
Abstract
Environmental archives, such as lake sediments, harbour DNA of past and present ecosystems. However, our understanding of the provenance, deposition and distribution of sedimentary DNA in lake systems is largely unknown, limiting the breadth of derived spatiotemporal inferences. By mapping the distribution of aquatic and terrestrial taxa in a large deep lake using metabarcoding, we characterise the spatial heterogeneity of sedimentary DNA and point to its potential driving factors. Taxa composition varies across geographic gradients in the lake, and spatial distribution of DNA is linked to the range and life mode of organisms. Exogenous taxa, such as alpine plants, have the most reliable detection near the mouth of the inflow. Our data reveal that sedimentary DNA is reflecting the mosaic distribution of organisms and organic remains in the environment, and a single location from lakes with watersheds across different elevations, biomes or other diversity boundaries does not capture the full dynamics in the surrounding area.
Similar content being viewed by others

Introduction
Sedimentary deposits are important archives for the understanding of past ecological and climatic conditions1,2. Lacustrine sediments contain mineral and organic matter from both the water body and the terrestrial surroundings, and sedimentary DNA (sedDNA) - DNA extracted from a bulk sediment sample rather than isolated organisms – has been used to identify organisms from all trophic levels even in the absence of fossils. With reliable dating, sedDNA has become a valuable tool for studying paleoecology3,4,5,6,7 and anthropogenic ecosystem changes8,9.
Arguably, paleo-ecological inferences made from lake sedDNA can be biased by the common practice of using single sediment cores or sites, which assumes that a single-location record is an ecological representation across both space and time. Several earlier studies have discussed the challenge inflicted by the heterogeneous feature of sediments due to, for example, non-linear and non-stationary response of lake systems to climatic forcing10 or changing sediment source11, which in turn affects lake sedimentary research12. For sedDNA, the knowledge on its immediate source and deposition pathway is lacking. Although it has been found to exist in different states: intracellular or extracellular13, dissolved or particle-absorbed14, it is still unclear whether sedDNA is deposited as free molecules, as mineral-bound, or linked to organismal remains.
There have been a few studies investigating inter- and intra-lake variabilities of sedDNA and influencing factors. For example, terrestrial plant DNA was found to represent local taxa15,16, related to organism abundance and distance from the source to the sampling sites17 and possibly linked to soil erosion18. For aquatic organisms, a multi-site study across a Siberian boreal lake showed that diatom sedDNA diversity varied with water depth19. Yet another study that sampled surface sedDNA from 12 sites in a small lake in New Zealand showed that most bacterial amplicon variants were shared among sites20. Aquatic plants can also be reliably detected as shown in 11 lakes in Norway17. Less is known about the spatial representation of aquatic animals in sediments or sedDNA, although animals such as crustacean zooplankton have been frequently used as paleo-ecological indicators21,22. Macroscopic animals such as fish are of high interest, but retrieval of their DNA has proven challenging23,24,25.
For freshwater environments in general, the distribution of aquatic environmental DNA (eDNA)26 of various organisms has been investigated, yet conclusions do not agree across studies. On the one hand, empirical and experimental data have shown that eDNA can travel long distances (up to 50 km) in streams and rivers27 (fish eDNA) and is evenly distributed in small lakes with low within-lake variation28 (plant and algae eDNA); on the other hand, eDNA in water bodies can be heterogenous in distribution29,30 (empirical data) or provides highly resolved spatiotemporal diversity patterns in river systems31 (simulation data of fish, invertebrates and bacteria). Although these studies hint that aquatic sedDNA is unevenly distributed in space, we currently lack such information for different organismal groups within lakes, especially large lakes, which nurture diverse ecosystems and accumulate deposits from larger surrounding areas32,33. Knowing these patterns and their geographic and biological driving factors will inform on future sampling strategy, data interpretation, and retrospectively, on the taphonomic processes causing these patterns.
Here we present the spatial distribution of sedDNA from a large deep perialpine lake, Upper Lake Constance, and associate the observed patterns to the characteristics of source organisms and the surrounding landscape. To cover a wide range of aquatic and terrestrial taxa, we performed four metabarcoding runs: three using published markers that target vascular plants34, general eukaryotes35,36, cyanobacteria37,38, and one using a marker for copepods designed in this study. This copepod marker allowed us to target an important group of zooplankton that is less resolved by the general eukaryote marker. We also investigated the association between DNA distribution and substrate type using sedimentological data.
Our study shows that surface sedDNA in Lake Constance has a heterogenous distribution, the degree of which varies both across geographic locations and among organismal groups. The distribution of DNA for different aquatic organisms is reflecting their natural occurrence and life mode in the lake, while that of common plants is more ubiquitous. We also find that the distribution of distant exogenous organisms such as alpine plants is determined by inflow water entering and settling in the lake and is most reliably detected close to the mouth of the inflow. Therefore, single sediment cores from large lakes likely do not capture the complete diversity of the surrounding area, but can be reliable for widely distributed local taxa. Our results further suggest that sedDNA is mainly released from sedimented organismal remains rather than molecules settling through the water column. The observed heterogeneity of sedDNA is therefore a result of both the mosaic occurrences of organisms and the varying deposition pathways thanks to the complex landscape.
Results
General characteristics of amplicon data
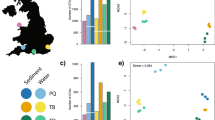
To study the spatial distribution of sedDNA within Upper Lake Constance, we collected surface sediments from 53 cores across 25 sites for DNA extraction (Fig. 1a). Sites were selected to cover different geographic gradients, including north-south and east-west transects and water depth. Each sediment sample was split in two and subsequently extracted for DNA. Three replicate PCR reactions were carried out for each DNA extract for all four genetic markers, resulting in a total of 1225 PCRs (including 181 extraction and PCR blanks). The final datasets of the plants, general eukaryotes, and cyanobacteria included all 53 sediment samples from 25 sites; the copepod dataset included 42 sediment samples from 25 sites.

a Twenty-five sampling sites of this study. Sites are numbered according to longitude (eastmost site S01 to westmost site S25). Site water depths are between 46 m and 251 m. Bathymetry data82 used in the map are from IGKB (2016)83. Spatial data of the catchment are from the Spatial Information and Planning System of the LUBW84. Map was created using the software ArcGIS85. b Alpha diversity: Shannon diversity of taxa from DNA extracts across all sites, for all four datasets. Each data point represents data from a DNA extract. Abundance of a taxon is the sum of ASVs identified to that taxon. Taxa are identified at least to family for all four datasets. Alpha diversity for all datasets plotted against longitude, latitude and water depth is further shown in Supplementary Fig. 3a. c Beta diversity: Across-site Sørensen dissimilarity of taxa presence/absence between any two DNA extracts for plants and eukaryotes, plotted against the horizontal distance between their location. Linear regression lines, R2 and p-values of regression are shown on the figure. Taxa are identified at least to family for plants and eukaryotes. Across-site dissimilarity of cyanobacteria and copepods is calculated using ASV presence/absence data and is shown in Supplementary Fig. 3b. For both b and c, ASVs detected in fewer than two PCR replicates are discarded. d PCA plots are based on normalised and log-chord transformed ASV count data. Each point represents one DNA extract, where ASVs from three PCR replicates are averaged. Points are coloured by longitude of sampling site (the strongest and most significant explanatory variable in RDA, see Supplementary Table 2) to show its relationship with sample variation.
