic and continental crust, is one of the most trailblazing research fields, as it provides critical insight into the evolution of our planet1. Its structure is typically shaped by mantle-derived melts reacting with upper mantle and lower crustal rocks2,3,4. These reactions inter alia drive the sulfide-controlled metal migration4,5. As the mantle-derived ascending melts have nearly constantly high Cu content (80–120 ppm)6,7,8 beneath the oceanic and continental crust, the markedly decreased Cu content of the lower continental crust (on average 26 ppm)9 indicates a crucial role of magmatic differentiation at the crust-mantle boundary in sulfide segregation. To explain the discrepancy in the Cu content between the primitive melts and the lower continental crust, the formation of deep-rooted sulfide-bearing cumulates was postulated6,10. However, field evidence for the occurrence of these sulfide-rich cumulates is still limited. For instance, the exhumed lower to middle crustal cumulates from the Kohistan island arc section might have originally contained abundant sulfides, but most of these sulfides seem to have been lost during metamorphism11,12,13.
Sulfide saturation in the melts stagnating at the crust-mantle boundary14,15 was proposed in studies of rocks sampled from both oceanic4,16 and continental lithosphere6,10,17. However, the paucity of large-scale exposures of the continental crust-mantle transition, which is mainly examined by remote investigations18, limits our knowledge of the sulfide distribution and migration of chalcophile metals in this sector of the planet. To elucidate the role of magma underplating on sulfide distribution, we investigated the external contact zone of the Balmuccia mantle massif (BM) and the lower crust, Permian Mafic Complex (MC) within the Ivrea-Verbano Zone (IVZ; Italian Alps; Fig. 1). The IVZ is one of the most complete and worldwide-known examples of the lower crust exhumed at the Earth’s surface18,19, and therefore it is continuously studied providing critical insights into metallogeny processes20,21. For instance, the MC was locally intruded by alkaline mafic-ultramafic pipes containing substantial amounts of disseminated sulfide mineralization of magmatic origin20,21,22,23,24. Specifically, Holwell et al.20 documented that crustal cumulates such as Isola sill and Sella Bassa, emplaced within MC, trapped a high amount of sulfides and associated metals. Therefore, in this contribution, we aim to verify if the IVZ underplated cumulates at the crust-mantle transition may constitute a Cu sulfide repository.

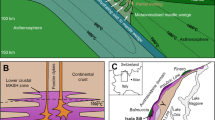
a Simplified geological map of the Ivrea-Verbano Zone, depicting its generalized stratigraphy and main mantle peridotite bodies localized mostly near the Insubric Line. The insert displays the close-up of the geological map of the Balmuccia mantle massif (BM) and the adjacent Mafic Complex (MC)65. b Geological map displaying the external margin of the BM with sampling sites corresponding to the lithological profile presented in Fig. 2. The background image has been produced using © 2023 Maxar satellite image. c Field image of the CS1 sampling site, displaying the primary contact between BM and Contact Series (CS) sequence. The insert reports a CS2 rock sample comprising a sharp pyroxenite-gabbronorite contact. The blue rectangles highlight the thin-sections locations (TS 62–63; Supplementary Data 1).
The studied contact zone (called here Contact Series, CS; Fig. 1b) represents a primary magmatic transition from mantle peridotites to lower crustal gabbroic rocks25,26,27,28, thereby implying that it may be considered as a fossil continental crust-mantle transition zone18,29,30. The BM-CS-MC association formed in the Permian at 0.8–0.9 GPa28,31 and was ultimately exposed in response to the Alpine orogeny (ref. 19 and references therein). Notably, although the CS origin is not fully established, a role of crustal assimilation in the differentiation of the CS-forming mantle magmas was hypothesized, based on limited Sr and Nd isotopic analyses27.
Previous studies of small-scale xenoliths, oceanic-cored samples, and ophiolites documented that melt-mantle reactions are the key differentiation mechanism that control the crystallization of sulfides and related chalcophile element enrichment (e.g., Cu, Ag) during mantle metasomatism17 and at the oceanic crust-mantle transition zone (ref. 32 and references therein). However, in the external zone of BM, the CS rocks revealed crustal signatures27, suggesting the importance of crustal contamination in the subcontinental magma differentiation and sulfide saturation33,34,35. Therefore, in this study, we investigated the CS sequence that constitutes an exceptional large-scale field laboratory, allowing us to determine the role of crustal contamination in sulfide segregation and advance the understanding of metallogeny processes shaping the subcontinental crust-mantle transition zone.
Results
Sulfide enrichment in the Contact Series
The studied contact zone formed at ~25–28 km depth, by magma ponding along the BM boundary27,28,36,37,38. The CS sequence is well-exposed along the eastern side of the BM massif, retaining primary contact relations with BM peridotites (Fig. 1). The most external sector of BM peridotites (mainly spinel lherzolites; Mg# = 91–90; Supplementary Data 2) is highlighted by the local occurrence of cm-scale gabbronorite and darkish pyroxenite veins. We sampled a 5-cm-thick gabbronorite (Mg# = 72) vein (VS8 site) enveloped by 2-cm-thick pyroxenite (Mg# = 75) and a ~80-m-thick CS sequence (Fig. 1b), whose thickness varies at the external contact of BM peridotites39. The CS sequence consists of interlayered pyroxenites and gabbronorites, typically few-tens of centimeters thick, with Mg# ranging from 51 to 71 (Figs. 1 and 2, and Supplementary Fig. 1). We observed an overall Mg# decrease across the CS, from BM to MC, and high contents of chalcophile elements (Cu, Ag) in the CS, especially within CS1 and CS2, relative to CS3, BM lherzolites and MC gabbronorites (Mg# of 65–58; Fig. 2).

a Simplified lithological profile presenting the distribution of the studied samples across the CS sequence, including BM peridotites and MC gabbronorites and corresponding silicate (b) and sulfide (c) modes determined for the studied thin-sections. The whole-rock geochemical data have been plotted for the entire CS sequence including (d) Mg# [molar Mg/(Mg+Fe2+)]), (e) selected chalcophile element (Cu, Ag) and S concentrations, (f) sulfides abundances and bulk S/Se ratios, and (g) bulk S isotope compositions. Note that sulfide contents correlate with bulk S/Se ratio and Cu as well as Ag contents. The violet box in panel f refers to the mantle range of S/Se ratios66,67, whereas the gray box in the bulk S isotopes column (g) indicates the reference values for mantle-derived sulfur of 0 ± 2‰48. In the case when the same sample had two or more overlapping thin-sections, the average value of sulfide contents (f) has been plotted and the error bars refer to 1 SD. The names of sampling sites refer to Fig. 1.
