
Abstract
Environmental phosphate concentrations are typically much lower (~10−6 M) than needed for prebiotic phosphorylation of nucleosides, critical for the origin of life. Here, we tested hypotheses explaining highly concentrated dissolved phosphate in carbonate-rich “soda” lakes by examining phosphorus and nitrogen cycling in Last Chance Lake and Goodenough Lake, Canada. We find a lack of geochemical phosphorus precipitation, that sedimentary calcium is in dolomite rather than apatite, and that N2-fixation rates, probably suppressed by high salinity, are too low to create significant biological phosphate demand. Thus, nitrogen-limitation of biological production and precipitation of calcium-rich carbonate instead of apatite combine to allow unimpeded evaporative phosphate buildup in Last Chance Lake to the highest known natural levels (37 mM) due to small biological and geochemical phosphorus sinks. Forming on basaltic rock, which was likely common on early Earth, evaporative soda lakes were consequently plausible origin-of-life settings with sufficient phosphate for prebiotic synthesis.
Similar content being viewed by others
Introduction
Evaporative lakes have been identified as promising settings for the origin of life on Earth during the Hadean (4.6–4.0 Ga BP) because they can concentrate the major biogenic elements (C, H, N, O, P, S) as NOx− or NH4+, sulfite or sulfide, cyanide, and phosphate1,2,3,4. Their concentration is crucial for enabling the prebiotic synthesis of nucleotides, amino acids, sugars, and lipid precursors5. “Soda lakes”, which are alkaline and saline water bodies dominated by sodium, (bi)carbonate, and chloride ions, are often rich in phosphate2,6,7,8 and may therefore resolve the “phosphate problem of the origin of life”2,9,10—a discrepancy between the typically low environmental phosphate concentrations (~10−6 M11,12) and the high levels (~0.1–1 M) used in prebiotic nucleotide-forming phosphorylation experiments13,14,15,16.
The phosphorylation reaction binding phosphate to a nucleoside is a crucial step toward prebiotic production of RNA, widely considered a key molecule for the origin of life17. Nucleosides can form in geochemically plausible reaction networks from hydrogen cyanide18, which would have been created in abundant amounts in the atmosphere after inevitable large impacts on the early Earth19,20. Cyanide could have been concentrated and stockpiled as ferrocyanide in evaporative lakes1,5. Nucleoside phosphorylation is a condensation reaction, so the dry phase of wet–dry cycles in evaporative lakes may have provided the physical environment to drive this reaction in addition to concentrating reactants14. The ability for phosphate to stay in solution to very high concentrations during dehydration is crucial for yielding the large nucleotide concentrations (tens of mM21) necessary for non-enzymatic RNA primer extension enabling abiogenesis. Prebiotic nucleoside phosphorylation has been demonstrated at sodium phosphate concentrations ranging from 40 mM prior to further concentration from the dry down initiating the reaction16 and up to 1 M in solution13. Additionally, phosphate at mM to tens of mM concentration acts as a catalyst for the prebiotic synthesis of nucleoside precursors22.
Environmental dissolved phosphate concentrations, however, are normally orders of magnitude lower (in the low µM or sub-µM range) because of (i) loss into insoluble calcium phosphate (apatite), and (ii) biological uptake with eventual sediment burial of part of the resulting biomass, which constitutes the largest (~50%) and second largest (~25%) oceanic phosphorus (P) sinks, respectively23,24. Yet, in soda lakes, the apatite sink has been hypothesized to be abated because calcium carbonate precipitation causes low calcium concentrations that prevent apatite from reaching saturation2,25,26. This might explain phosphate concentrations ≥1 mM in some soda lakes (Supplementary Table 1). However, the hypothesis, which is supported by a correlation between dissolved inorganic carbon and dissolved phosphate in soda lakes2, has not been proven by examining sedimentary calcium phases and demonstrating that phosphate concentrates with a degree of evaporation in the environment. Also, the role of biological phosphate uptake in this hypothesized predicament in modern settings has not been assessed either.
Understanding the biological phosphate sink, while obviously absent at the origin of life, is key to interpreting P cycling in modern soda lakes if they are to serve as effective analogs to the origin-of-life environment and for more accurate inference of phosphate levels in prebiotic soda lakes. P and inorganic nitrogen (mainly nitrate and ammonium) are the two macronutrients sustaining biological production. Following uptake into biomass, both nutrients can be recycled within the biosphere by remineralization or lost to sediment burial. But whereas diazotrophic (N2-fixing) microbes can make atmospheric N2 bioavailable, physical and geochemical factors (e.g., transport and mixing, solubility) largely determine the sources of dissolved P available to the biosphere. Thus, P is often considered the ultimate limiting nutrient because ecological selection for biological N2-fixation (BNF) can overcome nitrogen (N) limitation and balance P and fixed N (=all N except N2) over sufficient temporal and spatial scales by negative feedback12. Indeed, experiments often find that low fixed N concentrations favor BNF, while many empirical challenges to the notion that BNF rates scale with fixed N and P concentrations remain27. Unlike the ocean’s “Redfield ratio” of N:P = 16:128, N:P ratios in lakes tend to be higher and more variable because of smaller volumes and more influential source water chemistry29,30. BNF was shown to balance a lake’s N:P ratio following experimental phosphate addition31 but did not balance high agricultural phosphate input in some other lakes32.
Primary productivity in different soda lakes has been reported to be limited by a variety of factors, including light33,34, nitrogen35,36, and phosphorus37. While this clearly suggests inconsistent control by localized factors, N loss due to NH3 volatilization at alkaline pH is likely a common factor affecting nutrient ratios in soda lakes36,38,39. BNF rate measurements in soda lakes are rare with almost none in their water columns40. Steward et al.41 found no detectable BNF rates in the water column of Mono Lake (California) despite phosphate (~0.1 mM) exceeding fixed N concentrations, whereas BNF by phototrophic mats was observed in Mono Lake’s littoral benthos42,43 and in hypersaline soda lakes in the Kulunda Steppe (Russia)44,45. To our knowledge, no BNF rate measurements have been reported from any lake with phosphate concentrations ≥1 mM (Supplementary Table 1). Yet, such measurements are crucial for understanding the extent of biological control on phosphate concentrations in modern soda lakes and, by inference, phosphate concentrations in prebiotic soda lakes that are potentially higher than in inhabited modern analogs.
Here, we present new field data from Last Chance Lake (LCL), which has the highest reported natural phosphate concentrations on Earth2,7. We compare LCL to Goodenough Lake (GL), which is only ~100 m from LCL, with the goal of understanding why it has substantially lower phosphate concentrations. We suspected that a key difference may lie in GL’s thick cyanobacterial mats (absent from LCL), which could create biological phosphate demand by N2-fixation. Combining geochemical measurements and biogeochemical approaches to measure BNF rates and identify N as a limiting nutrient to primary productivity, we field-tested the hypothesis that soda lakes can concentrate phosphate to extreme levels because of reduced apatite precipitation in the presence of high carbonate concentrations2,25,26 and showed why high accumulation of the macronutrient phosphate in LCL occurs despite biology’s presence.
Results and discussion
The physical and biological context of Last Chance and Goodenough soda lakes
LCL and GL sit on a thin layer (1–5 m) of glacial till overlying flood basalts of the Cariboo Plateau, British Columbia46,47 and have various physical and biogeochemical similarities and differences. These adjacent but unconnected closed-basin (=no outflow) lakes (Fig. 1) are predominantly fed by direct precipitation and groundwater discharge48, which therefore are also the likely main inputs of P to the lakes (water and sediments). We sampled the lakes in November 2021, June 2022, and September 2022 (Supplementary Fig. 1). In June, the lakes reached maximum depths of ~25 cm (LCL) and ~110 cm (GL), with evaporites mostly absent. LCL’s water column was opaque (Supplementary Table 2; Supplementary Fig. 2a), probably because of wind-stirred sediments rather than abundant phototrophs given that chlorophyll (Chl) a was about as low as the much less turbid GL (Supplementary Table 2). By September, LCL had dried up while GL’s water depth had fallen by some decimeters. LCL water samples in September were dense brines enclosed between salt crusts (>300 g/L; Supplementary Fig. 2d) or less saline (~120 g/L) rainwater–brine mixtures sampled after strong rainfall (Supplementary Fig. 1b, c). The latter includes the samples used to determine September’s BNF rates. In November, GL’s near-shore was covered by thin ice, while LCL’s concentrated brine and partially frozen salt crust underwent daily freeze-thaw cycles, which expanded the water volume enough during the daytime to cover most of the mudflat.