PCRs were pooled by marker type and converted into four Illumina libraries. Due to longer marker sizes, libraries of eukaryotes and cyanobacteria were each sequenced on an Illumina MiSeq 2 × 250 bp run (12–15 million clusters). Libraries of plants and copepods were sequenced together on a mutualised Novaseq 2 × 100 bp run (100 million clusters). This resulted in a total of 106.3 million paired reads, which after quality filtering, read alignment and amplicon identification, yielded 2526, 17841, 9576 and 2401 amplicon sequence variants (ASVs) for the plant, eukaryote, cyanobacteria and copepod datasets respectively (detailed data description is in Supplementary Table 1). For each marker, we plotted the read count data of all PCR reactions in respective ordination plots to examine if the PCR replicates cluster together (Supplementary Fig. 1). Relative to the variation of the whole dataset, variations of PCR replicates for eukaryotes copepods are smaller, indicating a more heterogeneous dataset. For the plants and cyanobacteria, larger variations of PCR replicates relative to the whole dataset indicates a less heterogeneous dataset. PCR replicates from the same DNA extract were merged for analyses hereafter.
Within- and across-site diversity
To quantify taxonomic diversity captured by metabarcoding, we calculated taxon-based biodiversity indices for each DNA extract. We find that alpha diversity measured from each DNA extract, calculated as the Shannon effective number of taxa, varies both within and across sites (Fig. 1b). Changes in alpha diversity across the lake do not show a specific trend except for an increase from northwestern to southeastern sites for cyanobacteria (p values < 0.001) (Supplementary Fig. 3a). However, across-site beta diversity (Sørensen dissimilarity) shows a significant linear increase with geographic distance in all datasets (p value < 0.001 for all datasets): R2 are 0.16 and 0.34 for plants and eukaryotes (taxa-based dissimilarity, Fig. 1c). Compared with eukaryotes, across-site taxa dissimilarity is generally lower for the plants, with 26.5% (standard error 0.12%) same-site dissimilarity and an increase of 0.16% (standard error 0.004%) for every 1 km increase in distance; for the eukaryotes, it is 36.7% (standard error 0.15%) same-site dissimilarity and an increase of 0.32% (standard error 0.005%) per 1 km increase in distance (Fig. 1c). At greater detail, we find most of the across-site dissimilarity is explained by the replacement of taxa rather than richness differences (Supplementary Fig. 3b). Another measure of beta diversity is to evaluate within-site dissimilarity by comparing all DNA extracts from the same site. Within-site Sørensen dissimilarity can be decomposed as the sum of a replacement dissimilarity score (beta.SIM) and a nestedness-resultant dissimilarity score (beta.NES) (Supplementary Fig. 3c). Overall, we find the within-site taxa-based dissimilarity among DNA extracts to range from 22% to 46% for plants, and from 21% to 53% for eukaryotes. beta.SIM is generally higher than beta.NES, indicating that newly detected taxa are responsible for most of the observed dissimilarity within the same site. Due to insufficient taxonomic assignment for cyanobacteria (e.g. only 339 out of 3354 cyanobacterial ASVs are identified at family level, Supplementary Table 1) and for copepods, we report beta diversity using presence/absence of ASVs for these datasets. Similar to the plants and eukaryotes, the across-site dissimilarity calculated in this way also increases with geographic distance (p values < 0.001), yet only a small amount of variation is explained (R2 = 0.24 for cyanobacteria and 0.1 for copepods) (Supplementary Fig. 3b). ASV-based within-site dissimilarity ranges from 42% to 73% for cyanobacteria and from 41% to 77% for copepods, to which ASV replacement is also the major contributor (Supplementary Fig. 3c).
Variability in sedDNA composition in relation to geographic variables
We then sought to visualise the variability in amplicon composition among DNA extracts and find its association with influencing environmental factors, i.e. geographical location, water depth and sedimentological properties. We performed principal component analyses (PCA) and redundancy analyses (RDA) using normalised and log-chord transformed amplicon counts (see Methods). The amplicon composition is found to vary along geographical gradients for all four datasets (Fig. 1d; Supplementary Table 2a). For plants and eukaryotes, we find samples close to the Rhine inflow clustering together, away from other samples in ordination space. We then used RDA to quantify the association between variation in DNA extracts and environmental factors. RDA Model 1 shows that longitude, latitude and water depth are significant explanatory factors for the variability among DNA extracts for all datasets (p values 0.001–0.018), accounting for 9.5% to 12.2% of variabilities altogether (Supplementary Table 2a). The vector representing water depth being almost vertical to that of longitude (or latitude) indicates that the effect of water depth on sample variability has little correlation with that of longitude (or latitude) (Supplementary Fig. 4a). In RDA Model 2, we used sedimentological properties as explanatory factors while controlling for longitude, latitude and water depth. A few factors appear as significant explanatory variables: chlorite content for plants, eukaryotes and copepods (p values 0.001, 0.001 and 0.058, respectively), and calcite content for plants, eukaryotes and cyanobacteria (p values 0.007, 0.001 and 0.001, respectively). Overall, sedimentological contents explain 1.5% to 9.3% of sample variabilities, but none is significant across all four datasets (Supplementary Fig. 4b, Supplementary Table 2b).
Taxon- and trait-associated distributions of sedDNA
We then grouped amplicon data by taxonomic affiliation, and for taxa with sufficient data and taxonomic resolution, we display the distribution of their frequency of detection at all sites (Figs. 2 and 3a). Across the lake, sedDNA shows varying spatial patterns by taxonomic groups. Overall, we find DNA from larger or less mobile aquatic organisms more scattered and uneven in distribution, while that from plants and smaller organisms at low trophic levels more ubiquitous. For example, orders Prolecithophora, Proseriata, and Tricladida of the flatworm phylum (Platyhelminthes), and orders Crassiclitellata, Enchytraeida and Lumbriculida of the ringed worm phylum (Annelida) are only detected at a few sites (Fig. 2a). DNA of the nematode (Nematoda), arthropod (Arthropoda) (Fig. 2a) and cercozoan (Supplementary Fig. 5) phyla is widespread across the lake but show differentiated spatial distribution among the subgroups. Specifically, detection of the orders Triplonchida (Nematoda), Haplotaxida (Annelida) and some unidentified cercozoan order(s) is clustered to the eastern sites, which are under stronger influence from the Rhine inflow; while orders Schizomida and Stomatopoda (Arthropoda), some unidentified nematode and cercozoan order(s) are detected mainly at central to western part of the lake (Fig. 2a, Supplementary Fig. 5). For zooplankton, copepods (Copepoda), cillates (Ciliophora) and other arthropod groups (Supplementary Fig. 5) as well as some nematodes (Fig. 2a), sedDNA is more homogeneous but taxon-specific distributions are still present. For the lower-trophic groups such as diatoms (Bacillariophyta), cyanobacteria, green algae (Chlorophyta), fungi (Fig. 2b) and plants (Fig. 3a), sedDNA is generally ubiquitous throughout the lake, except at sites close to the Rhine inflow. In Lake Überlingen, frequency of detection is lower for some taxa, most pronounced for nematodes (Fig. 2a), cyanobacteria (Fig. 2b) and some arthropod groups (Supplementary Fig. 5).


a Heatmaps showing the frequency of detection of MOTUs (rows) across all sampling sites (columns) for selected larger-bodied animals. b Heatmaps showing the frequency of detection of MOTUs (rows) across all sampling sites (columns) for selected planktonic and/or lower-trophic groups. In each heatmap, MOTUS are arranged by a chosen taxonomic rank: family for Cyanobacteria, genus for Copepoda, and order for the other groups. Identification at the chosen rank is labelled at the first appearing MOTU of this taxon, next to the heatmap. For some taxonomic groups, we also plot MOTUs that are only identified one rank higher and marked them as unidentified. c, d Clark & Evans aggregation index of MOTUs, plotted against the taxonomic identity or life mode of the organism. In each box plot, the thick dark line indicates median, the box represents the range from the first quartile (Q1) to the third quartile (Q3), the whisker marks the range from minimum (Q1−1.5·(Q3-Q1)) to maximum (Q3 + 1.5·(Q3-Q1)). The jitter plot shows the data points and the violin plot indicates the density of data points. The closer the index value is to 0, the more aggregated a distribution the MOTU has. ASVs detected in fewer than two PCR replicates and MOTUs detected in fewer than two sites are discarded from the calculation of aggregation index. Taxon names and trait information are listed in Supplementary Data 2.