Sulfides in the studied rocks occur as polyphasic grains forming irregular (~82%), rounded (~15%), or elongated (~3%) grains, mainly interstitial to silicates (~80% vs. enclosed 20%; Supplementary Data 3). The Balmuccia lherzolite-hosted sulfides are pentlandite-rich (Pn), whereas those in CS comprise pyrrhotite (Po) with local troilite (Tro) exsolutions (Fig. 3a), massive chalcopyrite crystals (Ccp) mostly at the sulfide rims, and Pn either as Po-enclosed lamellae or irregular crystals (Fig. 3b). These magmatic-origin sulfide assemblages are scarcely (CS1), almost entirely (CS2), or completely (CS3) replaced by pyrite (Py) throughout the CS profile (Fig. 3c and Supplementary Fig. 2). In particular, the abundant Ccp crystals are unevenly dispersed within Py grains in CS2 (Fig. 3c and Supplementary Fig. 2f, g). The inherent sulfides display different mean assemblages: 1) Pn71-Po18-Ccp11 in Balmuccia lherzolites, 2) Po81-Pn18-Ccp1 in gabbronorite VS8 vein within Balmuccia lherzolites, 3) Po84-Pn13-Ccp3 in CS1; 4) Py95-Po3-Ccp2 in CS2; 5) Py99-Ccp1 in CS3, and 6) Py78-Po21-Ccp1 in adjacent MC gabbronorites (Supplementary Data 3). The sulfide mineralogy is reflected in different bulk sulfide chemical compositions (Supplementary Fig. 3). Lherzolite-hosted sulfides are Ni- and Co-rich, as they are dominated by Pn. The sulfides from the gabbronorite vein within BM (sample VS8) and CS1 rocks are distinct within the CS rocks in slightly higher contents of Ni and Co, and lower S content compared with CS2 and CS3 sulfides, which are in turn dominated by Py. In addition, the CS and vein VS8 sulfides are typically associated with interstitial-to-silicates, irregular-shaped ilmenite and magnetite (Fig. 3d). In vein VS8, sulfides predominate over oxides (Supplementary Data 3), whereas the ratios between the sulfides and oxides are typically similar in CS1 and CS2 rocks. Close to MC, in CS3 rocks, oxides predominate over sulfides.

a Back-scattered (BSE) image of a primary sulfide grain hosted by CS1 gabbronorite (Gab) domain displaying (lighter) lamellae of troilite (Tro) exsolved from pyrrhotite (Po; darker) associated with small elongated crystals of magmatic low-temperature (LT) pentlandite (Pn). b The typical sulfide assemblage in a CS1 pyroxenite (Px) including Po, massive grains of chalcopyrite (Ccp) and high-temperature Pn (HT-Pn). c Sulfide assemblage in a CS2 Px displaying partial replacement of primary grains (relict Po) by massive pyrite (Py) with Ccp crystals or blebs enclosed within Py. d Association of sulfides and massive, irregular-shaped ilmenite (Ilm) and magnetite (Mag) in a CS1 Px. Images b to d are reflected light images. e Binary diagram of bulk S versus Se indicating S addition to CS1 and CS2, and S loss in the top CS3 site. The mantle S/Se range is adapted from Eckstrand and Hulbert66. The black dashed lines denote the S/Se ratios. f Binary plot of Sb/Se and As/Se ratios in selected Pn. The inset indicates the effect of processes controlling these ratios. The colored fields are adapted from Mansur et al.35. High Sb/Se and As/Se values suggest an external input of As and Sb in the sulfide liquid by crustal assimilation. To plot Pn composition from the VS8 site, the As and Sb concentrations are calculated as 0.75 of the detection limit. g Binary diagram of Cd/Se versus Ni/Se in Ccp. h Binary diagram of Co/Sb versus Se/As in Py, enabling to discriminate between magmatic and hydrothermal Py. The black dashed lines in diagrams (g) and (h) separate the magmatic and hydrothermal compositional fields68,69, respectively; the colored fields correspond to the Ccp and Py compositions from the magmatic deposits35. All sampling sites abbreviations refer to Fig. 1.
The mantle peridotites are sulfide-poor (on average 0.1 vol.‰; Supplementary Data 3) and have on average 125 ppm S and 19 ppm Cu, in agreement with previously published data5. In CS1 and CS2 rocks, the bulk Mg# are lower than in mantle peridotites, but the modal sulfide abundances are higher, as reflected by elevated S, Cu, and Ag contents (Fig. 2 and Supplementary Fig. 4). In particular, the gabbronorite-pyroxenite vein VS8 (~2.0 vol.‰ of sulfides) and CS1 and CS2 rocks (~2.5 vol.‰ of sulfides) contain ~1740 ppm S and ~80 ppm Cu, and ~2780 ppm S and ~141 ppm Cu, respectively (Fig. 2). Conversely, the Py-bearing CS3 rocks (0.2 vol.‰ of sulfides) reveal ~170 ppm S and ~11 ppm Cu (Fig. 2). The MC gabbronorites are sulfide-poor (Supplementary Data 3) and contain only ~1 ppm Cu. The bulk S/Se ratios for BM peridotites range from 70 to 200 (n = 6). Within the CS zone, CS1 and CS2 rocks display higher S/Se values (1380–24200; n = 10) than CS3 rocks (380–920; n = 5; Supplementary Data 2). The S/Se ratio roughly correlates with Cu and Ag contents and sulfide modes (Fig. 2).
Based on in-situ LA-ICPMS measurements (see Methods; Supplementary Data 4–7), the S/Se ratios of sulfides in BM peridotites are consistently lower (1090–8300; on average 3630; n = 30) compared to CS pyroxenites (2940-26900; on average 9310; n = 85) and gabbronorites (1360–12600; on average 7440; n = 130). The LA-ICPMS results also revealed that all sulfide phases, especially Ccp and Pn, are enriched in highly chalcophile elements (from 101 to 105) compared to the primitive mantle (PM; Supplementary Fig. 5). In particular, Ccp and Pn display a similar range of PM-normalized values for platinum group elements (PGEs), Au, Te, Re, and Se, with higher In, Tl, Sb, Zn, Pd, and Ag values in Ccp. Considering the distribution of PGEs among the sulfide phases, we provide evidence for Ccp containing the highest concentrations of Pd-PGE (with Pt negative anomaly similar to other sulfides), whereas Po and Py display similar, near-constant PM-normalized contents ranging from 100 to 102 (Supplementary Fig. 5). The high values of Pd in Ccp are related to peaks in the LA-ICPMS time resolved spectra and correlate to the detection of Te, suggesting the plausible presence of micro- or nano-scale Pd-Te inclusions. Gold (from 0.1 to 2.1 ppm) and Ag (from 0.1 to 18.1 ppm) were also measured within all sulfide phases (Supplementary Data 4–7), and have PM-normalized values ranging from 101 to 103. Importantly, EPMA measurements (Supplementary Data 8; Supplementary Fig. 6) documented that the CS1-hosted Py is Co-poor (avg. 0.1 wt.%) compared to the Co-bearing Py from CS2 and CS3 rocks (avg. 0.4–1.2 wt.%). This observation was confirmed by LA-ICPMS measurements (Supplementary Fig. 5d). The opposite trend is observed for Ni, as CS1-hosted Py is Ni-rich (up to 0.9 wt.%), whereas most CS2- and CS3-hosted Py is almost Ni-free (Supplementary Fig. 6).