Markers indicate the location of the lakes (51°19’44N, 121°38’18W) and sampling stations GL1–GL5 and LC1–LC10. © Mapbox, © OpenStreetMap.
The biology of LCL and GL differs. We observed thin, green microbial mats only at the fringes of LCL during the wet season (Supplementary Fig. 2a). These mats are dominated by photoautotrophic eukaryotes incapable of N2-fixation49 and may be confined near-shore because water turbidity blocks sunlight at depth (Supplementary Table 2). In GL, thick perennial microbial mats (~10 cm) cover most of the benthos and are dominated near-surface by photo-diazotrophic cyanobacteria49,50.
The geochemistry underlying extreme phosphate concentration in the soda lakes
Sodium (Na), dissolved inorganic carbon (DIC) and chloride (Cl-) were dominant ions in both lakes with higher concentrations in LCL (Table 1, Supplementary Fig. 3). These are typical major ions of soda lakes developed on basaltic rock46,51. Sulfate (SO42−), potassium (K), phosphate (PO43−), and magnesium (Mg) were also abundant, whereas calcium (Ca), iron (Fe), and dissolved inorganic nitrogen (DIN = NO3− + NO2− + NH3 + NH4+) were present at very low concentrations. LCL salinity (total dissolved solids) had stronger seasonal variations and was generally higher than in GL (Table 1), reaching up to 462 g/L (≈13 × seawater salinity). Both lakes had pH ≥ 9.9 and perennially oxic water columns (Supplementary Table 2).
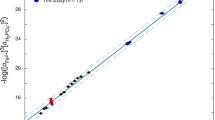
Across sampling stations (Fig. 1) and seasons (June, September, November), phosphate in LCL varied linearly with chloride (Fig. 2), which behaves conservatively during soda lake evaporation and can therefore be used as a salinity proxy (Supplementary Fig. 4f)7,52. The highest total dissolved phosphorus (tdP), phosphate, and chloride concentrations were measured in LCL’s concentrated brines in September 2022 (tdP = 37 ± 9.0 mM, PO43− = 37 ± 0.37 mM, Cl− = 2.5 ± 0.0041 M), while spring water feeding LCL had much lower concentrations (tdP = 21 ± 15 µM, PO43− = 5.7 ± 0.057 µM, Cl− = 4.5 ± 3.2 mM). Phosphate concentrations in Table 1 are seasonal averages. Mean PO43:Cl− ratios in LCL water and sediment porewater (GL water: 0.002 ± 0.001; GL sediment: 0.006 ± 0.005; LCL water: 0.02 ± 0.006; LCL sediment: 0.03 ± 0.009) were an order of magnitude higher than in GL suggesting that LCL’s higher phosphate concentrations are not simply because it is more evaporated (i.e., more saline and so more Cl−-rich) than GL. Instead, GL’s lower PO43−:Cl− ratios and lack of correlation between chloride and phosphate (Fig. 2) or tdP (Supplementary Fig. 4a) suggest stronger P sinks, i.e., biological uptake (supported by findings described below) and/or P mineral precipitation, in GL compared to LCL.

Linear regression of phosphate vs. chloride across seasons and sampling sites in Last Chance Lake (LCL) and Goodenough Lake (GL) water column (crosses: gray = LCL; pink = GL) and sediment porewater (inverted triangles: black = LCL; red = GL). The inset shows the much lower levels of phosphate and a lack of correlation with chloride in GL.
In both lakes, chloride correlated with monovalent cations (Na, K) but not with divalent (Ca, Mg) ones (Supplementary Fig. 4), suggesting that the latter precipitated at comparatively low salinities. Furthermore, bulk sediment Ca substantially exceeded dissolved Ca concentrations, especially in LCL, showing early Ca precipitation into the sediments (Supplementary Figs. 5 and 6). Authigenic calcium carbonate formation was substantiated by X-ray diffraction (XRD), showing dolomites (22.3 ± 8.21%) predominating in the non-clay phase of LCL sediments after plagioclase (38.5 ± 13.4% anorthite and 31.5 ± 20.3% albite; Supplementary Table 3), consistent with previous measurements46. Plagioclase weathering produces Na–HCO3–CO3 dominated water chemistry from which calcite precipitates early and completely upon evaporation53.
Unlike sediments, evaporites from the surface or shores of LCL (Supplementary Table 3; ref. 46) did not contain detectable dolomites or other authigenic calcium minerals and consisted of a distinct set of mostly sodium (bi)carbonate minerals, predominantly thermonatrite (Na2CO3·H2O; 66.1 ± 20.5%), which likely represented natron (Na2CO3·10H2O) that dehydrated after sampling. GL’s sediment and evaporite mineralogy only differed marginally from LCL’s, apart from thermonatrite, which occurred abundantly in GL sediments (Supplementary Table 3). Evaporite minerals containing phosphorus or authigenic iron were not found with XRD and their low abundance in LCL evaporites was confirmed by ICP–MS (P: 0.13 ± 0.08%; Fe: 0.11 ± 0.19%; Ca: 0.10 ± 0.17%) (Supplementary Fig. 6). Consistent with Toner and Catling’s hypothesis2, these data suggest that early calcium carbonate precipitation into the sediments depletes dissolved Ca in both lakes (Table 1), causing negligible P precipitation that allows for extreme phosphate accumulation in LCL.
Low N2-fixation limits biological phosphate uptake in Last Chance Lake
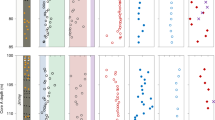
To address why biology does not deplete phosphate in LCL, we tested a hypothesis that primary production could be N-limited. DIN was several orders of magnitude lower than phosphate in LCL (Table 1) with DIN:PO43− ratios far below the Redfield ratio ([16N:1P]; ln(16) = 2.77; Fig. 3b). By contrast, DIN:PO43− ratios in GL approximated the Redfield ratio (Fig. 3a). Accordingly, particulate N:P ratios in GL were close to the Redfield ratio and within the typical range of lakes29,30, but LCL ratios were far lower (Fig. 3c, d).

Data from different seasons are shown for Goodenough (top row) and Last Chance (bottom row) lake water (blue shading) and sediment (brown shading), excluding data from Last Chance Lake’s spring. a, b Dissolved inorganic N:P ratios (DIN:PO43− in water, [NH4+ + NH3]:PO43− in sediment). c, d N:P ratios in suspended water particles and bulk sediment. e, f δ15N in suspended water particles and bulk sediment. Different marker symbols and line types indicate different sampling stations across each lake as specified in Fig. 1 (see Supplementary Fig. 5 for legend). Vertical dotted lines indicate the arithmetic mean of water and sediment measurements, respectively. DIN = dissolved inorganic nitrogen = NH4+ + NH3 + NO2− + NO3−.
We further demonstrated N-limitation in LCL by the addition of an ammonium-nitrate mixture to bottles filled with lake water. For LCL, this N-fertilization led to doubled Chl a concentration within ~48 h of incubation in natural sunlight, while controls that were untreated or amended with sodium-phosphate showed no increase (Fig. 4b). By contrast, the ammonium-nitrate fertilization did not stimulate growth in GL (Fig. 4a), suggesting N-limitation in LCL but not GL.