a Heatmap showing the frequency of detection for selected plant species, ordered by their traits. Species names and trait information are listed in Supplementary Data 3. b The relationship between the number of sites a plant species or genus was detected in the sediment, elevation of its natural occurrence and abundance in the catchment area. Occurrence data is from InfoFlora76, therefore restricted to the Swiss part of the catchment area: elevation is the medium elevation of each 5 × 5 km sampling grid, number of observations (a proxy for abundance) is the summed count within each sampling grid. Plant species/genera are not labelled on the chart except for whether they are aquatic plants. c Example distributions that show the occurrence and abundance of three selected plant species in comparison to the abundance of their sedimentary DNA. Size of red violet circles indicates the number of observations within in a 5 × 5 km sampling grid (a proxy for abundance). Size of indigo squares indicates the log count of ASVs at each site in the lake. The approximate range of the Swiss catchment, where occurrence data are available in InfoFlora76, is marked with dashed lines. Terrain map is retrieved from Stamen Maps86 using the R package ggmap87.
To quantify the observed connection between sedDNA distribution and the biological characteristics of taxa, we calculated an aggregation index (Clark and Evans 1954) for each Molecular Operational Taxonomic Units (MOTUs, definition see Material & Methods) in the eukaryote and cyanobacteria datasets and plotted its values against the taxonomic identity and life mode of respective organisms (Fig. 2c). This index measures how much the distribution of a MOTU departs from randomness, and both taxonomic affiliation and life mode can be seen associated with the index. We find that larger and less mobile groups such as flatworms and ringed worms (and to a lesser extent, nematodes and fungi) show a strong signal of aggregation, as most of the index values are close to zero. In comparison, arthropods, diatoms, green algae and cyanobacteria have a wider range in index values, indicating less clustered occurrences in the lake. Figure 2d further shows that when grouped by life mode, benthic, parasitic and terrestrial organisms all show a strong signal of aggregation, while the planktonic, planktonic-benthic and epiphytic/epizoic organisms are less aggregated. It should be noted that due to irregular sampling in space, the calculated Clark and Evans index is a systematic underestimation (i.e. an overestimation of aggregation), therefore comparing the aggregation index across groups is more meaningful than interpreting its absolute value.
sedDNA distribution of plants in relation to their natural occurrences in catchment
We examined the distribution of plant sedDNA separately from other datasets, because unlike the eukaryote, cyanobacteria and copepod datasets, most plants we detected are of terrestrial origin. We show that DNA from common plants is ubiquitous with high frequency of detection across the lake, while rarer taxa are scattered with lower frequency of detection (Fig. 3a, and Supplementary Fig. 6a for key taxa). Furthermore, we find that more alpine species are detected at the central-to-east part of the lake, with the highest frequency of detection at sites close to the mouth of the Rhine (Supplementary Fig. 6b). This phenomenon is not limited to alpine taxa such as the glacier buttercup (Ranunculus glacialis) (Fig. 3c), but also present for other species like the common kidneyvetch (Anthyllis vulneraria) and the mild stonecrop (Sedum sexangulare) (Supplementary Fig. 6a), indicating a localised transport and deposition of plants occurring in the upper-stream catchment. Out of the 131 species shown on Fig. 3a, only eight species register as aquatic plants. The number of aquatic species detected is similar across the lake except at sites S02-04, S20 and S21 where the river Rhine flows in and out of the Upper Lake (Supplementary Fig. 6b). Plants that are not alpine or aquatic show consistent detection across the lake (Supplementary Fig. 6b). When grouping species by growth form, it is evident that the dataset consists mostly of non-grass herbs, shrubs and trees, and to a lesser extent of ferns, grass and sedges. However, we found no association between plant growth form and distribution of sedDNA (Supplementary Fig. 6c).
Lastly, we evaluated the relationship between plant sedDNA distribution and their natural occurrences in the catchment. Due to inconsistencies in sampling and recording techniques among existing surveys, we restricted our analysis to the Swiss catchment, where spatially resolved plant survey data are provided by Info Flora (infoflora.ch). Despite relative high variation, association between the natural occurrences of plants and their sedDNA distribution can be summarised as follows: plants that occur at higher altitudes or with lower abundance are often only detected at a few sites, for example close to the mouth of Rhine (Fig. 3b, Supplementary Fig. 6b), while abundant and lower-altitude plants are ubiquitous across the lake. Aquatic plants are scarce both in survey data and sedDNA data, hence the link between altitude of occurrence and number of sites detected cannot be deduced. Yet although three species/genera occur as high as 2000 m in the catchment, most occurrences are below 600 m (Fig. 3b). As an illustration of how natural occurrences are related to the distribution of sedDNA, we display the mapped occurrences and sedDNA detection of three plant species with contrasting distributions in Fig. 3c.
Discussion
Focusing on a single large lake system, our findings add to the growing body of evidence that sedDNA is heterogeneously deposited and distributed in various types of environments39,40,41,42. By showing the distinct distributions among highly diverse organisms both of aquatic and of terrestrial origins, we point to the factors governing these distributions and provide an outlook for the use of sedDNA in contemporary and paleo ecological research.
Overall, different than the far-travelling eDNA in river networks27,43, our data suggest more complex transport and deposition pathways of lake sedDNA. Although Upper Lake Constance has frequent waves and vertical mixing44,45, water movement seems to have limited effect on the (re)distribution of endogenous organism remains in the sediment, which presents varying spatial patterns linked to their range of occurrence and life mode. For exogenous organisms, while common plants are ubiquitously detected with high read number and frequency, rare and distant taxa such as alpine and aquatic plants are showing the effect of carriage by water. DNA of alpine plants (and other distant plant species) primarily aggregate near the inflow, yet at (but not beyond) the deepest site some 20 km from the mouth of inflowing Rhine, they are still detected with similar diversity although fewer reads and lower rates of detection (Supplementary Fig. 6b). Aquatic plants have a drop in diversity near inflow and outflow, where water speed is fast. We speculate that it is the passiveness and rarity of those plants that make their organic remains susceptible to water movement, and consequently their sedDNA. Overall, our results imply that the immediate source of sedDNA is the settled organismal remains or living benthic organisms, rather than water eDNA molecules which are more easily carried away by water flow. Living or dead, the final settled place of an organism determines the vicinity where the respective sedDNA will be found.
Across the lake, we observed highly variable alpha diversity and high dissimilarity (i.e., high beta diversity) among samples. This could be caused by the low detectability of rare DNA (Supplementary Fig. 2), heterogeneity of sediment DNA at small scales, and the natural variation in the lake biome along geographic gradients. The importance of these three factors on the variability of retrieved data should also increase in this order: in Fig. 1d and Supplementary Fig. 1, we show it is generally the trend that PCRs vary primarily across geographic locations, with PCRs from extracts of the same site (biological replicates), and PCRs from the same extracts (technical replicates) clustering together with smaller variations. If more biological or technical replicates were added, it would be reasonable to expect that they would have similar variations as the existing ones. Therefore, if the variations of PCR replicates are small compared with the variation of PCRs across sites, it is unlikely that more replicates will reduce the heterogeneity in the whole dataset. This is the case for the eukaryote dataset (Supplementary Fig. 1). However, for the plant and cyanobacteria datasets, where the variations across sites relative to those of PCR replicates are smaller, more replicates might capture more variation across sites, hence reduce the overall heterogeneity in the dataset. In general, we think increasing PCR replication, extracting DNA from higher amount of sediment, or collecting more samples from a site can alleviate heterogeneity to a certain extent, but spatial heterogeneity would still be present in a dataset collected from a large and complex area.