The bulk S isotope signature (δ34S) of mantle peridotites ranges from –0.7 to +0.7‰ (Supplementary Data 9). The δ34S of the gabbronorite-pyroxenite vein VS8 is –0.7‰, and the CS rocks have δ34S of –1.6 to +0.5‰, with one sample displaying an exceptionally low δ34S of –3.3‰. The bulk δ34S of reference MC gabbronorites is +2.0 to +3.1‰ (Fig. 2).
Discussion
Control of crustal contamination on sulfide saturation
The CS sequence was formed by mantle-derived melts that stagnated and evolved at the crust-mantle boundary27, as documented by the Mg# variations (Fig. 2). In addition, the CS growth involved multiple injections of primitive melts (as indicated by elevated Mg# values in CS2 and CS3), which were inferred to have assimilated crustal material27 based on the Nd-Sr isotopic signature (εNd = −1.0, 87Sr/86Sr = 0.7059) of a pyroxenite. This complex evolution of the parental magma evidences that CS recorded an open-system fractionation of mantle-derived underplated melts. Here, we demonstrate that the CS sequence is overall enriched in sulfides and sulfide-loving elements compared to BM and MC (Fig. 2). The subcontinental lithospheric P-T conditions40 allowed the melts to cool and enabled the segregation of typical magmatic (Po-Ccp-Pn) sulfide35 and oxide41 assemblages. The Tro exsolutions within Po evidence low-temperature equilibration of metal-rich Po, suggesting a low S fugacity of the original sulfide melt23. The Tro exsolutions together with co-occurrence of crystals (HT) and oriented-lamellae (LT) of Pn indicate42 stable and long-lasting conditions with a slow-cooling rate.
The interstitial and enclosed textural positions of sulfides hosted by the CS rocks (Fig. 3 and Supplementary Fig. 2) and the same bulk composition of sulfides in both textural positions provide evidence that underplated melts might have become supersaturated in sulfides early upon the emplacement at the base of the crust20,43. Based on sulfide compositions, we thus aimed to decipher the potential role of crustal contamination in sulfide saturation and chalcophile element trapping.
The low S/Se ratios of BM lherzolites (Fig. 2) indicate that BM has become depleted by melt extraction (19 and references therein) without any involvement of crustal components, as documented by bulk-rock chemical and Nd-Sr isotopic compositions44. The S/Se ratios obtained from the gabbronorite-pyroxenite vein VS8 and from CS1 rocks are within or close to the typical mantle values (Fig. 2), pointing to a limited extent of melt-crust interaction. On the other hand, the Cu-enriched samples reveal, independently from the location within CS (Fig. 2 and Supplementary Data 2), elevated S/Se ratios suggesting assimilation of crustal components45 (Figs. 3e and 4). This interpretation is sustained by the trace element Pn signature, which documents an increase of As/Se and Sb/Se values, from BM peridotites towards the CS sequence (Fig. 3f). In particular, the CS Pn has higher As/Se and Sb/Se ratios than those from Duluth and Mirabela Complex deposits35 (Fig. 3f), which were interpreted to have crystallized from crustally contaminated melts46,47. The enrichment in slightly to moderately chalcophile element concentrations of sulfides relative to PM (Supplementary Fig. 5) is consistent with the influence of crustal components. Similar trends were also observed for worldwide Ni-Cu deposits recording the effect of crustal contamination35. In general, as analyzed sulfide phases have relatively homogenous trace element compositions between the CS sites (Supplementary Fig. 5), the entire CS sequence must have assimilated the same crustal material. However, based on the bulk-rock S/Se ratios and Cu contents the amount of assimilated crustal material had to differ locally throughout the CS sequence.

Plot displaying Cu content versus bulk S/Se ratios indicating a positive correlation (R2 = 0.91; linear trend line). The extent of crustal contamination likely controlled the sulfide precipitation resulting in the enrichment of sulfur-loving metals. The dashed lines represent the referenced Cu contents9. The inset presents a simplified reconstruction of the processes responsible for the sulfide differentiation along the subcontinental crust-mantle transition zone (see details in the text).
Evidence of crustal contamination (i.e., S/Se ratios of up to ~11,000, slightly elevated δ34S of up to +2.9 ‰, high initial 87Sr/86Sr of 0.7113 and the low initial εNd of −7.3) was also provided for a several m-thick pyroxenitic layer (Isola sill) exposed near a granulite-facies metasedimentary septum, at deep-levels of the Mafic Complex20,21. The CS rocks may similarly record a process of crustal contamination by metasedimentary material, as indicated by the bulk rock S/Se values and trace element signature of Pn. However, this interpretation is challenged by the bulk rock CS δ34S values (–1.6 to +0.5‰) that are within typical mantle values48 and lower than those observed (up to ~+10 ‰ δ34S) for the basement adjacent to the Mafic Complex23, thereby arguing against a substantial assimilation of the metasedimentary S. Hence, we propose that the sulfides in the CS precipitated in response to a magmatic differentiation associated with variable content of assimilated crustal material, from either the MC gabbronorites or the pre-existing metamorphic basement. We argue that crustal contamination was an important mechanism driving sulfide saturation and therefore chalcophile element trapping in CS. As samples characterized by the highest sulfur and metal contents are dispersed through the CS1 and CS2 sequences, we conclude that crustal assimilation operated on a localized scale and/or was related to the individual injections of magma.