Time series of chlorophyll a concentration during incubation in natural light following experimental amendment of a Goodenough Lake, and b Last Chance Lake water with no fertilizer (“control”), 100 µmol N L−1 of a 1:1 (NH4)2SO4 + NaNO3 mixed solution (“+nitrate + ammonium”), or a 100 µmol P L−1 of a Na3PO4 solution (“+phosphate”). Error bars indicate the standard deviation from replicate chlorophyll a analyses.
Next, we examined why N-limitation in LCL was not overcome by biological N2-fixation (BNF). The stable isotope composition of particulate organic nitrogen, δ15NPON, ranged from 6.9‰ to 16.4‰ in LCL (Fig. 3f) and 2.0–12.4‰ in GL (Fig. 3e). The high values in LCL and much of GL can be explained by ammonium being a nitrogen source for primary producers that was enriched in 15N from NH3-volatilization at high pH39. N2-fixing diazotrophs produce biomass with δ15NPON ≤ 1.2‰54, which was only approximated in the surface stratum of GL’s microbial mats (≥2.0‰), suggesting BNF as the locally predominant N-assimilation pathway there (Fig. 3e). Indeed, GL sediments (including microbial mats) showed a negative correlation (R2 = 0.48) between bulk N:P (Fig. 3c) and δ15NPON (Fig. 3e), which is consistent with BNF causing both lower δ15NPON values and higher (more balanced) N:P ratios.
To quantify BNF rates, we experimentally enriched lake water with 15N2. Measured rates were similar between the two lakes with higher values in June versus September (Table 2). Rates fell within a range typical of surface waters of large lakes55,56 and the ocean57,58. However, much higher BNF rates (>1 µmol N L−1 d−1) are possible in small to medium-sized, low-N:P, or mesotrophic to eutrophic lakes32,59,60. Thus, water-column rates from our two lakes fell into the low- to mid-range of BNF rates in aquatic systems.
Because the δ15NPON minimum in GL identified the photo-diazotrophic cyanobacteria dominated49,50 surface stratum of the microbial mats rather than the water column as the BNF hotspot, we additionally incubated slurries from the surface of GL’s microbial mats under elevated 15N2, comparing to incubations of slurries from LCL’s sediment surface (n = 4 per lake). These experiments allowed us to calculate areal rates and then estimate the effect of biological phosphate demand created by BNF. The 15N2 was incorporated into biomass at much higher rates in GL than LCL: 1674.3 ± 263.9 nmol N gdry-slurry−1 d−1 versus 2.7 ± 1.4 nmol N gdry-slurry−1 d−1. Conversion into volumetric rates using the slurries’ measured wet densities yielded average rates of 115.3 µmol N L−1 d−1 (GL) and 0.8 µmol N L−1 d−1 (LCL), i.e., 4–5 (GL) and 1.5 (LCL) orders of magnitude higher than water-column based rates (Table 2). In LCL, the higher slurry-based rates may be because diazotrophs concentrated onto the sediment surface during the dry season (Supplementary Fig. 2b) or because BNF rates by heterotrophic sediment bacteria exceeded those by pelagic phototrophs43,44. In GL slurries, the much higher rates compared to the water column are readily explained by cyanobacterial photo-diazotrophs dominating these mats49,50. Converted to an areal rate by integration of the volumetric rate over the surface 1-cm of the mat (48.1 µmol N m−2 h−1), GL slurry rates were similar to rates in cyanobacterial mats from a salt marsh (19–636 µmol N m−2 h−1)61, littoral zone sediments of Mono Lake covered by algal aggregates (11 µmol N m−2 h−1)43, and littoral, phototrophic microbial mats from Mono Lake (46 µmol N m−2 h−1)42.
Assuming that mat-based cyanobacteria are GL’s predominant diazotrophs49 and that they are confined to a photic zone comprising the top 1-cm of the mat62, how does the measured volumetric rate of 115.3 µmol N L−1 d−1 (=42.1 µmol N cm−3 yr−1) relate to the P pool of the entire lake? Further assuming 0.5 m mean water depth in GL and that the top 0.5 m of GL sediments (including microbial mats) are affected by exchange with the surface microbial mats, we take the sum of the mean tdP measured cross-seasonally in GL water (0.122 µmol cm−3) and porewater (0.854 µmol cm−3) and integrate over 1 m depth. This yields an inventory of 98 µmol P cm−2, which is the same order of magnitude as the estimated 42.1 µmol N cm−3 yr−1 fixed by the top centimeter of the mats. This similarity suggests a significant role for BNF in lowering phosphate in GL by creating biological P-demand63. In LCL, however, both the sediment-associated slurry rates (0.8 µmol N L−1 d−1 = 0.30 mmol N L−1 yr−1) and the even lower pelagic rates (Table 2) are orders of magnitude smaller than the lake’s mM-level phosphate pool (Table 1), suggesting that BNF does not create significant biological P demand. High BNF rates on the order of ~10 mM N L−1 yr−1 as encountered in microbial mats from GL (this study), Mono Lake42,43, or some eutrophic lakes59 would allow microbes to decrease phosphate concentrations on the order of mM. Therefore, the limitation of biological productivity by low BNF (see also Supplementary Note 1) likely contributes significantly to the world record phosphate concentrations in LCL.
What inhibits BNF in LCL? One plausible factor is limited availability of iron or molybdenum, which are needed for the nitrogenase enzyme64 but, with the caveat that our data were not produced using strictly trace-metal free protocols, concentrations of both metals were higher in LCL compared to GL (Table 1), which is inconsistent with trace metal limitation of BNF in LCL but not GL. Instead, the most parsimonious explanation for low BNF rates in LCL is high salinity, which is known to repress nitrogenase synthesis and divert energy away from N2-fixation65. Moreover, the negative effect of high salinity on N2-solubility66 plausibly lowers BNF efficiency in LCL. Across all seasons (Table 1) and within the specific samples incubated for BNF rates, salinity in LCL was ≥100–150 g/L, the range above which BNF inhibition is consistently indicated by a lack of the high BNF rates observed at lower salinities in soda lake sediments and phototrophic mats (Fig. 5)42,44,45. However, more research is needed to identify why BNF is inhibited in LCL. An alternative or additional inhibitor could be sulfide, given that sediments of LCL compared to GL contain much higher dissolved sulfur concentrations (Supplementary Fig. 4i). Under the reducing sediment conditions some of this dissolved S likely represents hydrogen sulfide produced by sulfate reduction. Sulfide is toxic to many organisms, but its effect on BNF is ambiguous, depending on microbial species, sulfide concentration, and pH67,68.

Data are from Goodenough and Last Chance lakes (this study) as well as from Mono Lake41,42 and the Kulunda Steppe (Russia) lakes Bitter-144,45, Cock Soda Lake, Bitter−3, Tanatar VI, Picturesque Lake, and Crooked Lake44. Except for the present study and the Mono Lake water column rates41, plotted rates are based on the acetylene assay, which measures nitrogenase activity via acetylene reduction to ethylene. The conversion factor relating acetylene reduction to actual N2-fixation rates is typically (including in this figure) assumed to be 4 on theoretical grounds100, but in reality can vary by at least one order of magnitude64,101. None of the cited studies determined the locally applicable acetylene-N2 conversion factors, so the plotted rates should be taken with caution, especially when comparing rates from different studies.
Extremely low N:P ratios in the water column of LCL or Mono Lake42 show that BNF cannot balance N:P ratios in lakes if geochemical (this study) or anthropogenic32 P input is high. Thus, LCL is an extreme example of a lake in which large P excess remains unbalanced by BNF, whereas balanced particulate N:P ratios in neighboring GL are associated with strong BNF. The difference may lie in a single, localized BNF-inhibiting environmental factor, probably high salinity, implying the absence of a general rule governing the ability of BNF to balance N:P ratios across different environments.