Although our data were collected from surface sediments where DNA is less degraded, the spatial heterogeneity we observe in surface sediments will likely pertain to ancient sedDNA data retrieved from a similarly complex area. This hence poses a challenge in drawing temporal inference from sedimentary data. Since one core only captures a subset of organisms in the area and one might argue that the representation of samples can further deteriorate when going back in time due to DNA degradation, it becomes possible that the recovered temporal signal is artefactual or biased. However, from empirical sedDNA studies that cross-checked other data sources, we see that sedimentary DNA does reflect the expected temporal change, be it major condition shifts due to climatic or anthropogenic impact46,47 or annual variations48. On the other hand, diversity metrics can be evaluated on widespread low-trophic-level organisms such as common plants and plankton19 to attain more reliable results, or applied in large scale multi-site analyses to obtain higher statistical power49.
The fact that endogenous and exogenous taxa have different DNA deposition pathways, and taxa with different life mode have distinct DNA distributions suggests a more organism-aware sampling scheme for future sedDNA studies. For exogenous organisms that are rare and occur far from the lake, sampling should be done near the mouth of the inflow. For aquatic animals, sampling within the range of occurrence would be more desirable. We did not find sediment composition to be associated with the general retrieval of sedDNA (Supplementary Table 2b), possibly due to the consistent sediment composition in Lake Constance (Supplementary Fig. 7a), yet certain elemental contents are associated with the detection of some organisms. For example, detection of annelid ASVs is associated with sediments higher in organic carbon and sulphur (Supplementary Fig. 7b) and detection of ASVs of epiphytic/epizoic or terrestrial organisms is associated with sediment higher in dolomite content and C/N ratio (Supplementary Fig. 7c). We speculate that sediment composition reflects the habitat characteristics that certain organisms prefer or are likely to be deposited in, yet the overall effect from sediment content is still weak.
While posing challenges, the observed heterogeneity and putative spatial fidelity of sedDNA opens its possibility for fine-scaled spatiotemporal mapping of species, even within waterbodies. By sampling at different sites, ancient sedDNA may shed light on the timing and location of diversification for species that are known to coexist as subpopulations, such as the Eurasian perch (Perca fuviatilis L.)50. As distant exogenous organisms are deposited within limited range in the lake, their changes in sedDNA detectability through time along an in-lake transect may reflect the causes and directions of colonisation. The influence of climate versus human settlement on the spreading of some plant species, such as the beeches (Fagus) and other trees51,52 may therefore be better resolved using ancient sedDNA data. To approach these questions, we would need not only high-throughput sequencing data with higher depth and better accuracy, but also the integration of archaeo-anthropological and digital geographic model of the surrounding terrain.
Although evidence is concurrent across the groups of organisms analysed, our study has possible methodological limitations due to biases in sampling, metabarcoding markers, limited taxonomic resolution and lack of comprehensive reference and trait databases. Due to lost cores from challenging sampling conditions, some sites ended up having two instead of four DNA extracts, which could have inflated the dissimilarity among sites. The cyanobacteria marker primers had ~45% unspecific amplification and those identified as cyanobacteria had poor taxonomic resolution despite the long marker size (~380 bp, Table 1). The differential representation and DNA decay rate among cyanobacteria taxa53,54 could further introduce biases. The copepod marker yielded sequence variants identified to copepods, but most could not be identified to genus level with existing reference databases; the 18S marker for eukaryotes also has limited taxonomic resolution in groups such as arthropods and vertebrates. Even after successful taxonomical classification, the challenges in assigning traits to identified organisms further downsized the total number of taxa that could be analysed. For comparing plant sedDNA and plant occurrences in the catchment area, we only used the database from InfoFlora and restricted to the Swiss catchment area, while in reality, plant sedDNA in Lake Constance has sources from the Austrian and German catchment as well, so the phenomenon we observed in the plant dataset could not be fully addressed with the survey and trait databases we used. Overall, since our study utilises multiple markers to compensate for biases from one, and the analyses are performed on ample data not restricted to specific taxa, we believe the main conclusions of our study to hold despite these limitations.
Methods
Surface sediment sampling and sedimentological data
In this study, sediment samples from a total of 76 short cores across 25 sites from the Upper Lake Constance, including Lake Überlingen (Fig. 1A) were analysed. Sediment coring at sites S01 to S22 took place in February 2019 and at sites S23 to S25 in March 2019. Sites were chosen in accordance with a previous sampling of the lake bottom55 after consultation with the Institute for Lake Research at the State Institute for Environment Baden-Württemberg (LUBW, Landesanstalt für Umwelt Baden-Württemberg). At sites S01 to S22, we attempted to extract three cores from the ship MS Kormoran with a gravity multicorer. Due to practical difficulties, overall two to three cores were successfully taken. One of these cores was used to retrieve sedimentological data and the other one or two were sampled for DNA extraction. At sites S23 to S25, four cores were taken with single coring at each site from the ship MS Lauterborn and one surface sediment sample was collected for each core. To take surface sediment samples, on the boat immediately after coring we first removed visible organism remains and then took sediment from top 1.5 cm of the core with a sterile syringe. The sterile syringe had the top of its barrel removed for the ease of sampling. Sediment samples were kept at −20 °C until DNA extraction.
Sedimentological data of surface sediment from sites S01 to S22 were collected at the Baden-Württemberg State Institute for the Environment (LUBW) (Supplementary Data 1). Sediment samples were first freeze-dried and weighed to measure water content. They were then milled, from which small quantities (in milligrams) were analysed to measure total carbon, total nitrogen, total sulphur and total organic carbon (after carbonates were removed by diluted HCl) with an element-analyser (Euro EA). Relative quantities of minerals (muscovite, chlorite, quartz, calcite, feldspar, dolomite, pyrite and amphibolite) in each sample were determined using a Rigaku X-ray diffractometer. Grain size distributions (sand, silt, clay) of a parallel sample were measured with a laser diffractometer (Saturn DigiSizer) after removing organic carbon using H2O2.
DNA extraction, metabarcoding and amplicon sequencing
All laboratory work was conducted in the DNA laboratories of the Limnological Institute at the University of Konstanz. DNA extraction and PCR set-up were carried out under respective designated UV-hoods for work with environmental DNA, located in an eDNA area of a DNA extraction laboratory kept separate from PCR products. PCRs were run and further processed in a PCR laboratory on the above floor.
Sediment DNA was extracted between April and June 2019, using ~1 g of surface sediment using the DNeasy PowerSoil Kit (Qiagen, Germany) under an environmental DNA designated hood. For each extraction, 1 g of sediment was divided in half, vortexed and lysed in two separate PowerBead Tubes. The two portions of lysate were then combined and processed according to the standard protocol of the manufacturer. Sediment DNA was eluted in 100 µL Elution Buffer. For each core from sites S01 to S22 we carried out two extractions, which gave us two or four extractions at each site. For each core from sites S23 to S25 we carried out one extraction. In total we generated 92 DNA extracts from 53 sediment cores and 10 extraction negative controls. The extraction negative controls were processed in the same way as the samples except that they had no sediment input.
Four metabarcoding runs were performed on DNA extracts in three replicates. These four runs (Table 1) targeted: (1) the V7 region of the 18 s rRNA gene of general eukaryotes; (2) the trnL gene of vascular plants; (3) the 16s rRNA gene of cyanobacteria; and 4) the 28s rRNA gene of copepods, respectively. The PCR targeting copepods used primers designed in this study and were run on a subset of 69 DNA extracts from 53 sediment cores. Methods to design the copepod primers are described in Supplementary Methods and Supplementary Table 4.