Evolution of CS sulfide mineralization
The studied CS transect reveals the transition expressed by sulfide contents and their mineralogy, from BM towards MC (Fig. 2). Primary magmatic sulfides35,49 occur within the BM-hosted veins and the CS1 sequence (Fig. 3a, b), whereas in CS2 and CS3 sequences, we found evidence that Py replaced pre-existing magmatic sulfide assemblages (Fig. 3c). Additionally, within CS2, we identified relics of primary magmatic sulfides such as Po and Ccp (Fig. 3c), which display similar trace element compositions to CS1 (Supplementary Fig. 5), indicating the same origin and likely contribution of the crustal components. Hence, we assert that the process of crustal assimilation, which most likely drove the sulfide segregation, was unrelated to the crystallization of Py. Furthermore, Po and Py provided similar patterns for PGEs and other highly chalcophile elements (e.g., Au, Te, and Ag; Supplementary Fig. 5b, d), thereby indicating that Py retains the trace element signature of primary magmatic sulfides. Therefore, the Py replacement (sulfurization reactions) might be caused by either re-equilibration with late-magmatic fluids, or processes associated with metamorphism, driving the stabilization of Py over Po35. Chalcopyrite from CS2 and CS3 rocks must have also recrystallized, as it occurs as inclusions within Py. The high ratios of Co/Sb and Ni/Se in Py and Ccp, respectively, indeed suggest the magmatic origin of their primary metal contents (Fig. 3g, h), similar to CS1-hosted sulfide assemblages.
Similar disclosures were made for Pyrenean peridotites in which Co-bearing Py coexists with magmatic sulfides50,51, and provide indisputable evidence for Py (Supplementary Fig. 2) replacing pre-existing mantle sulfides50,52. Additionally, the chalcophile element contents provided evidence for the assimilation of external sulfur, most likely driven from the metamorphosed sedimentary country rocks during the emplacement of peridotite bodies into the upper crust50. In this study, we propose that the original magmatic sulfides were mostly replaced in CS2 and CS3 sequences, whereas the metal contents in newly formed Py and Ccp are mainly inherited from precursors. Following this model, the Py-dominated alteration resulted in minor trace element changes and re-homogenization of the original S isotope signature (Fig. 2). This process may be related to interaction with either deep late-magmatic fluids in the Permian, fluids associated with the late Triassic to early Jurassic rifting evolution of the IVZ, or low-temperature metamorphism related to the exhumation of IVZ (ref. 19 and references therein).
To track sulfide saturation during magma differentiation and subsequent sulfide replacement, we used geochemical data and reconstructed the evolution of subcrustal metallogeny of the CS sequence (Fig. 4). Firstly, upon subcrustal conditions, the differentiation of mantle-derived melts (Fig. 2) resulted in enhanced sulfide segregation (Stage 1; Fig. 3a). Concurrently, sulfur and crustal components addition to CS sequence from the crust (Stage 2) affected the melts, which reached saturation in S due to lowering of the FeO content associated with crystallization of ilmenite and magnetite (Fig. 3d;42,53). This process enhanced the proportion of segregated sulfides, which incorporated the melt-incompatible chalcophile metals (e.g., Cu, Ag). The structurally higher levels of the CS sequence indicate the influence of late fluids leading to the replacement of primary sulfides by Py. Despite the CS2 and CS3 sites are adjacent to each other, they record different degree of pyritization. Holwell et al.54 noted that the alteration of primary magmatic sulfides is ultimately associated with Fe and S losses (Fig. 2e) which are documented for CS3. Upon complete alteration of the primary sulfide assemblages (Stage 3), as observed for CS3, where sulfides are rimmed by hydroxides (Supplementary Fig. 2), the base and precious metals are easily remobilized (ref. 54 and references therein). However, importantly for our study, the pyritization left relics of magmatic sulfides (Po-Ccp) in CS2. The composition of newly crystallized sulfides resembles that of the relics, suggesting a substantial lack of metal mobilization. Furthermore, in the proposed model, we should also consider an alternative/complementary scenario in which the sulfides from the uppermost CS sequence have been mobilized before the pyritization due to reheating20 by ascending melts.
Implications for Cu missing reservoir
The largest Cu mineralizing systems on Earth are related to volcanic arc activity and associated migration of metals and S through the crust forming porphyry-epithermal deposits20,55,56. Accumulation of Cu-rich sulfides within the underplated cumulates has been already postulated as critical for the formation of these deposits6,57. Because the upper crust adjacent to the IVZ is devoid of the Cu porphyry deposits that might have potentially been sourced by the remobilization of sulfides from the deep-seated Cu sulfide reservoirs, we can provide insights into undisturbed sulfide distribution within the lower crustal cumulates and their metal budget. A recent investigation of the sulfide-rich, lower crustal cumulates20 has demonstrated that these rocks trapped large amount of Cu sulfides. Specifically, the several m-thick Isola sill emplaced near the base of MC retains ~308 ppm of Cu20. Importantly, the present study provides a similar sulfide-controlled Cu metal enrichment (up to ~380 ppm Cu) for the CS1 and CS2 underplated cumulates. Our detailed sampling throughout the CS sequence provided evidence for the crystallization of ~80-m-thick CS zone of sulfide- and Cu-rich underplated cumulates which constitute the Cu repository situated at the crust-mantle transition zone.
The identification of sulfide-rich cumulates throughout the IVZ lower crust, starting from the base of underplated cumulates of CS through stratigraphically higher cumulates of the MC20, indicate a critical role of the ascending mantle-derived melts in the upper lithospheric metallogeny. This hypothesis may operate also for other geotectonic settings such as the Kohistan island arc, where modeled metal content of the parental magma that formed the lower crustal cumulates contained up to 140 ppm Cu12. These cumulates could represent an original Cu repository that could be subsequently mobilized, thereby transferring Cu to porphyry Cu deposits in the shallow crust12. In addition, similar Cu contents (up to 112 ppm) were observed for the lower crustal pyroxenitic xenoliths brought to the surface in cratonic margins58, suggesting that the mafic lower crustal rocks have also experienced sulfide segregation and accumulation. Therefore, our studies and the available literature data are consistent with the idea of a global Cu enrichment at the crust-mantle transition, by sulfide entrapment in underplated cumulates.
The sulfide segregation upon magmatic differentiation may be critical for understanding crustal metallogeny. As the CS sequence formed by stagnating mantle-derived melts (80–120 ppm Cu) building the continental crust-mantle transition18, its significantly higher Cu content (up to 14 times) compared to that of the lower continental crust (26 ppm Cu9) implies that Cu is trapped by underplated subcrustal cumulates6,10. Although the discovered Cu enrichment in the CS sequence constitutes an important Cu repository for the lower continental crust, it still doesn’t fully explain the discrepancy between the estimated Cu content in the bulk continental crust and the parental melts. A fraction of the missing Cu content may be also distributed within sulfide-rich ultramafic magmatic rocks dispersed through the MC20,21,23,59. However, the contribution of these rocks to the Cu content of the lower continental crust remains puzzling, due to their unequal distribution and compositions60. As the CS sequence is expected to be a continuous fossil crust-mantle transition layer18, and the ultramafic cumulates mainly form laterally discontinuous or discordant pipe-like bodies, the discovery of the Cu sulfide-rich CS provides critical pieces of evidence for sulfide segregation during the magmatic differentiation responsible for making the continental crust.