Implications for the origin of life
The up to 37 mM phosphate found in LCL water and sediment porewaters far exceeds P concentrations in other natural waters and reaches a minimal tens of mM phosphate that is sufficient for non-enzymatic phosphorylation reactions16. This implies that shallow, carbonate-rich lakes on early Earth could have provided sufficiently phosphate-rich conditions for the origin of life, adding to an ability of such waterbodies to concentrate carbon, nitrogen, and sulfur feedstocks1,3,4 and facilitate CO2 reduction in the presence of ultraviolet light and volcanic SO2 as sulfite to organic compounds that are important in central carbon metabolism69. High carbonate concentrations and resulting low calcium concentrations appear to be the key requirements for extreme phosphate accumulation2,25,26, which also apply to the subsurface ocean of Saturn’s moon, Enceladus70,71. On Earth, soda lakes are commonly created by evaporative concentration within closed-basin lakes fed by carbonate-rich (ground)water leaching cations from basaltic rock where the divalent cations are removed into carbonates and clays51. Lakes geochemically similar to LCL probably occurred on Hadean Earth, because there was likely some basaltic land72 and relatively carbon-rich air73. For similar reasons, such lakes may have also occurred on early Mars74.
One key geochemical difference between LCL and a prebiotic soda lake is LCL’s oxic water. Minor levels of P were found in LCL sediments (0.10 ± 0.01% [w/w]; Supplementary Fig. 6) but more detailed analysis is required to determine how much of this would represent a P sink also under anoxic early-Earth conditions. Calcite-adsorbed P would represent a potentially relevant sink also under prebiotic conditions and so would P bound as vivianite (Fe(II)-phosphate) due to higher Fe2+ solubility under anoxic conditions. By contrast, P bound to organics (from biological production), or ferric iron (due to oxic conditions in water and surface sediment) would only represent a P sink under modern-day (biotic and oxic) conditions and detrital P (e.g., non-authigenic apatite from surrounding rock) would not represent a P sink under any conditions.
Another potential difference between LCL and a prebiotic lake is sulfide content. Sulfide was likely rare in Hadean ponds3 but is biologically produced from sulfate (Table 1) in the anoxic sediments of modern soda lakes6. High sulfide concentrations can suppress vivianite precipitation by forming insoluble FeS275. However, vivianite was probably not a significant phosphate sink in anoxic Hadean soda lakes because high carbonate concentrations favor siderite (Fe(II)-carbonate) over vivianite precipitation76. Moreover, vivianite solubility in Hadean seawater at equilibrium with a 0.1-bar pCO2 atmosphere may have been in the mM-range77 and this solubility should only increase with carbonate concentration in more concentrated lake waters. Hence, because calcite solubility exceeds that of siderite, phosphate concentrations were likely controlled by Ca2+ rather than Fe2+ even in anoxic soda lakes2.
Even higher phosphate concentrations than observed in LCL are plausible under anoxic and prebiotic conditions. Lower Hadean ferric iron concentrations78 could increase phosphate concentrations relative to modern LCL because phosphate can be adsorbed onto iron oxides in modern lakes79,80. Additionally, some biological production in modern LCL is evident from the presence of chlorophyll and near-shore biofilms. Even slow biological production could create a constant biological P demand, decreasing dissolved P concentrations (and lowering the Cl–PO43− slope; Fig. 2) over long (century to millennia) time scales as a portion of organic-bound P sinks and is buried in the sediment63. Biological production in excess of that enabled by the small BNF rates could be fueled by ammonium (from organic matter mineralization) diffusing into modern LCL from sediments. Depending on the size of such fluxes and the rate of organic P burial, future studies using LCL as a Hadean analog system would be expected to show that phosphate concentrations in a prebiotic analog lake could be even higher than those in LCL.
Finally, soda lakes on early Earth may have received higher influxes of P than modern ones. A more CO2-rich atmosphere would have enabled faster weathering of apatite in igneous rocks81. Additional P may have been added to individual soda lakes during impact bombardment in the Hadean and early Archean82 by meteorites containing the P-rich mineral schreibersite (Fe3P)83. Schreibersite corrosion could have added phosphate as well as reduced and reactive P compounds favoring prebiotic chemistry in early-Earth soda lakes84,85. Plausible photo-geochemistry can oxidize reduced P compounds derived from schreibersite to phosphate favoring prebiotic phosphorylation86 and soda lakes could have provided the calcium-poor conditions for this phosphate to stay in solution.
By examining the world’s most phosphate-rich lake, we demonstrated with new field data from sediments and from the water column the previously hypothesized2 lack of P precipitation in natural carbonate-rich, calcium-depleted waters. In LCL, phosphate accumulates to concentrations of tens of mM instead of precipitating as apatite or being biologically consumed. Low BNF rates and N-limitation are essential for also decreasing the biological P sink, which distinguishes LCL from the less phosphate-rich GL and other soda lakes with high benthic BNF rates, such as Mono Lake (<1 mM PO43−) or Bitter-142,44,45. As BNF was absent on prebiotic Earth, LCL is a remarkably promising analog to a “Hadean soda pond” that can be used to understand plausible environments for prebiotic synthesis more deeply.
Methods
Collection and treatment of lake water, sediment and evaporite samples
Evaporites were collected at multiple locations around Last Chance and Goodenough lakes using vinyl gloves and stored in plastic bags and small plastic vials at freezing temperatures until characterization. Water and sediment samples were taken from geographically fixed stations distributed over Goodenough and Last Chance lakes (Fig. 1) during three field campaigns in mid-November 2021, mid-June 2022, and mid-September 2022. Care was taken to avoid contamination of sampled water with disturbed sediments by using either Niskin bottles from a small boat or a several-meter-long pole with clean (sample-flushed several times) vinyl-tubing that allowed sampling pristine water while wading in the lake. Upon sampling, water samples for dissolved geochemical concentration analyses were filtered using 0.2-µm syringe filters (cellulose-acetate, Puradisc, Whatman) after flushing with a few milliliters of sample. Filtered water was filled into clean Falcon tubes that were first rinsed with a few milliliters of sample. For analysis of the particulate phase (POC, PON, POP, chlorophyll), water was collected, stored cool and dark until vacuum filtration (described below) within several hours of sampling.
Sediment cores were taken in PVC pipes (10-cm diameter) that were pushed into the sediment manually and sealed air-tight upon retrieval. Within several hours of sampling, sediment cores were subsampled vertically by slicing and the subsamples were then stored inside centrifuge tubes. Prior to slicing, porewater was extracted from some cores taken in PVC pipes with pre-drilled holes using rhizons (5 cm porous length, 0.15 µm pore size, 19.21.23F, Rhizosphere). For other cores, porewater was later extracted from thawed subsamples using centrifugation and syringe-filtration (0.2-µm, as above) of the supernatant. All samples (porewater, bulk sediment slices, filtered water, filtered particulates) were stored at −20 °C until laboratory treatment or analysis within minutes (especially all filtered particulates) to hours of (sub)sampling. Acid-washed (10% HCl, 24 h) plastic or glassware was used for transfer and storage of samples.
Dissolved geochemical concentrations
Dissolved O2, pH, and turbidity were measured using ProDSS sensors (YSI). Total salinity was determined by drying filtered lake water at 60 °C and subtracting dry weight from wet weight standardized to water volume. Water density was measured by weighing a specified volume of filtered water at 22 °C. Ionic (SO42−, Cl−; detection limits not relevant) and elemental (Ca, Fe, K, Mg, Mo, Na, P, and S) concentrations in filtered lake and sediment porewater were analyzed using ion chromatography and ion-coupled plasma mass spectrometry (ICP–MS, ICP 61E, Thermo Scientific), respectively. Dilution-dependent detection limits for ICP–MS were 0.0014–0.84 mM (Ca), 0.00016–0.097 mM (Fe), 0.0045–2.7 mM (Mg), 0.0015–0.87 mM (P), 0.00019–0.11 mM (Mo), or not relevant given high concentrations in all samples. Total dissolved inorganic carbon (DIC) was analyzed on a Shimadzu TOC/TN-V CSH by the difference between total dissolved carbon before and after acidification and sparging87. Phosphate concentrations in water and sediment porewater were analyzed using the standard spectrophotometric “molybdate blue” method with a precision of ≤1%88. Lake water samples were additionally analyzed with a nutrient autoanalyzer (Seal Analytical AA3), using standard methods to quantify ammonium ([NH4+ + NH3]), nitrite, and nitrate concentrations following Gordon et al.89 at dilution-dependent detection limits 0.09–0.9 µM (ammonium), 0.01–0.1 µM (nitrite) and 0.18–1.8 µM (nitrate). Ammonium concentrations in sediment porewaters were analyzed by the indophenol blue method88,90. Nutrient ratios are presented after logarithmic transformation91.