To distinguish each PCR reaction after sequencing, primers had unique 8 bp tags added on the 5’ end and further three random base pairs (NNN) on the 5’ end of the tags56. These tags varied from each other in at least five base pairs. Information of PCR reaction conditions and marker primers (without tags) used in this study are listed in Table 1. PCR reactions included the following reagents: Platinum Taq DNA Polymerase High Fidelity (Thermo Fisher Scientific), dNTP Mix (25 mM each, Thermo Fisher Scientific), BSA (20 mg/mL, molecular biology grade, New England Biolabs; processed under UV for 10 min before use), MgSO4 solution (50 mM, provided together with Taq Polymerase), DEPC-treated water (Carl Roth). Primers were ordered from Integrated DNA Technologies, using standard desalting purification. PCR reactions were set up according to Supplementary Table 3.
We purified PCR products with the HighPrep PCR Clean-up System (MagBio Genomics, USA), measured DNA concentration using the AccuBlue Broad Range dsDNA Quantitation Kit (Biotium, USA) on 96-well Optical-Bottom plates using the CLARIOstar Plus microplate reader and pooled PCR products in equal DNA quantity. Pooling volumes were at least 1 µL for non-control PCR products and 10 µL for extraction and PCR negative controls. The pool of PCR products was concentrated with MinElute PCR Purification Kit (Qiagen, Germany) and sent to Fasteris SA (Switzerland) for amplicon library preparation and sequencing. Libraries of eukaryote and cyanobacteria amplicons were each sequenced on a Miseq 2 × 250 bp run (12–15 million cluster); libraries of vascular plant and copepod amplicons were sequenced together on a mutualised Novaseq 2 × 100 bp run (100 million cluster).
Amplicon data processing and taxonomic identification
After receiving the raw sequence reads, we first removed the adapters using the ILLUMINACLIP programme in Trimmomatic57, followed by read alignment, demultiplexing, dereplicating and filtering using OBITools58 release 3 (https://metabarcoding.org/obitools3). Specifically, we discarded reads whose alignment scores (score_norm in ObiTools3) were below 0.5 and whose total count were fewer than 10. To remove single base pair errors presumably induced in PCR, we used obi clean (OBITools3) with parameter r set to 0.5 (for copepod dataset) or 0.2 (for other datasets). We then detected chimeras with programmes VSEARCH uchime2_denovo, VSEARCH uchime_ref59 and DADA260. After comparison, only the output from VSEARCH uchime2_denovo was used. We did not use outputs from VSEARCH uchime_ref and DADA2 because they marked over 60% reads as chimeras and the geographical gradient present in the original dataset, which we believe is authentic, was disrupted. Lastly, we performed amplicon taxonomic identification to the lowest common ancestor (LCA) using the ecotag programme (OBITools3). The eukaryote and cyanobacteria datasets were identified at 97% identity threshold with the SILVA database61 (SSU and LSU Ref NR 138.1); the plant dataset was identified at 98% threshold with the EMBL Nucleotide Sequence Database62 (release March 2020); the copepod dataset was identified at 85% threshold using a customised reference database consisting of all 28s rRNA gene sequences archived in SILVA and EMBL databases and sequences from collected copepod specimens (Supplementary Methods). Sequences that were not assigned to any taxonomic rank were discarded.
We then examined sequencing depth, contamination and PCR replicability of identified sequences using the R package metabaR63. All samples in the plant and copepod datasets had around 104 reads or more, therefore were kept for analysis. Samples having more than 1000 reads were kept for the eukaryote dataset. The cyanobacteria dataset had overall a low sequencing depth due to unspecific amplification, and we kept samples that had more than 250 reads. We removed contaminant sequences, i.e. any sequences having the highest frequency in at least one negative control (function contaslayer, method = “max”). Next, we adjusted sequence counts in consideration of potential tag-jumps with function tagjumpslayer. Lastly, we identified and discarded dysfunctional PCRs (outlier replicates or singleton replicates) using function pcrslayer (method = “centroid”) and aggregated remaining replicates by summing up read counts using the function aggregate_pcrs. Cleaned datasets consisted of all unique sequences, also referred to as Amplicon Sequence Variants or ASVs.
For downstream analyses, we either directly used the ASVs as input data or used Molecular Operational Taxonomic Units (MOTUs) aggregated from the ASVs (considering many ASVs are potentially erroneous sequences rather than authentic unique variants). We also used three types of abundance measures in different analyses. Data preparation is described in the following two paragraphs:
To construct MOTUs for the eukaryote, cyanobacteria and copepod datasets, we aggregated closely related ASVs based on phylogenetic relatedness. First, we ran multiple alignments of the sequences and made neighbour-joining phylogenetic trees under the Tamura-Nei genetic distance model using Geneious Prime 2022.1.1 (https://www.geneious.com). Using the phylogenetic tree, we marked clades whose branch lengths were under a value set by visual examination (0.007 for the eukaryote and cyanobacteria datasets, and 13.3 for the copepod dataset) and collapsed all sequences having the same taxonomic identification in the same clade. Collapsed sequences were considered as one MOTU and their read counts were summed. Since the trnL gh marker is not suitable for making phylogenies at interspecies level across many families64, we created MOTUs for the plant dataset by summing up ASVs having the same taxonomic identity.
Two types of ASV/MOTU abundance measures were used in the analyses. (1) Normalised and log-chord transformed read counts, which were used in ordination analyses such as principal component analysis (PCA) and redundancy analysis (RDA). Amplicon counts in PCR samples were normalised with function estimateSizeFactors from the R package DESeq265, where size factors were estimated using the shorth function. Normalised amplicon counts were then log-chord transformed by the decostand function in the R package vegan66. We chose log-chord transformation because it reduces skewness of sequence counts through log-transformation, and then enables the calculated Euclidean distance to be double-zero asymmetric through chord-transformation67. (2) Frequency of detection68 in PCRs at sites, which was used in heat map visualisation (Figs. 2a, b and 3a). We calculated this as the frequency of an ASV or a MOTU among all individual PCRs using all DNA extracts of sediment samples at same site. For example, an ASV detected in two out of 12 individual PCRs at a site (3 PCR replicates per extract x 2 extracts per sediment sample x 2 sediment samples per site) has a frequency of detection of 2/12. The rationale behind using frequency of detection as a proxy for abundance is that sampling a site with n PCRs can be viewed as an experiment of n binomial trials, thus linking detection probability to abundance69. We find that frequency of detection data correlates to read counts in our datasets (Supplementary Fig. 2) and avoids extreme values. Additionally, we used normalised and log-transformed read counts in direct comparison of ASV/MOTU abundance, such as shown in the example distributions of Fig. 3b and Supplementary Figs. 2 and 5. When analyses were PCR based, such as assessing PCR replicability (Supplementary Fig. 1), we did not merge PCRs; when analyses were DNA extract based, such as calculating alpha and beta diversity (Fig. 1b, c), we merged PCRs from the same DNA extract; when analyses were site based, such as calculating frequency of detection at a site (Fig. 2a, b, Fig. 3a), we used all PCRs from extracts sampled from the same site.