The data presented in this study, together with previously published results documenting sulfide-rich cumulates20,21,22,23,24 dispersed through the entire lower IVZ crust, indicate that Cu is heterogeneously distributed through the lower continental crust. Although the paradox of missing Cu reservoir for the continental crust composition has been currently addressed and investigated6,10, it has not been fully solved. We assert that IVZ constitutes an ideal exposure for defining the substantial contribution of mafic-ultramafic cumulates in the Cu metal budget of the lower continental crust, which was previously overlooked and not accounted for global calculations. Such calculations are required to balance the presence of sulfide-rich underplating cumulate complexes and resolve the missing Cu paradox.
Methods
Sampling and bulk rock geochemistry
Sulfide-bearing rock samples were collected from the external margin of the Balmuccia massif (BM), along an >100-m-long transect that comprise ~7-cm-thick mafic/ultramafic VS8 vein (~5-cm-thick gabbronoritic core rimmed by ~2-cm-thick pyroxenite domain) and ~80-m-thick zone composed of interlayered pyroxenites and gabbronorites (called Contact Series; CS). The Contact Series zone comprises three sampling sites located 0–5 m (site CS1), 65–70 m (CS2), and 75–80 m (CS3) from the external margin of the BM peridotites. In addition, reference peridotites (BQ) and gabbronorites (VS9) were collected from outcrops and dumps of the Balmuccia quarry and the Mafic Complex close to Isola village, respectively (Fig. 1). The entire set of samples (thin-sections and whole rocks) is presented in Supplementary Data 1. All samples were subdivided into two portions including smaller portion for thin-section preparation and larger portion destined for whole-rock analyses. Whole-rock samples for major and trace element analysis were carefully cleaned with SiC sandpaper (grit size P120) to remove potential saw contamination and washed three times with deionized water in an ultrasonic bath for 12 min. Subsequently, these samples were crushed to gravel fraction using a Retsch BB51 tungsten carbide jaw crusher. Then, the representative portion (~100–250 g) of the crushed material was grinded by tungsten swing mill at the Institute of Geological Sciences, University of Wroclaw, Poland.
Major element data (Supplementary Data 2) were obtained on pressed powder pellets that comprise 1 g of sample powder and 5 g of LiBO2, which were mixed and melted for 20 min at 1200 °C to yield pellets for the analyses. The pellets were analyzed with a wavelength dispersive PANalytical AXIOS X-ray spectrometer equipped with a Rh X-ray tube at the Federal Institute for Geosciences and Natural Resources (BGR), Hannover, Germany. Calibration of the X-ray spectrometer was performed using a total of ~150 certified international reference materials. Data were automatically and internally corrected for matrix effects and spectral interferences using the de Jongh method. The accuracies and precisions for the whole-rock major element analysis are reported in Supplementary Data 10. Total loss-on-ignition (LOI) was measured on pre-dried powders and after ignition at 1030 °C in a muffle furnace for 10 min. The concentrations of trace elements (Supplementary Data 2) were obtained using the Ultratrace 5 analytical package which is a combination of Instrumental Neutron Activation Analysis (INAA) and 4-acid digestion in a sequence of perchloric, hydrofluoric, hydrochloric, and nitric acids further followed by analysis by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) at the Activation Laboratories Ltd., Canada. If available, >30 g of sample was encapsulated in a polyethylene vial for INAA measurements to counteract the heterogeneous distribution of precious metals. Detailed description of the multi-method analysis is available in the Ultratrace 5 - Total Digestion - ICPMS, INAA subsection of the Methods section of the Actlab website (www.actlabs.com). Accuracies and precisions for trace element data are presented in Supplementary Data 11.
The whole-rock S concentrations (Supplementary Data 2) were measured using an ELTRA CS580 carbon-sulfur analyzer at the Institut of Geology and Paleontology, University of Münster, Germany. Approximately 75–100 mg of sample powder and 0.4–0.9 g of tungsten (V) oxide were placed in a porcelain crucible, which was beforehand heated to 1200 °C in a muffle furnace for several hours to remove potential sulfur compounds, and combusted in an oxygen atmosphere at 1350 °C and analyzed via infrared spectroscopy. We achieved precision of 3.3% (double relative standard deviation; 2RSD) and accuracy 3.6% (2RSD) based on the reference material with 1.41% S. Eleven sample replicates indicate a precision of 2.0% (2RSD). Sulfur extraction and isotopic analyses were carried out at the same laboratory (Supplementary Data 9). Acid volatile sulfides (AVS) and Cr-reducible sulfur (CRS) were extracted sequentially or together as Ag2S, using a modified version of the Canfield’s method61 described by Pedersen et al.62. The extraction was performed in a closed, all-glass digestion vessel, using 6 M HCl and a 1 M CrCl2-HCl solutions. All reactions were conducted in a nitrogen atmosphere. The 34S/32S ratios are reported in the standard delta notation (δ34S) as per mil difference to the Vienna Canyon Diablo Troilite (V-CDT) and measured using a Thermo Finnigan Delta Plus mass spectrometer (Thermo Fisher Scientific, United States) interfaced to an elemental analyzer. The relative uncertainty of ±0.06‰ (2σ) for δ34S was achieved. The measurements were monitored using the IAEA-S1, IAEA-S2, IAEA-S3, and NBS 127 reference materials.
Quantitative mineralogy
The mineral composition of silicates and sulfide grains and their abundances (Supplementary Data 3 and 12) in polished (50-µm-thick) thin sections were determined under transmitted and reflected light using Zeiss Axioplan 2 imaging optical microscope at the Institute of Geology, Adam Mickiewicz University in Poznań, Poland. The silicate modes were calculated based on the point-counting method (Supplementary Data 12), whereas the sulfide contents for the investigated samples were estimated by calculating cumulative areas of sulfide grains. The individual sulfides’ areas were calculated as the area of the most congruent geometric figure. The modal content of sulfide phases was determined using the same method. For most of the described sulfides, we have taken images under reflected light that were used in further analyses and calculations.