Particulate biogeochemical concentrations and stable isotope analysis
Water column particulates were concentrated onto 0.7-µm, 0.47-mm glass-fiber filters (pre-combusted for 4.5 h at 450 °C) using vacuum filtration. Chl a concentrations were determined after acetone extraction of filtered particulates on a fluorometer (TD-700, Turner Designs). Other filters containing particulates were oven-heated (50–60 °C) to dryness, exposed to a fuming hydrochloric acid atmosphere for 8 h to remove carbonates, oven-dried again, and finally stored in a desiccator until analysis. Particulate phosphorus was acid-extracted from some of these filters and measured as described below for dried sediments. Filters for PON concentrations and stable isotopes were packed in tin cups and analyzed by elemental-analysis/mass-spectrometry (EA-MS; Finnigan MAT253) as previously described92. For standardization to the commonly used stable isotope reference air–N2 (15N/14N2 = 0.0036765), we used an international standard (USGS41: δ15N = +47.57) as well as in-house glutamic-acid or salmon-biomass standard materials (δ15N = −4.6‰; −5.7‰; 11.3‰), which were calibrated against the international standards USGS40 (δ15N = −4.52‰) and USGS41. The δ notation was used to express natural abundance stable isotope signatures, where R is the stable isotope ratio (15N/14N):
All δ15N values were blank-corrected. Additionally, samples with N amounts below an empirically determined linear range, which is the threshold below which a size effect was observed on the post-calibration δ15N of standards, were linearity-corrected proportionally for this empirically determined offset.
Frozen bulk sediment subsamples were briefly thawed and centrifuged to measure wet-volume and wet-weight, then frozen again for freeze-drying. Upon drying, dry-weight was measured to calculate dry density [gdry-weight Lwet-volume−1], which was later used to convert dry-weight-based bulk sediment concentrations to volumetric units. Dried sediments were sieved through a 2-mm mesh to remove any granules and pebbles and then ground to ≤125 µm grain size. This sediment was then further processed for X-ray diffraction (see below), weighed into tin cups for EA-MS (as above), or weighed in for acid-digestion for total phosphorus extraction according to EPA-3050B93,94 followed by ICP–MS analysis of the extract. The elemental composition of evaporites was determined by dissolving a weighed amount of the homogenized salt in a known volume of deionized water and subsequent ICP-MS analysis of this solution.
X-ray diffraction (XRD) of evaporite salts and sediments
Evaporites were prepared for analysis by isolating visually pure salt phases from the samples and drying them in a fume hood for multiple days, then grinding with quartz mortar and pestle to a grain size ≤200 µm. Ground sediment samples (see above) were processed to remove clays prior to XRD analysis. This involved suspending ~1 g of the powdered sample per 30 mL deionized water in a 50 mL Falcon tube and disaggregating the sample for 10−20 min at ~200 W using an ultrasonic probe. The sediment was then allowed to gravitationally settle for 47 min per centimeter water depth (~5 h), after which the supernatant, containing the majority of the ≤2 µm fraction95, was decanted. The clay-reduced sediment pellet was then dried at 60 °C.
The mineralogy of the lakeshore evaporites and lake sediments from various sediment depths (0–55 cm) was analyzed on a Bruker D8 X-ray diffractometer with a high-efficiency Cu anode microfocus x-ray source and Pilatus 100 K large-area 2D detector (Molecular Engineering and Sciences Institute, University of Washington). Diffractograms were acquired at 50 kV acceleration voltage and 1000 µA current, with 2θ scans ranging from 16° to 91° with a 2θ step of 5° at 30 s each. The samples were analyzed by either (i) dissolving the ground sample in a small amount of ethanol, dropping it onto a silicon wafer, and allowing it to dry, or (ii) loading them onto a Si crystal zero diffraction plate sample holder with a 5 × 0.2 mm diameter well and using a blade to level. Bruker EVA software (V4.2.1) and the ICCD PDF-4+ (2023) database were used for phase identification and exporting CIF files for refinement.
GSAS-296 was used to perform the Rietveld refinements on the XRD patterns. The analytic Hessian method was used for least-squares curve fitting (Levenberg–Marquardt singular value decomposition algorithm), and a Chebyschev function with three coefficients was used to fit the background, with individual background peaks added as needed for sediment samples with potential amorphous content. Means and standard deviations are presented here with sediment and evaporite samples from the same lakes regarded as replicates, respectively.
Fertilization experiment
Lake water for the fertilization experiment was sampled on September 13, 2022, from Goodenough and Last Chance Lake, respectively, and sample volumes of 200 mL were filled into clear glass bottles with a headspace of 65 mL filled with ambient air. Three treatments were made with n = 3 bottles (=time points) per treatment and lake: (1) a Na3PO4 solution was added to a total surplus concentration of 100 µmol PO43− L−1 to the “+phosphate” treatment bottles. (2) a (NH4)2SO4 + NaNO3 mixed solution (1:1 N-molar mixing ratio) was added to a total surplus concentration of 100 µmol N L−1 to the “+ammonium+nitrate” treatment bottles. (3) No additions were made to the “control” bottles. Each treatment was subsequently incubated under natural light in an unshaded location near the lakes and at the bottom of a ~30 cm deep water column inside a plastic box. The water bath was used to avoid unnaturally large temperature variations inside the incubation bottles from solar radiation and recreate the light conditions in the shallow lake water columns. During the incubation, the weather was mostly sunny. Approximately 0 h, 1 day, and 2 days after treatment, the experiment was terminated in one bottle per treatment by vacuum-filtering its content as described above. The filters were then treated and analyzed for Chl a as described above.
15N2 and stable isotope labeling experiment
Lake water as well as slurries of microbial mat and sediment were incubated in the presence of 15N-enriched N2 to quantify rates of N2-fixation in each lake, following the premise of previously described experiments57,97,98. Samples for these experiments were taken in mid-June (water samples only) and mid-September 2022 from two stations in each lake. Water samples were taken manually as described above. Slurries were sampled by manually scooping up surface sediment or microbial mat directly into the incubation bottle and filling the rest of the bottle with in-situ water. For each station and type (water and slurry), duplicate samples were incubated inside clear glass bottles with septum lids, while a third sample (natural-abundance reference) was immediately preserved (water by vacuum filtration and freezing, slurries by freezing) for EA-MS analysis.
A fourth parallel sample from each lake (September experiment) or water from each lake sampled in November and frozen in the meantime (June experiment) was used to prepare 15N2 enriched lake water. This water was first vacuum-filtered (0.7 µm) and the filtrate was then moved into pre-combusted glass bottles that were subsequently crimp-shut gastight. Next, the water was stripped of dissolved N2 by helium-bubbling, degassed using a vacuum, and amended with pure 15N2 gas (≥98% 15N, Cambridge Isotope Laboratories Inc., lot number I−25854/X732957) using a gastight syringe: 0.075% (v/v) mL in June and 0.101% (v/v) or 0.052% (v/v) in September for Goodenough and Last Chance, respectively. Equilibration of the added 15N2 gas was enhanced by ≥1 h of shaking.