General scheme of statistical analysis and data visualisation
For each of the four datasets, we reported the within- and across-site diversity using common diversity metrics. We then characterised the variability of ASV composition among samples in relation to geographic factors and visualised the distribution of different taxonomic or trait groups. In addition, we linked the plant dataset with plant occurrences in the lake catchment area by including vegetation survey data into the analysis. Except in reporting diversity metrics, when comparing samples/sites we favoured using normalised and transformed read counts or frequency of detection data (both described in the paragraph above) over summary distance metrics (such as Hellinger distance and Bray-Curtis dissimilarity) to minimise information loss. Amplicon count data, or transformed forms of them, preserve abundance and taxonomic diversity information in the raw dataset, while distance metrics tend to reduce the high-dimensional data to a single distance measure between two samples.
Measuring within- and across-site diversity in DNA samples
For plants, eukaryotes and cyanobacteria, we calculated taxon-based biodiversity indices for each DNA extract to quantify the taxonomic diversity captured by metabarcoding. These indices measure alpha diversity in a DNA extract, within-site beta diversity (by comparing data from all DNA extracts from the same site), and across-site beta diversity (by comparing any two DNA extracts). First, we used the effective number of taxa based on Shannon index as a measure of alpha diversity (function diversity, index = “shannon”, package vegan). Normalised (but not transformed) taxon abundance data were used for this calculation. Richness of taxa was calculated as the count of unique taxa. For beta diversity, we first measured Sørensen dissimilarity by comparing all DNA extracts within a site, which can be partitioned into the replacement dissimilarity and nestedness-resultant dissimilarity (βSIM and βNES from Baselga70). We then measured across-site dissimilarity by calculating Sørensen dissimilarity between every two DNA extracts across the lake (function beta.div.comp(coef = “BS”, quant = F) from Legendre71). This index can be decomposed into replacement index and richness difference index. We used presence/absence data to calculate beta diversity and to reduce noise, we removed ASVs that appeared in fewer than two out of three PCR replicates before aggregating them by taxonomic affiliation. R codes that calculate these indices were from source literature.
Evaluating sample variability in relation to geographic variables
To visualise the variability in amplicon composition among DNA extracts and its association with potential influencing factors, we conducted principal component analyses (PCA) using normalised, log-chord transformed ASV counts (function prcomp, package stats). We then ran redundancy analyses (RDA) to quantify the strength and significance of such associations (function rda, package vegan). Input data for RDA was normalised and log-chord transformed amplicon counts in each DNA extract. We built two RDA models to characterise the effect of geographic location and of sedimentological property separately. In Model 1, we included geographical coordinates and sampling water depth as explanatory variables. All samples across the lake were included. In Model 2, we included sedimentological data as potential explanatory variables and conditioned on longitude, latitude and water depth. To avoid extreme confounding effects from the irregular shape of the lake and the effect from its in- and outflows, in Model 2 we excluded the eastern-most samples close to the Rhine inflow and the western-most samples in Lake Überlingen. Collinearity between explanatory variables was assessed using the vif.cca function (package vegan), and variables with an output value larger than 10 were removed. Coordinates and water depth were kept in Model 2 regardless of their output values from vif.cca, as we believe they are intrinsic factors contributing to the variability of sequence read composition in samples and therefore need to be controlled for. Adjusted R2 was computed using function RsquareAdj (package vegan) to measure the amount of variance explained by explanatory variables. Statistical significance was assessed through permutation test using function anova.cca (package vegan, number of permutations = 10000).
Visualisation of DNA distribution for different taxonomic groups
To visualise the spatial distribution of reads assigned to specific taxonomic groups, we constructed heat maps associated to the sites across the lake. For cyanobacteria, copepod and some non-plant phyla in the eukaryote dataset that have the most MOTU counts, heat maps were constructed in the following way: Input data were the frequency of PCR detection at sites of MOTUs. Each MOTU was shown on the heat map as a row of pixels, the colour of which indicates frequency of detection at a site (0 to 1). Vertically, sequences were ordered first by phylogenetic relatedness and then by similarity in distribution across the lake. Distribution similarity among MOTUs was found through hierarchical clustering (function hclust, R package stats).
For the plant dataset, we constructed the heat maps similarly, but using normalised log-transformed MOTU counts as input. MOTUs were displayed based on types (alpine, aquatic, cultivated, other) or growth forms (trees and shrubs, forbs, grass and sedges, fern) instead of taxonomic or phylogenetic relatedness. We annotated plant taxa using the Info Flora database (infoflora.ch) and the Categorical Traits Lookup Table from the TRY Database72 (2012 release).
Trait assignment and quantification of randomness of DNA distribution
We conducted exploratory analyses to establish whether DNA distribution is related to specific traits of organisms. We assigned the traits body size, trophic level and life mode to aquatic organisms that were identified at least to family level. We also assigned growth forms and types (alpine, aquatic, cultivated, other) to plants that were identified at least to genus level. Multiple trait databases for aquatic organisms73,74,75 and for plants72,76 were used. However, due to lack of information for most identified taxa, our final analysis addresses the relation between life mode and DNA distribution. Taxa were assigned life mode categories as in the Bundestaxaliste der Gewässerorganismen Deutschlands (BTL)75: planktonic (P), benthic (B), parasitic (Pa), epiphytic / epizoic (E), submersed/emerged (SE), nekton (NK), neuston (NS) and terrestrial (T). If a taxon was identified only to genus level, life mode was assigned only if all species under that genus in the database have the same life mode. Similar was done for taxa identified only to family level. Taxa that were not listed in these databases but have sufficient information available were processed individually, with the assistance of TaxonKit77 to parse taxonomic information. Taxa with no information were dropped.
To quantify how the distribution of DNA of different taxa deviates from randomness, we adopted the aggregation index R introduced by Clark and Evans (1954)78 (function clarkevans from the R package spatstat.explore79). For each MOTU, R was calculated as the ratio of its mean observed distance to the nearest sample of presence, to what would be expected in a random distribution of the same density. As required by the calculation, distances between surface sediment samples collected from separate cores at the same site were estimated to be 35 cm apart, and samples from the surface sediment of the same core were estimated to be 2 cm apart. R has a limited range of from 0 to 2.1491. The smaller R is, the more aggregated a distribution it indicates. Although in theory under a random distribution R = 1 and under an even distribution R = 2.1491, in our irregular sampling space this would not be the case. Therefore, comparing R value across groups is more meaningful than interpreting its absolute value.
Linking plant occurrence in the catchment area to sedDNA detection
To characterise the association between the natural occurrences of plants and sediment DNA distribution, we acquired plant occurrence data in the catchment area of Lake Constance (Swiss side) from InfoFlora76. Catchment area was defined according to Der Bodensee (2004; p. 8)80. Growth locations were standardised into 5 × 5 km grids, and occurrence was recorded as the number of observations. We used the medium elevation within each grid as a proxy for growth elevation. Elevation data were extracted using the get_elev_raster function (package elevatr) and rounded to the multiple of 50 m so that occurrences at similar elevation could be aggregated. We chose plant taxa that were non-cultivated, identified to species level or to genus level but for which only one species was known to occur in the region. To account for survey bias, we excluded plant species whose recorded occurrence in the Info Flora database deviated too much from general knowledge. A total of 77 plant taxa was kept for this analysis.
Data availability
Raw sequence data are deposited to the European Nucleotide Archive (ENA) under the project accession PRJEB61101. Supplementary Data 1–4 and are deposited together with custom code at: https://github.com/wangyi91/spatial-heterogeneity81 (https://doi.org/10.5281/zenodo.7817775).
Code availability
Data analyses and plotting used R (version 4.0.5). Custom code and files necessary to process data, run analyses and produce results are available at: https://github.com/wangyi91/spatial-heterogeneity81, (https://doi.org/10.5281/zenodo.7817775).
References
Smol, J. P. & Cumming, B. F. Tracking long-term changes in climate using algal indicators in lake sediments. J. Phycol. 36, 986–1011 (2000).
Arnaud, F. et al. Erosion under climate and human pressures: An alpine lake sediment perspective. Quat. Sci. Rev. 152, 1–18 (2016).