In-situ sulfide analysis
Major element compositions of sulfides (Supplementary Data 4–8) were determined using Cameca SX-Five electron microprobe (EMPA) at the Laboratory of Electron Microscopy and Electron Probe Micro-Analysis of the Faculty of Geology, University of Warsaw, Poland. Standard materials for sulfides include oxides (CoO, NiO and Fe2O3), sphalerite (S), chalcopyrite (Cu) and tellurites (Ag). The analyses were carried out with an acceleration voltage of 25 kV, a current of 15 nA, and a focused beam. The detection limits, standard deviations (error, 1 SD), accuracy, and relative precision of the EPMA measurements are reported in Supplementary Data 13. To calculate the bulk composition of sulfides in the Cu-S-Fe-/Ni+Co/ system, the EPMA-based measured concentrations of major elements were recalculated into bulk rock concentrations for the investigated samples. The bulk contents of selected elements were calculated by multiplying the averaged concentration of an element in all analyzed grains from a certain sample (thin-section) [wt.%] with modal content of sulfide in the thin-section [vol.‰], and normalized to 100% to plot them on the Cu-S-Fe-/Ni+Co/ system diagram.
To determine the trace elements concentrations of sulfides (Supplementary Data 4–7) we used an ELEMENT-XR (Thermo Fisher Scientific, Germany) fast-scanning sector field ICPMS coupled to a femtosecond laser ablation (fs-LA) system (Solstice, Spectra-Physics, USA) at the Institute of Mineralogy, Leibniz University of Hannover, Germany. We used instrumental setting and measurement conditions reported by Ciazela et al.4. During tuning, a low ThO/Th of <0.4% was set to limit the rate of oxide formation. At the beginning and end of each session, the NIST610 and PGE-A reference materials were measured as external standards. We used laser beam diameters of 60 µm for the standards and 30–60 µm for sulfide grains. The repetition rate was 10 Hz for the standards and 29–71 Hz for the sulfides. The Fe, Cu, and Ni contents measured with EPMA were used as internal standards for pyrrhotite/pyrite, chalcopyrite/cubanite, and pentlandite, respectively. Data were reduced using the Matlab-based SILLS software63, including drift correction. Furthermore, the light PGEs (that is 99Ru, 101Ru, 103Rh, 104Ru, 105Pd, 106Pd, and 108Pd) isotopes required interference correction for the argides of 59Co, 61Ni, 62Ni, 63Cu, 64Ni, 64Zn, 65Cu, 66Zn, 68Zn. The interferences were corrected by measuring the native Ni, native Cu, as well as ZnS (MAC) reference materials and comparing the pure metal signals to the metal argide signals. Detection limits and sample errors of the LA-ICPMS measurements are presented in Supplementary Data 14–18. The accuracy and precision of the measurements was calculated based on elements contained in both NIST 610 and PGE-A standards. To achieve this, we used Se, and Rh contents occurring in both NIST 610 and PGE-A standards. For example, we achieved accuracy for Se of ~4.8% (247 ± 27 ppm, 1 SD, n = 72) in PGE-A against NIST-610 vs. 236 ppm obtained by Lorand and Alard64. For Au, we achieved an accuracy of ~2.9% (230 ± 21 ppm, 1 SD, n = 72) in PGE-A against NIST-610 vs. 223 ppm obtained by Lorand and Alard64.
Data availability
The dataset that supports the findings of this study is available in the Supplementary Information file. In addition, tables of analytical results are uploaded separately as Supplementary Data files and can be found in the online open-access repository at https://doi.org/10.6084/m9.figshare.24850092.
References
Pistone, M. et al. Joint Geophysical-Petrological Modeling on the Ivrea Geophysical Body Beneath Valsesia, Italy: Constraints on the Continental Lower Crust. Geochem. Geophys. Geosyst. 21, e2020GC009397 (2020).
Hermann, J., Müntener, O. & Günther, D. Differentiation of Mafic Magma in a continental crust-to-mantle transition zone. J. Petrol. 42, 189–206 (2001).
Abily, B. & Ceuleneer, G. The dunitic mantle-crust transition zone in the Oman ophiolite: Residue of melt-rock interaction, cumulates from high-mgo melts, or both? Geology 41, 67–70 (2013).
Ciazela, J. et al. Sulfide enrichment at an oceanic crust-mantle transition zone: Kane Megamullion (23°N, MAR). Geochim. Cosmochim. Acta 230, 155–189 (2018).
Wang, Z. & Becker, H. Abundances of Ag and Cu in mantle peridotites and the implications for the behavior of chalcophile elements in the mantle. Geochim. Cosmochim. Acta 160, 209–226 (2015).
Lee, C. T. A. et al. Copper systematics in arc magmas and implications for crust-mantle differentiation. Science (80). 335, 64–66 (2012).
Jenner, F. E. et al. Chalcophile element systematics in volcanic glasses from the northwestern Lau Basin. Geochem. Geophys. Geosyst. 13, Q06014 (2012).
Ding, S. & Dasgupta, R. The fate of sulfide during decompression melting of peridotite – implications for sulfur inventory of the MORB-source depleted upper mantle. Earth Planet. Sci. Lett. 459, 183–195 (2017).
Rudnick, R. L. & Gao, S. Composition of the continental crust. In Treatise on Geochemistry, vol. 3 (ed. Rudnick, R. L.) https://doi.org/10.1016/B0-08-043751-6/03016-4 (2003).
Chen, K. et al. Sulfide-bearing cumulates in deep continental arcs: The missing copper reservoir. Earth Planet. Sci. Lett. 531, 115971 (2020).
Zhang, J., Chang, J., Wang, R. & Audétat, A. Can Post-Subduction Porphyry Cu Magmas Form by Partial Melting of Typical Lower Crustal Amphibole-Rich Cumulates? Petrographic and Experimental Constraints from Samples of the Kohistan and Gangdese Arc Roots. J. Petrol. 63, 1–22 (2022).
Ahmad, I. et al. Fractionation of Sulfide Phases Controls the Chalcophile Metal Budget of Arc Magmas: Evidence from the Chilas Complex, Kohistan Arc, Pakistan. SEG Spec. Publ. 2, 297–310 (2022).
Luo, C. H., Wang, R., Zhao, Y., Huang, J. & Evans, N. J. Mobilization of Cu in the continental lower crust: A perspective from Cu isotopes. Geosci. Front. 14, 101590 (2023).
Jenner, F. E. Cumulate causes for the low contents of sulfide-loving elements in the continental crust. Nat. Geosci. 10, 524–529 (2017).
Chiaradia, M. Copper enrichment in arc magmas controlled by overriding plate thickness. Nat. Geosci. 7, 43–46 (2014).
González-Jiménez, J. M. et al. Precious metals in magmatic Fe-Ni-Cu sulfides from the Potosí chromitite deposit, eastern Cuba. Ore Geol. Rev. 118, 103339 (2020).
Patkó, L. et al. Iron isotope and trace metal variations during mantle metasomatism: In situ study on sulfide minerals from peridotite xenoliths from Nógrád-Gömör Volcanic Field (Northern Pannonian Basin). Lithos 396–397, 106238 (2021).