Marking the start of the incubation, the 15N2 enriched lake water was then added to the closed incubation bottles, which were completely filled with sample water (20–125 mL in June, 45– 315 mL in September) or slurry (6–42 mL in June, 18–125 mL in September). The differences in the exact amounts of added 15N served the purpose of achieving a final labeling percent of 1–10% 15N2 in incubation bottles containing water of different salinity and temperature and, therefore, N2 solubility. Incubation took place under natural sunlight and daylight length inside a water bath as described above for the fertilization experiment (weather: cloudy in June, sunny in September). After approximately 48 h, incubation was terminated by vacuum-filtration (water incubations) or freezing (slurries). Immediately prior to termination, subsamples for the determination of 15N% of dissolved N2 were taken by subsampling the incubation bottles, using needles and cut-off 1-mL syringes to fill 12-mL exetainers (Labco) with minimal gas-intrusion and headspace-free. These N2 subsamples were poisoned with 100 µL concentrated HgCl2. A 5-mL helium headspace was created and allowed to equilibrate for 24 h prior to analysis using a Delta-XP and a gas-bench II (Thermo Finnigan, Germany). The main samples (filters or frozen slurries) were analyzed for PON and δ15N-PON by EA-MS as described above following drying and acid-fumigation of the water-incubation-derived filters and freeze-drying, weighing, and homogenization of the slurries. Additionally, prior to freeze-drying, slurries and their natural abundance references were briefly centrifuged to remove excess water. This supernatant was also used to measure salinity after filtration (as described above).
N2-fixation rates were calculated from the stable isotope composition in the filtered particulates or slurries as follows57,99:
where atom%post-incubation is the atom-percent 15N of particulate matter after incubation, atom%nat-ab-ref is the atom-percent 15N of the particulate matter prior to incubation (natural abundance reference), atom%substrate is the atom percent 15N (measured) of the process substrate (N2) at the end of the incubation, [Particulate] is the particulate nitrogen concentration in units [µmol L−1] for water samples or [µmol gdry-slurry−1] for slurries at the end of the incubation period (Δt [days]). Therefore, the units of Eq. (2) are [µmol L−1 d−1] for water-incubations and [µmol gdry-slurry−1 d−1] for slurry-incubations.
Rate detection limits were defined as an increase in atom% 15N of the particulate matter equal to the three-fold standard deviation of parallel reference material measurements57, which were determined to be 3 × 0.000044 15N% (slurries) or 3 × 0.000032 15N% (water). Minimum detectable uptake rates calculated based on [Particulate], Δt and atom%substrate values in a given incubation bottle, ranged from 0.16 to 3.18 nmol N Lwater−1 d−1 and 0.3–106.4 nmol N gdry-slurry−1 d−1. Rate uncertainties (shown in Table 2) were calculated by propagating the uncertainties associated with [Particulate] and atom% measurements according to Fonseca-Batista et al.57.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are available in the main text or supplementary information, or in the figshare repository (https://doi.org/10.6084/m9.figshare.23907951).
References
Toner, J. D. & Catling, D. C. Alkaline lake settings for concentrated prebiotic cyanide and the origin of life. Geochim. Cosmochim. Acta 260, 124–132 (2019).
Toner, J. D. & Catling, D. C. A carbonate-rich lake solution to the phosphate problem of the origin of life. Proc. Natl Acad. Sci. USA 117, 883–888 (2020).
Ranjan, S., Todd, Z. R., Sutherland, J. D. & Sasselov, D. D. Sulfidic anion concentrations on early earth for surficial origins-of-life chemistry. Astrobiology 18, 1023–1040 (2018).
Ranjan, S., Todd, Z. R., Rimmer, P. B., Sasselov, D. D. & Babbin, A. R. Nitrogen oxide concentrations in natural waters on early earth. Geochem. Geophys. Geosyst. 20, 2021–2039 (2019).
Sasselov, D. D., Grotzinger, J. P. & Sutherland, J. D. The origin of life as a planetary phenomenon. Sci. Adv. 6, eaax3419 (2020).
Imhoff, J. F., Sahl, H. G., Soliman, G. S. & Trüper, H. G. The Wadi Natrun: chemical composition and microbial mass developments in alkaline brines of eutrophic desert lakes. Geomicrobiol. J. 1, 219–234 (1979).
Hirst, J. F. Sedimentology, Diagenesis and Hydrochemistry of the Saline, Alkaline Lakes on the Cariboo Plateau, Interior British Columbia, Canada (University of Saskatchewan, 1995).
Friedman, I., Smith, G. I. & Hardcastle, K. G. Studies of quaternary saline lakes—II. Isotopic and compositional changes during desiccation of the brines in Owens Lake, California, 1969–1971. Geochim. Cosmochim. Acta 40, 501–511 (1976).
Schwartz, A. W. Phosphorus in prebiotic chemistry. Philos. Trans. R. Soc. B: Biol. Sci. 361, 1743–1749 (2006).
Pasek, M. A., Gull, M. & Herschy, B. Phosphorylation on the early earth. Chem. Geol. 475, 149–170 (2017).
Hudson, J. J., Taylor, W. D. & Schindler, D. W. Phosphate concentrations in lakes. Nature 406, 54–56 (2000).
Tyrrell, T. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400, 525–531 (1999).
Powner, M. W., Gerland, B. & Sutherland, J. D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 459, 239–242 (2009).
Morasch, M. et al. Heated gas bubbles enrich, crystallize, dry, phosphorylate and encapsulate prebiotic molecules. Nat. Chem. 11, 779–788 (2019).
Gull, M., Cafferty, B. J., Hud, N. V. & Pasek, M. A. Silicate-promoted phosphorylation of glycerol in non-aqueous solvents: a prebiotically plausible route to organophosphates. Life 7, 29 (2017).
Xu, J., Green, N. J., Gibard, C., Krishnamurthy, R. & Sutherland, J. D. Prebiotic phosphorylation of 2-thiouridine provides either nucleotides or DNA building blocks via photoreduction. Nat. Chem. 11, 457–462 (2019).
Szostak, J. W. The narrow road to the deep past: in search of the chemistry of the origin of life. Angew. Chem. 129, 11182–11189 (2017).
Patel, B. H., Percivalle, C., Ritson, D. J., Duffy, C. D. & Sutherland, J. D. Common origins of RNA, protein and lipid precursors in a cyanosulfidic protometabolism. Nat. Chem. 7, 301–307 (2015).
Wogan, N. F., Catling, D. C., Zahnle, K. J. & Lupu, R. Origin-of-life molecules in the atmosphere after big impacts on the early Earth. Planet. Sci. J. 4, 169 (2023).
Zahnle, K. J., Lupu, R., Catling, D. C. & Wogan, N. Creation and evolution of impact-generated reduced atmospheres of early earth. Planet. Sci. J 1, 11 (2020).
Walton, T. & Szostak, J. W. A highly reactive imidazolium-bridged dinucleotide intermediate in nonenzymatic RNA primer extension. J. Am. Chem. Soc. 138, 11996–12002 (2016).
Ritson, D. J., Battilocchio, C., Ley, S. V. & Sutherland, J. D. Mimicking the surface and prebiotic chemistry of early Earth using flow chemistry. Nat. Commun. 9, 1821 (2018).
Ruttenberg, K. C. & Berner, R. A. Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochim. Cosmochim. Acta 57, 991–1007 (1993).
Slomp, C. P. & Van Cappellen, P. The global marine phosphorus cycle: sensitivity to oceanic circulation. Biogeosciences 4, 155–171 (2007).
Gulbrandsen, R. A. Physical and chemical factors in the formation of marine apatite. Bull. Soc. Econ. Geol. 64, 365–382 (1969).
Nathan, Y. & Sass, E. Stability relations of apatites and calcium carbonates. Chem. Geol. 34, 103–111 (1981).
Knapp, A. N. The sensitivity of marine N2 fixation to dissolved inorganic nitrogen. Front. Microbiol. 3, 374 (2012).
Redfield, A. C. The biological control of chemical factors in the environment. Am. Sci. 46, 205–221 (1958).
Guildford, S. J. & Hecky, R. E. Total nitrogen, total phosphorus, and nutrient limitation in lakes and oceans: is there a common relationship? Limnol. Oceanogr. 45, 1213–1223 (2000).