Willerslev, E. et al. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science 300, 791–795 (2003).
Zimmermann, H. et al. Sedimentary ancient DNA and pollen reveal the composition of plant organic matter in Late Quaternary permafrost sediments of the Buor Khaya Peninsula (north-eastern Siberia). Biogeosciences 14, 575–596 (2017).
Crump, S. E. et al. Ancient plant DNA reveals High Arctic greening during the Last Interglacial. Proc. Natl. Acad. Sci. 118, e2019069118 (2021).
Lejzerowicz, F. et al. Ancient DNA complements microfossil record in deep-sea subsurface sediments. Biol. Lett. 9, 20130283 (2013).
Kjær, K. H. et al. A 2-million-year-old ecosystem in Greenland uncovered by environmental DNA. Nature 612, 283–291 (2022).
Monchamp, M.-E. et al. Homogenization of lake cyanobacterial communities over a century of climate change and eutrophication. Nat. Ecol. Evol. 2, 317–324 (2018).
Barouillet, C. et al. Investigating the effects of anthropogenic stressors on lake biota using sedimentary DNA. Freshw. Biol. 00, 1–19 (2022).
Fritz, S. C. Deciphering climatic history from lake sediments. J. Paleolimnol. 39, 5–16 (2008).
Dearing, J. A. & Jones, R. T. Coupling temporal and spatial dimensions of global sediment flux through lake and marine sediment records. Glob. Planet. Change 39, 147–168 (2003).
Beaudoin, A. B. & Reasoner, M. A. Evaluation of differential pollen deposition and pollen focussing from three Holocene intervals in sediments from Lake O’Hara, Yoho National Park, British Columbia, Canada: intra-lake variability in pollen percentages, concentrations and influx. Rev. Palaeobot. Palynol. 75, 103–131 (1992).
Corinaldesi, C., Danovaro, R. & Dell’Anno, A. Simultaneous recovery of extracellular and intracellular DNA suitable for molecular studies from marine sediments. Appl. Environ. Microbiol. 71, 46–50 (2005).
Mauvisseau, Q. et al. The multiple states of environmental DNA and what is known about their persistence in aquatic environments. Environ. Sci. Technol. 56, 5322–5333 (2022).
Niemeyer, B., Epp, L. S., Stoof-Leichsenring, K. R., Pestryakova, L. A. & Herzschuh, U. A comparison of sedimentary DNA and pollen from lake sediments in recording vegetation composition at the Siberian treeline. Mol. Ecol. Resour. 17, e46–e62 (2017).
Sjögren, P. et al. Lake sedimentary DNA accurately records 20th Century introductions of exotic conifers in Scotland. New Phytol 213, 929–941 (2017).
Alsos, I. G. et al. Plant DNA metabarcoding of lake sediments: How does it represent the contemporary vegetation. PLOS ONE 13, e0195403 (2018).
Giguet-Covex, C. et al. New insights on lake sediment DNA from the catchment: importance of taphonomic and analytical issues on the record quality. Sci. Rep. 9, 14676 (2019).
Stoof-Leichsenring, K. R., Dulias, K., Biskaborn, B. K., Pestryakova, L. A. & Herzschuh, U. Lake-depth related pattern of genetic and morphological diatom diversity in boreal Lake Bolshoe Toko, Eastern Siberia. PLOS ONE 15, e0230284 (2020).
Weisbrod, B. et al. Is a central sediment sample sufficient? Exploring spatial and temporal microbial diversity in a small lake. Toxins 12, 580 (2020).
Jeppesen, E. et al. Sub-fossils of cladocerans in the surface sediment of 135 lakes as proxies for community structure of zooplankton, fish abundance and lake temperature. Hydrobiologia 491, 321–330 (2003).
Jiang, X. et al. Increasing dominance of small zooplankton with toxic cyanobacteria. Freshw. Biol. 62, 429–443 (2017).
Buxton, A. S., Groombridge, J. J. & Griffiths, R. A. Seasonal variation in environmental DNA detection in sediment and water samples. PLOS ONE 13, e0191737 (2018).
Olajos, F. et al. Estimating species colonization dates using DNA in lake sediment. Methods Ecol. Evol. 9, 535–543 (2018).
Thomson-Laing, G., Howarth, J. D., Vandergoes, M. J. & Wood, S. A. Optimised protocol for the extraction of fish DNA from freshwater sediments. Freshw. Biol. 67, 1584–1603 (2022).
Taberlet, P., Coissac, E., Hajibabaei, M. & Rieseberg, L. H. Environmental DNA. Mol. Ecol 21, 1789–1793 (2012).
Nevers, M. B. et al. Influence of sediment and stream transport on detecting a source of environmental DNA. PLOS ONE 15, e0244086 (2020).
Drummond, J. A. et al. Diversity metrics are robust to differences in sampling location and depth for environmental DNA of plants in small temperate lakes. Front. Environ. Sci. 9, 617924 (2021).
Beentjes, K. K., Speksnijder, A. G. C. L., Schilthuizen, M., Hoogeveen, M. & van der Hoorn, B. B. The effects of spatial and temporal replicate sampling on eDNA metabarcoding. PeerJ 7, e7335 (2019).
Troth, C. R., Sweet, M. J., Nightingale, J. & Burian, A. Seasonality, DNA degradation and spatial heterogeneity as drivers of eDNA detection dynamics. Sci. Total Environ. 768, 144466 (2021).
Carraro, L., Blackman, R. C. & Altermatt, F. Modelling eDNA transport in river networks reveals highly resolved spatio-temporal patterns of freshwater biodiversity. 2022.01.25.475970 Preprint at https://doi.org/10.1101/2022.01.25.475970 (2022).
Vadeboncoeur, Y., McIntyre, P. B. & Vander Zanden, M. J. Borders of biodiversity: Life at the edge of the World’s large lakes. BioScience 61, 526–537 (2011).
Tolotti, M. et al. Large and deep perialpine lakes: a paleolimnological perspective for the advance of ecosystem science. Hydrobiologia 824, 291–321 (2018).
Taberlet, P. et al. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 35, e14 (2007).
Gast, R. J., Dennett, M. R. & Caron, D. A. Characterization of protistan assemblages in the ross sea, antarctica, by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 70, 2028–2037 (2004).
Van de Peer, Y., De Rijk, P., Wuyts, J., Winkelmans, T. & De Wachter, R. The European small subunit ribosomal RNA database. Nucleic Acids Res 28, 175–176 (2000).
Nübel, U., Garcia-Pichel, F. & Muyzer, G. PCR primers to amplify 16S rRNA genes from cyanobacteria. Appl. Environ. Microbiol. 63, 3327–3332 (1997).
Boutte, C., Grubisic, S., Balthasart, P. & Wilmotte, A. Testing of primers for the study of cyanobacterial molecular diversity by DGGE. J. Microbiol. Methods 65, 542–550 (2006).
Andersen, K. et al. Meta-barcoding of ‘dirt’ DNA from soil reflects vertebrate biodiversity. Mol. Ecol 21, 1966–1979 (2012).
Edwards, M. E. et al. Metabarcoding of modern soil DNA gives a highly local vegetation signal in Svalbard tundra. Holocene 28, 2006–2016 (2018).
Jeunen, G.-J. et al. Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol. Ecol. Resour. 19, 426–438 (2019).
Massilani, D. et al. Microstratigraphic preservation of ancient faunal and hominin DNA in Pleistocene cave sediments. Proc. Natl. Acad. Sci. 119, e2113666118 (2022).
Barnes, M. A. & Turner, C. R. The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 17, 1–17 (2016).