Ryberg, T. et al. 3D imaging of the Balmuccia peridotite body (Ivrea-Verbano zone, NW-Italy) using controlled source seismic data. Geophys. J. Int. ggad182, https://doi.org/10.1093/gji/ggad182 (2023).
Decarlis, A., Ceriani, A., Zanetti, A., Abimbola, C. O. & Tribuzio, R. The Ivrea-Verbano tectonic evolution: The role of the crust- mantle interactions in rifting localization. Earth Sci. Rev., https://doi.org/10.1016/j.earscirev.2023.104318 (2023).
Holwell, D. A. et al. Mobilisation of deep crustal sulfide melts as a first order control on upper lithospheric metallogeny. Nat. Commun. 13, 1–12 (2022).
Fiorentini, M. L. et al. Post-collisional alkaline magmatism as gateway for metal and sulfur enrichment of the continental lower crust. Geochim. Cosmochim. Acta 223, 175–197 (2018).
Locmelis, M. et al. Sulfur and metal fertilization of the lower continental crust. Lithos 244, 74–93 (2016).
Garuti, A., Fiandri, G. & Rossi, A. P. Sulfide composition and phase relations in the Fe-Ni-Cu ore deposits of the Ivrea-Verbano basic complex (western Alps, Italy). Miner. Depos. 34, 22–34 (1986).
Garuti, G., Naldrett, A. J. & Ferrario, A. Platinum-Group Elements in Magmatic Sulfides from the Ivrea Zone: Their Control by Sulfide Assimilation and Silicate Fractionation. Econ. Geol. 85, 328–336 (1990).
Quick, J. E. et al. Geologic Map of the Southern Ivrea-Verbano Zone, Northwestern Italy. USGS Geol. Investig. Ser. 27, 128 (2003).
Quick, J. E., Mainz, D. & Sinigoi, S. Emplacement of mantle peridotite in the lower continental crust, Ivrea-Verbano zone, northwest Italy. Geology 23, 739–742 (1995).
Shervais, J. W. & Mukasa, S. B. The Balmuccia Orogenic Lherzolite Massif, Italy. J. Petrol. 2, 155–174 (1991).
Mukasa, S. B. & Shervais, J. W. Growth of subcontinental lithosphere: evidence from repeated dike injections in the Balmuccia lherzolite massif, Italian Alps. Lithos 48, 287–316 (1999).
Shervais, J. W. Thermal emplacement model for the alpine lherzolite massif at Balmuccia, Italy. J. Petrol. 20, 795–820 (1979).
Rivalenti, G., Garuti, G., Rossi, A., Siena, F. & Sinigoi, S. Existence of Different Peridotite Types and of a Layered Igneous Complex in the Ivrea Zone of the Western Alps. J. Pet. 22, 127–153 (1981).
Kunz, B. E. & White, R. W. Phase equilibrium modelling of the amphibolite to granulite facies transition in metabasic rocks (Ivrea Zone, NW Italy). J. Metamorph. Geol. 37, 935–950 (2019).
Ciazela, J. et al. Thin crust and exposed mantle control sulfide differentiation in slow-spreading ridge magmas. Geology 45, 935–938 (2017).
Wang, C. et al. Constraints on sulfide saturation by crustal contamination in the Shitoukengde Cu-Ni deposit, East Kunlun orogenic belt, northern Qinghai-Tibet Plateau, China. Geosci. J. 25, 401–415 (2021).
Tang, D. M., Qin, K. Z., Sun, H., Su, B. X. & Xiao, Q. H. The role of crustal contamination in the formation of Ni-Cu sulfide deposits in Eastern Tianshan, Xinjiang, Northwest China: Evidence from trace element geochemistry, Re-Os, Sr-Nd, zircon Hf-O, and sulfur isotopes. J. Asian Earth Sci. 49, 145–160 (2012).
Mansur, E. T., Barnes, S. J. & Duran, C. J. An overview of chalcophile element contents of pyrrhotite, pentlandite, chalcopyrite, and pyrite from magmatic Ni-Cu-PGE sulfide deposits. Miner. Depos. 56, 179–204 (2021).
Demarchi, G., Quick, J. E., Sinigoi, S. & Mayer, A. Pressure gradient and original orientation of a lower-crustal intrusion in the Ivrea-Verbano Zone, northern Italy. J. Geol. 106, 609–622 (1998).
Peressini, G., Quick, J. E., Sinigoi, S., Hofmann, A. W. & Fanning, M. Duration of a Large Mafic Intrusion and Heat Transfer in the Lower Crust: a SHRIMP U–Pb Zircon Study in the Ivrea–Verbano Zone (Western Alps, Italy). J. Petrol. 48, 1185–1218 (2007).
Karakas, O. et al. The pace of crustal-scale magma accretion and differentiation beneath silicic caldera volcanoes. Geology 47, 719–723 (2019).
Beltrando, M. Can the scale of observation hide complexities in the deformation history of a terrane? An examples from Balmuccia peridotite massif, Ivrea Zone (NW Italy). Ofioliti 46, 107–116 (2021).
Smythe, D. J., Wood, B. J. & Kiseeva, E. S. The S content of silicate melts at sulfide saturation: New experiments and a model incorporating the effects of sulfide composition. Am. Mineral. 102, 795–803 (2017).
Pang, K. N., Zhou, M. F., Lindsley, D., Zhao, D. & Malpas, J. Origin of Fe-Ti oxide ores in mafic intrusions: Evidence from the Panzhihua intrusion, SW China. J. Petrol. 49, 295–313 (2008).
Pieterek, B. et al. Sulfide enrichment along igneous layer boundaries in the lower oceanic crust: IODP Hole U1473A, Atlantis Bank, Southwest Indian Ridge. Geochim. Cosmochim. Acta 320, 179–206 (2022).
Patten, C., Barnes, S. J., Mathez, E. A. & Jenner, F. E. Partition coefficients of chalcophile elements between sulfide and silicate melts and the early crystallization history of sulfide liquid: LA-ICP-MS analysis of MORB sulfide droplets. Chem. Geol. 358, 170–188 (2013).
Hartmann, G. & Hans Wedepohl, K. The composition of peridotite tectonites from the Ivrea Complex, northern Italy: Residues from melt extraction. Geochim. Cosmochim. Acta 57, 1761–1782 (1993).
Smith, J. W., Holwell, D. A., McDonald, I. & Boyce, A. J. The application of S isotopes and S/Se ratios in determining ore-forming processes of magmatic Ni-Cu-PGE sulfide deposits: A cautionary case study from the northern Bushveld Complex. Ore Geol. Rev. 73, 148–174 (2016).