They, N. H., Amado, A. M. & Cotner, J. B. Redfield ratios in inland waters: higher biological control of C:N:P ratios in tropical semi-arid high water residence time lakes. Front. Microbiol. 8, 1505 (2017).
Schindler, D. W. et al. Eutrophication of lakes cannot be controlled by reducing nitrogen input: results of a 37-year whole-ecosystem experiment. Proc. Natl Acad. Sci. USA 105, 11254–11258 (2008).
Hayes, N. M. et al. Spatial and temporal variation in nitrogen fixation and its importance to phytoplankton in phosphorus-rich lakes. Freshw. Biol. 64, 269–283 (2019).
Oduor, S. O. & Schagerl, M. Phytoplankton primary productivity characteristics in response to photosynthetically active radiation in three Kenyan Rift Valley saline alkaline lakes. J. Plankton. Res. 29, 1041–1050 (2007).
Boros, E., V.-Balogh, K., Vörös, L. & Horváth, Z. Multiple extreme environmental conditions of intermittent soda pans in the Carpathian Basin (Central Europe). Limnologica 62, 38–46 (2017).
Axler, R., Gersberg, R. & Paulson, L. Primary productivity in meromictic Big Soda Lake, Nevada. Great Basin Nat. 38, 187–192 (1978).
Boros, E., Takács, A., Dobosy, P. & Vörös, L. Extreme guanotrophication by phosphorus in contradiction with the productivity of alkaline soda pan ecosystems. Sci. Total Environ. 793, 148300 (2021).
Melack, J. M., Kilham, P. & Fisher, T. R. Responses of phytoplankton to experimental fertilization with ammonium and phosphate in an African Soda Lake. Oecologia 52, 321–326 (1982).
Clarisse, L. et al. Atmospheric ammonia (NH3) emanations from Lake Natron’s saline mudflats. Sci. Rep. 9, 4441 (2019).
Menzel, P. et al. Influence of bottom water anoxia on nitrogen isotopic ratios and amino acid contributions of recent sediments from small eutrophic Lonar Lake, central India. Limnol. Oceanogr. 58, 1061–1074 (2013).
Sorokin, D. Y., Banciu, H. L. & Muyzer, G. Functional microbiology of soda lakes. Curr. Opin. Microbiol. 25, 88–96 (2015).
Steward, G. F., Zehr, J. P., Jellison, R., Montoya, J. P. & Hollibaugh, J. T. Vertical distribution of nitrogen-fixing phylotypes in a meromictic, hypersaline lake. Microb. Ecol. 47, 30–40 (2004).
Herbst, D. B. Potential salinity limitations on nitrogen fixation in sediments from Mono Lake, California. Int. J. Salt Lake Res. 7, 261–274 (1998).
Oremland, R. S. Nitrogen fixation dynamics of two diazotrophic communities in Mono Lake, California. Appl. Environ. Microbiol. 56, 614–622 (1990).
Samylina, O. S. et al. The patterns of nitrogen fixation in haloalkaliphilic phototrophic communities of Kulunda Steppe soda lakes (Altai, Russia). FEMS Microbiol. Ecol. 95, fiz174 (2019).
Namsaraev, Z. et al. Effect of salinity on diazotrophic activity and microbial composition of phototrophic communities from Bitter-1 soda lake (Kulunda Steppe, Russia). Extremophiles 22, 651–663 (2018).
Renaut, R. W. & Long, P. R. Freeze-out precipitation of salts in saline lakes—examples from Western Canada. Cryst. Precip. 33–42 https://doi.org/10.1016/B978-0-08-035751-5.50011-8 (1987).
Mathews, W. H. Neogene Chilcotin basalts in south-central British Columbia: geology, ages, and geomorphic history. Can. J. Earth Sci. 26, 969–982 (1989).
Renaut, R. W. & Long, P. R. Sedimentology of the saline lakes of the Cariboo Plateau, Interior British Columbia, Canada. Sediment. Geol. 64, 239–264 (1989).
Zorz, J. K. et al. A shared core microbiome in soda lakes separated by large distances. Nat. Commun. 10, 4230 (2019).
Schultze-Lam, S., Ferris, F. G., Sherwood-Lollar, B. & Gerits, J. P. Ultrastructure and seasonal growth patterns of microbial mats in a temperate climate saline-alkaline lake: Goodenough Lake, British Columbia, Canada. Can. J. Microbiol. 42, 147–161 (1996).
Cerling, T. E. Chemistry of closed basin lake waters: a comparison between African Rift Valley and some central North American rivers and lakes. In The Global Geological Record of Lake Basins (eds. Gierlowski-Kordesch, E. & Kelts, K.) Vol. 1 29–30 (Cambridge University Press, 1994).
Eugster, H. P. & Jones, B. F. Behavior of major solutes during closed-basin brine evolution. Am. J. Sci. 279, 609–631 (1979).
Garrels, R. M. & Mackenzie, F. T. Origin of the chemical compositions of some springs and lakes. In Equilibrium Concepts in Natural Water Systems (ed. Stumm, W.) 222–242 (American Chemical Society, 1967).
McRose, D. L. et al. Effect of iron limitation on the isotopic composition of cellular and released fixed nitrogen in Azotobacter vinelandii. Geochim. Cosmochim. Acta 244, 12–23 (2019).
Natwora, K. E. & Sheik, C. S. Assessment of nitrogen fixation rates in the Laurentian Great Lakes. J. Great Lakes Res. 47, 1288–1295 (2021).
Gondwe, M. J., Guildford, S. J. & Hecky, R. E. Planktonic nitrogen fixation in Lake Malawi/Nyasa. Hydrobiologia 596, 251–267 (2008).
Fonseca-Batista, D. et al. Nitrogen fixation in the eastern Atlantic reaches similar levels in the Southern and Northern Hemisphere. J. Geophys. Res. Oceans 122, 587–601 (2017).
Gradoville, M. R. et al. Diversity and activity of nitrogen-fixing communities across ocean basins. Limnol. Oceanogr. 62, 1895–1909 (2017).
Scott, J. T. & Grantz, E. M. N2 fixation exceeds internal nitrogen loading as a phytoplankton nutrient source in perpetually nitrogen-limited reservoirs. Freshw. Sci. 32, 849–861 (2013).
Horne, A. J. & Galat, D. L. Nitrogen fixation in an oligotrophic, saline desert lake: Pyramid Lake, Nevada. Limnol. Oceanogr. 30, 1229–1239 (1985).
Whitney, D. E., Woodwell, G. M. & Howarth, R. W. Nitrogen fixation in Flax Pond: a Long Island salt marsh. Limnol. Oceanogr. 20, 640–643 (1975).
Oren, A., Kühl, M. & Karsten, U. An endoevaporitic microbial mat within a gypsum crust: zonation of phototrophs, photopigments, and light penetration. Mar. Ecol. Prog. Ser. 128, 151–159 (1995).
Katsev, S. & Crowe, S. A. Organic carbon burial efficiencies in sediments: the power law of mineralization revisited. Geology 43, 607–610 (2015).
Falkowski, P. G. Enzymology of nitrogen assimilation. In Nitrogen in the Marine Environment (eds Carpenter, E. J. & Capone, D. G.) 839–868 (Academic Press, 1983).
Apte, S. K. Coping with salinity/water stress: cyanobacteria show the way. Proc. Natl Acad. Sci. India Sect. B: Biol. Sci. 67, 285–310 (2001).
Zheng, J. & Mao, S. A thermodynamic model for the solubility of N2, O2 and Ar in pure water and aqueous electrolyte solutions and its applications. Appl. Geochem. 107, 58–79 (2019).
Villbrandt, M. & Stal, L. J. The effect of sulfide on nitrogen fixation in heterocystous and non-heterocystous cyanobacterial mat communities. Algolog. Stud./Arch. Hydrobiol. Suppl. Vol. 83, 549–563 (1996).