Appt, J., Imberger, J. & Kobus, H. Basin-scale motion in stratified Upper Lake Constance. Limnol. Oceanogr. 49, 919–933 (2004).
Preusse, M., Peeters, F. & Lorke, A. Internal waves and the generation of turbulence in the thermocline of a large lake. Limnol. Oceanogr. 55, 2353–2365 (2010).
Epp, L. S. et al. Lake sediment multi-taxon DNA from North Greenland records early post-glacial appearance of vascular plants and accurately tracks environmental changes. Quat. Sci. Rev. 117, 152–163 (2015).
Ibrahim, A. et al. Anthropogenic impact on the historical phytoplankton community of Lake Constance reconstructed by multimarker analysis of sediment-core environmental DNA. Mol. Ecol. 30, 3040–3056 (2021).
Capo, E. et al. How does environmental inter-annual variability shape aquatic microbial communities? A 40-Year annual record of sedimentary DNA from a boreal lake (Nylandssjön, Sweden). Front. Ecol. Evol. 7, 245 (2019).
Wang, Y. et al. Late Quaternary dynamics of Arctic biota from ancient environmental genomics. Nature 600, 86–92 (2021).
Behrmann-Godel, J., Gerlach, G. & Eckmann, R. Postglacial colonization shows evidence for sympatric population splitting of Eurasian perch (Perca fluviatilis L) in Lake Constance. Mol. Ecol 13, 491–497 (2004).
Gardner, A. R. & Willis, K. J. Prehistoric farming and the postglacial expansion of beech and hornbeam: a comment on Küster. The Holocene 9, 119–121 (1999).
Gömöry, D. & Paule, L. Reticulate evolution patterns in western-Eurasian beeches. Bot. Helvetica 120, 63–74 (2010).
Nwosu, E. C. et al. From water into sediment—tracing freshwater cyanobacteria via DNA analyses. Microorganisms 9, 1778 (2021).
Mejbel, H. S., Dodsworth, W. & Pick, F. R. Effects of temperature and oxygen on cyanobacterial DNA preservation in sediments: A comparison study of major taxa. Environ. DNA 4, 717–731 (2022).
Güde, H. et al. Bodensee-Untersuchung-Seeboden. Forschungsprojekt von 2003 bis 2006. IGKB Blaue Reihe 56, 1–108 (2009).
De Barba, M. et al. DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet. Mol. Ecol. Resour. 14, 306–323 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Boyer, F. et al. obitools: a unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 16, 176–182 (2016).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–D596 (2013).
Kanz, C. et al. The EMBL nucleotide sequence database. Nucleic Acids Res 33, D29–D33 (2005).
Zinger, L. et al. metabaR: An r package for the evaluation and improvement of DNA metabarcoding data quality. Methods Ecol. Evol. 12, 586–592 (2021).
Gielly, L. & Taberlet, P. The use of chloroplast DNA to resolve plant phylogenies: noncoding versus rbcL sequences. Mol. Biol. Evol. 11, 769–777 (1994).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Oksanen, J. et al. vegan: Community Ecology Package. (2022).
Legendre, P. & Borcard, D. Box–Cox-chord transformations for community composition data prior to beta diversity analysis. Ecography 41, 1820–1824 (2018).
Ficetola, G. F. et al. DNA from lake sediments reveals long-term ecosystem changes after a biological invasion. Sci. Adv. 4, eaar4292 (2018).
Ficetola, G. F. et al. Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 15, 543–556 (2015).
Baselga, A. Partitioning the turnover and nestedness components of beta diversity. Glob. Ecol. Biogeogr 19, 134–143 (2010).
Legendre, P. Interpreting the replacement and richness difference components of beta diversity. Glob. Ecol. Biogeogr. 23, 1324–1334 (2014).
Kattge, J. et al. TRY plant trait database - enhanced coverage and open access. Glob. Change Biol. 26, 119–188 (2020).
Rimet, F. & Druart, J.-C. A trait database for Phytoplankton of temperate lakes. Ann. Limnol. - Int. J. Limnol. 54, 18 (2018).
Laplace-Treyture, C., Derot, J., Prévost, E., Le Mat, A. & Jamoneau, A. Phytoplankton morpho-functional trait dataset from French water-bodies. Sci. Data 8, 40 (2021).
Schilling, P. Bundestaxaliste der Gewässerorganismen Deutschlands (BTL) - Stand Mai 2020. Herausgegeben im Auftrag der Bund/Länder-Arbeitsgemeinschaft Wasser (LAWA) - Ausschuss Oberirdische Gewässer und Küstengewässer (AO) und des Umweltbundesamtes (UBA). https://www.gewaesser-bewertung.de/ (2020).
InfoFlora. https://www.infoflora.ch/en/.
Shen, W. & Ren, H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J. Genet. Genomics 48, 844–850 (2021).
Clark, P. J. & Evans, F. C. Distance to nearest neighbor as a measure of spatial relationships in populations. Ecology 35, 445–453 (1954).
Baddeley, A. et al. spatstat.explore: Exploratory Data Analysis for the ‘spatstat’ Family. (2023).
Der Bodensee: Zustand, Fakten, Perspektiven. (IGKB, 2004).
Wang, Y. spatial-heterogeneity. https://doi.org/10.5281/zenodo.7817775 (2023).
Internationale Gewässerschutzkommission für den Bodensee (IGKB) (2015). IGKB-Tiefenschärfe-Bodensee digitale Geländemodelle mit 10 m und 3 m Auflösung. PANGAEA https://doi.org/10.1594/PANGAEA.855987 (2015).
Wessels, M. et al. Tiefenschärfe – Hochauflösende Vermessung Bodensee. IGKB Blaue Reihe 61, 1–110 (2016).
Räumliches Informations- und Planungssystem der LUBW. https://www.lubw.baden-wuerttemberg.de/umweltinformationssystem/raeumliches-informations-und-planungssystem-rips.
GIS Mapping Software, Location Intelligence & Spatial Analytics | Esri. https://www.esri.com/en-us/home.
Map tiles by Stamen Design, under CC BY 3.0. Data by OpenStreetMap, under ODbL. http://maps.stamen.com/terrain.
Kahle, D. & Wickham, H. ggmap: Spatial Visualization with ggplot2. R J 5, 144 (2013).
Acknowledgements
This work was supported by the Baden-Württemberg Foundation through the Elite Programme for Postdocs and the Young Scholar Fund of the University of Konstanz, both granted to Laura S. Epp. Yi Wang is funded by the graduate school scholarship programme (ID: 57450037) of the German Academic Exchange Service (DAAD), and by the Research Training Group R3 - Resilience of Lake Ecosystems (GRK 2272, DFG award number 298726046) at the University of Konstanz. We thank the Scientific Compute Cluster at University of Konstanz for providing computation resources. We thank Myriam Schmid, Lisa Gutbrod, Iljas Müller, Kurt Sarembe and Andreas Schießl for assistance in sampling and in the laboratory, as well as Alfred Sulger, Charo López-Blanco and Paula Kankaala for providing copepod specimens.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Yi Wang: conceptualisation, methodology, DNA data acquisition, data curation, formal analysis, visualisation, writing – original draft, writing – review and editing. Martin Wessels: sedimentological data acquisition, visualisation, writing – review and editing. Mikkel W. Pedersen: methodology, writing – review and editing. Laura S. Epp: conceptualisation, methodology, project administration, funding acquisition, writing – original draft, writing – review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Inger Alsos and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Erin Bertrand and Clare Davis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Y., Wessels, M., Pedersen, M.W. et al. Spatial distribution of sedimentary DNA is taxon-specific and linked to local occurrence at intra-lake scale. Commun Earth Environ 4, 172 (2023). https://doi.org/10.1038/s43247-023-00829-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-023-00829-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