Samalens, N., Barnes, S. J. & Sawyer, E. W. A laser ablation inductively coupled plasma mass spectrometry study of the distribution of chalcophile elements among sulfide phases in sedimentary and magmatic rocks of the Duluth Complex, Minnesota, USA. Ore Geol. Rev. 90, 352–370 (2017).
Knight, R. D., Prichard, H. M. & Filho, C. F. F. Evidence for as contamination and the partitioning of Pd into pentlandite and Co + platinum group elements into pyrite in the fazenda mirabela intrusion, Brazil. Econ. Geol. 112, 1889–1912 (2017).
Seal, R. R. Sulfur isotope geochemistry of sulfide minerals. Rev. Mineral. Geochemistry 61, 633–677 (2006).
Holwell, D. A. & McDonald, I. A review of the behaviour of platinum group elements within natural magmatic sulfide ore systems. Platin. Met. Rev. 54, 26–36 (2010).
Lorand, J. P. & Alard, O. Pyrite tracks assimilation of crustal sulfur in Pyrenean peridotites. Mineral. Petrol. 101, 115–128 (2011).
Lorand, J. P. Mineralogy and chemistry of Cu-Fe-Ni sulfides in orogenic-type spinel peridotite bodies from Ariege (Northeastern Pyrenees, France). Contrib. to Mineral. Petrol. 103, 335–345 (1989).
Lorand, J. P. & Alard, O. Determination of selenium and tellurium concentrations in Pyrenean peridotites (Ariege, France): New insight into S/Se/Te systematics of the upper in mantle samples. Chem. Geol. 278, 120–130 (2010).
Liu, Y., Samaha, N. T. & Baker, D. R. Sulfur concentration at sulfide saturation (SCSS) in magmatic silicate melts. Geochim. Cosmochim. Acta 71, 1783–1799 (2007).
Holwell, D. A. et al. Low temperature alteration of magmatic Ni-Cu-PGE sulfides as a source for hydrothermal Ni and PGE ores: A quantitative approach using automated mineralogy. Ore Geol. Rev. 91, 718–740 (2017).
Wilkinson, J. J. Triggers for the formation of porphyry ore deposits in magmatic arcs. Nat. Geosci. 6, 917–925 (2013).
Richards, J. P. Postsubduction porphyry Cu-Au and epithermal Au deposits: Products of remelting of subduction-modified lithosphere. Geology 37, 247–250 (2009).
Sillitoe, R. H. Copper Provinces. In Geology and Genesis of Major Copper Deposits and Districts of the World: A Tribute to Richard H. Sillitoe (eds. Hedenquist, J. W., Harris, M. & Camus, F.) 1–18 (Society of Economic Geologists Special Publication, 2012).
Zhang, G. et al. Copper mobilization in the lower continental crust beneath cratonic margins, a Cu isotope perspective. Geochim. Cosmochim. Acta 322, 43–57 (2022).
Tribuzio, R. et al. The peridotite-pyroxenite sequence of Rocca d’Argimonia (Ivrea-Verbano Zone, Italy): Evidence for reactive melt flow and slow cooling in the lowermost continental crust. Chem. Geol. 619, 121315 (2023).
Garuti, G., Bea, F., Zaccarini, F. & Montero, P. Age, geochemistry and petrogenesis of the ultramafic pipes in the Ivrea Zone, NW Italy. J. Petrol. 42, 433–457 (2001).
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M. & Berner, R. A. The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155 (1986).
Pedersen, L. E. R., Staudigel, H., McLoughlin, N., Whitehouse, M. J. & Strauss, H. A multiple sulfur isotope study through the volcanic section of the Troodos ophiolite. Chem. Geol. 468, 49–62 (2017).
Guillong, M., Meier, D. L., Allan, M. M., Heinrich, C. A. & Yardley, B. W. D. SILLS: A Matlab-Based Program for the Reduction of Laser Ablation ICP–MS Data of Homogeneous Materials and Inclusions. Mineral. Assoc. Canada Short Course 40, 328–333 (2008).
Lorand, J. P. & Alard, O. Platinum-group element abundances in the upper mantle: New constraints from in situ and whole-rock analyses of massif central xenoliths (France). Geochim. Cosmochim. Acta 65, 2789–2806 (2001).
Sinigoi, S., Quick, J. E., Demarchi, G. & Peressini, G. The Sesia magmatic system. In The Geology of Italy (eds. Beltrando, M., Peccerillo, A., Mattei, M., Conticelli, S. & Doglioni, C.) 1–33 (Jour. Virtual Explorer, 2010).
Eckstrand, O. R. & Hulbert, L. J. Selenium and the source of sulfur in magmatic nickel and platinum deposits [abs.]. Geol. Assoc. Can. Mineralogical Assoc. Can. Progr. Abstr. 12, 40 (1987).
Hattori, K. H., Arai, S. & Clarke Barrie, D. B. Selenium, tellurium, arsenic and antimony contents of primary mantle sulfides. Can. Mineral. 40, 637–650 (2002).
George, L. L., Cook, N. J., Crowe, B. B. P. & Ciobanu, C. L. Trace elements in hydrothermal chalcopyrite. Mineral. Mag. 82, 59–88 (2018).
Duran, C. J., Barnes, S. J. & Corkery, J. T. Chalcophile and platinum-group element distribution in pyrites from the sulfide-rich pods of the Lac des Iles Pd deposits, Western Ontario, Canada: Implications for post-cumulus re-equilibration of the ore and the use of pyrite compositions in exploration. J. Geochemical Explor. 158, 223–242 (2015).
Acknowledgements
We thank editors Renbiao Tao and Joe Aslin for their guidance as well as David Holwell and Rui Wang for their thorough and insightful comments that significantly improved the overall quality of the manuscript. This research was funded by the National Science Centre of Poland project no. 2018/31/N/ST10/02146. B. Pieterek was financially supported by the Adam Mickiewicz University Foundation in the academic year 2022/2023. No permissions were required for sampling campaign.
Author information
Authors and Affiliations
Contributions
Conceptualization: B.P., R.T., M.M-M., and J. C. Methodology: B.P., I.H., S.W., H.S., and T.K. Data acquisition and curation: B.P., I.H., and T.K. Investigation: B.P., R.T., and M.M-M. Visualization: B.P. Writing—original draft: B.P. Writing—review and editing: B.P., R.T., M.M-M., and A.M. Project administration: B.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Rui Wang, David Holwell and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Renbiao Tao, Joe Aslin, Clare Davis and Martina Grecequet. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pieterek, B., Tribuzio, R., Matusiak-Małek, M. et al. Underplated melts control sulfide segregation at the continental crust-mantle transition. Commun Earth Environ 5, 45 (2024). https://doi.org/10.1038/s43247-024-01218-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-024-01218-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