Tam, T.-Y., Mayfield, C. I., Inniss, W. E. & Knowles, R. Effect of sulfide on nitrogen fixation in a stream sediment–water system. Appl. Environ. Microbiol. 43, 1076–1079 (1982).
Liu, Z. et al. Prebiotic photoredox synthesis from carbon dioxide and sulfite. Nat. Chem. 13, 1126–1132 (2021).
Hao, J. et al. Abundant phosphorus expected for possible life in Enceladus’s ocean. Proc. Natl Acad. Sci. USA 119, e2201388119 (2022).
Postberg, F. et al. Detection of phosphates originating from Enceladus’s ocean. Nature 618, 489–493 (2023).
Korenaga, J. Was there land on the early earth? Life 11, 1142 (2021).
Kadoya, S., Krissansen-Totton, J. & Catling, D. C. Probable cold and alkaline surface environment of the Hadean Earth caused by impact ejecta weathering. Geochem. Geophys. Geosyst. 21, e2019GC008734 (2020).
Hurowitz, J. A., Catling, D. C. & Fischer, W. W. High carbonate alkalinity lakes on mars and their potential role in an origin of life beyond earth. Elements 19, 501–508 (2023).
Kraal, P., Dijkstra, N., Behrends, T. & Slomp, C. P. Phosphorus burial in sediments of the sulfidic deep Black Sea: key roles for adsorption by calcium carbonate and apatite authigenesis. Geochim. Cosmochim. Acta 204, 140–158 (2017).
Postma, D. Formation of siderite and vivianite and the pore-water composition of a recent bog sediment in Denmark. Chem. Geol. 31, 225–244 (1981).
Brady, M. P., Tostevin, R. & Tosca, N. J. Marine phosphate availability and the chemical origins of life on Earth. Nat. Commun. 13, 5162 (2022).
Konhauser, K. O. et al. Decoupling photochemical Fe(II) oxidation from shallow-water BIF deposition. Earth Planet. Sci. Lett. 258, 87–100 (2007).
Einsele, W. Über die Beziehungen des Eisenkreislaufs zum Phosphatkreislauf im eutrophen See. Hydrobiologie 29, 664–686 (1936).
Blomqvist, S., Gunnars, A. & Elmgren, R. Why the limiting nutrient differs between temperate coastal seas and freshwater lakes: a matter of salt. Limnol. Oceanogr. 49, 2236–2241 (2004).
Hao, J., Sverjensky, D. A. & Hazen, R. M. Mobility of nutrients and trace metals during weathering in the late Archean. Earth Planet. Sci. Lett. 471, 148–159 (2017).
Zellner, N. E. B. Cataclysm no more: new views on the timing and delivery of lunar impactors. Orig. Life Evol. Biosph. 47, 261–280 (2017).
Pasek, M. A. Rethinking early Earth phosphorus geochemistry. Proc. Natl Acad. Sci. USA 105, 853–858 (2008).
Gull, M., Feng, T., Cruz, H. A., Krishnamurthy, R. & Pasek, M. A. Prebiotic chemistry of phosphite: mild thermal routes to form condensed-P energy currency molecules leading up to the formation of organophosphorus compounds. Life 13, 920 (2023).
Pasek, M. A. & Lauretta, D. S. Aqueous corrosion of phosphide minerals from iron meteorites: a highly reactive source of prebiotic phosphorus on the surface of the early earth. Astrobiology 5, 515–535 (2005).
Ritson, D. J., Mojzsis, S. J. & Sutherland, J. D. Supply of phosphate to early Earth by photogeochemistry after meteoritic weathering. Nat. Geosci. 13, 344–348 (2020).
Breitmeyer, S., Ray, J., Harris, C. & Miller, H. New Instrument Validation of Shimadzu TOC-L Carbon Analyzer Using Acidification with NDIR Method for Analysis of Dissolved Inorganic Carbon (DIC) in Water Samples (U.S. Geological Survey, 2019).
Strickland, J. D. H. & Parsons, T. R. A Practical Handbook of Seawater Analysis Vol. 167 Bulletin (Fisheries Research Board of Canada, 1972).
Gordon, L. I., Jennings, J. C., Andrew Ross, J. A. & Krest, J. M. A Suggested Protocol for Continuous Flow Automated Analysis of Seawater Nutrients (Phosphate, Nitrate, Nitrite and Silicic Acid) in the WOCE Hydrographic Program and the Joint Global Ocean Fluxes Study (College of Oceanic and Atmospheric Sciences Oregon State University: Corvallis, Oregon, 1993).
O’Dell, J. W. Determination of ammonia nitrogen by semi-automated colorimetry. Methods for the Determination of Inorganic Substances in Environmental Samples, Method 350 Vol. 2 (U.S. Environmental Protection Agency, 1993).
Isles, P. D. F. The misuse of ratios in ecological stoichiometry. Ecology 101, e03153 (2020).
Fry, B., Brand, W., Mersch, F. J., Tholke, K. & Garritt, R. Automated analysis system for coupled δ13C and δ15N measurements. Anal. Chem. 64, 288–291 (1992).
USEPA. Method 3050B: Acid Digestion of Sediments, Sludges, Sand Soils, Revision 2 (USEPA,1996).
Church, C., Spargo, J. & Fishel, S. Strong acid extraction methods for “total phosphorus” in soils: EPA method 3050B and EPA method 3051. Agric. Environ. Lett. 2, 160037 (2017).
Środoń, J. Identification and quantitative analysis of clay minerals. In Handbook of Clay Science Vol. 1 (eds Bergaya, F., Theng, B. K. G. & Lagaly, G.) 765–787 (Elsevier Ltd, 2006).
Toby, B. H. & Von Dreele, R. B. GSAS-II: the genesis of a modern open-source all purpose crystallography software package. J. Appl. Crystallogr. 46, 544–549 (2013).
Klawonn, I. et al. Simple approach for the preparation of 15-15N2-enriched water for nitrogen fixation assessments: evaluation, application and recommendations. Front. Microbiol. 6, 769 (2015).
Mohr, W., Großkopf, T., Wallace, D. W. R. & LaRoche, J. Methodological underestimation of oceanic nitrogen fixation rates. PLoS ONE 5, e12583 (2010).
Dugdale, R. C. & Wilkerson, F. P. The use of 15N to measure nitrogen uptake in eutrophic oceans; experimental considerations. Limnol. Oceanogr. 31, 673–689 (1986).
Schwintzer, C. R. & Tjepkema, J. D. Factors affecting the acetylene to 15N2 conversion ratio in root nodules of Myrica gale L. Plant Physiol. 106, 1041–1047 (1994).
Moisander, P., Lehtimäki, J., Sivonen, K. & Kononen, K. Comparison of 15N2 and acetylene reduction methods for the measurement of nitrogen fixation by Baltic Sea cyanobacteria. Phycologia 35, 140–146 (1996).
Acknowledgements
This work was funded by a grant from the Simons Foundation (511570FY20, D.C.C.). We would like to thank Zack Cohen, Ardith Bravenec, Benjamin Mousseau, and the participants of the September 2022 University of Washington (UW) Astrobiology field workshop for invaluable support in the field, Andrew Schauer (Isolab, UW) and Zoe Todd for help in the laboratory, as well as Shelly Carpenter (Demming Lab) and Andrea Ogsten (all UW) for lending us field equipment.
Author information
Authors and Affiliations
Contributions
D.C.C. and S.H. conceptualized the study and D.C.C. acquired funding, supervised, and administered the research. S.H. developed methodology and conducted field sampling, measurements, data analysis, and visualization, to which K.P.S. contributed the XRD and evaporite work. S.H. wrote the original draft of the paper and all authors contributed to reviewing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Craig Walton and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Mojtaba Fakhraee and Clare Davis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Haas, S., Sinclair, K.P. & Catling, D.C. Biogeochemical explanations for the world’s most phosphate-rich lake, an origin-of-life analog. Commun Earth Environ 5, 28 (2024). https://doi.org/10.1038/s43247-023-01192-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-023-01192-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
