
Abstract
Orogenic degassing is emerging as a potentially relevant source of carbon dioxide (CO2) from the continental crust. However, the processes of carbon mobilization are still poorly explored. Here, we use thermodynamic modeling to investigate the decarbonation of sediments metamorphosed under high geothermal gradients. Our modeling shows that immiscible CO2-rich vapors and hydrosaline brines are generated at these conditions, with different properties and mobility through the crust. The CO2-rich fluid fraction could rapidly rise toward the surface without interacting with the host rocks by carbo-fracturing the host rocks or through deep faults. The denser hydrosaline brines likely permeate the source rocks. When applied to the active Himalayan orogen, these observations reconcile measured CO2 fluxes at the surface and positive conductivity anomalies associated with micro-seismicity at depth. Our modeling shows that the continental crust represents a relevant reservoir of CO2 that can be efficiently degassed during hot collisions.
Similar content being viewed by others

Introduction
Although the global Earth’s natural CO2 degassing is considered irrelevant compared to anthropogenic emissions, much effort is devoted to assessing our planet contribution to the global carbon cycle. Knowing the rate of the Earth’s natural CO2 emissions is, in fact, crucial for quantifying and predicting the influence of anthropogenic perturbations on climate processes1. It is well known that metamorphism of crustal rocks in collisional orogens produces carbon-bearing fluids over geologic time scales, primarily through decarbonation reactions2,3,4,5,6,7. However, the potential contribution of orogenic processes to the global Earth’s carbon cycle is a long-standing open question5,8,9,10,11,12. The efficiency of metamorphic CO2 degassing in orogenic settings is primarily controlled by the ease at which the CO2-bearing aqueous fluids produced at depth are transported upward without interacting with the host rocks. Fluid-rock interactions following prograde metamorphic devolatilization reactions can lead to carbon re-precipitation typically as carbonate13,14,15,16,17,18,19,20,21 or graphite22,23, thus hampering significant CO2 outgassing at the surface. The ability (or inability) of deep CO2 to migrate upward and reach the Earth’s surface opens up two possible scenarios. If retrograde metamorphism represents a sink for CO212, collisional orogens’ contribution to the global Earth’s CO2 degassing would be minimal. Conversely, if metamorphic CO2-bearing fluids do not react with the host rocks, orogenic decarbonation would represent an important source of CO2 at the global scale. Recent research7 balancing estimates of decarbonation rates from deeply exhumed rocks24,25,26 and CO2 fluxes measured at the surface9,27,28, support the second hypothesis. However, neither were the physical and chemical characteristics of the released metamorphic fluids studied in detail nor was the transport path of deep CO2 fully elucidated.
Here, we study decarbonation reactions occurring in carbonate-bearing sediments along high geothermal gradients to define the physical and chemical nature of fluids that transport carbon in large hot orogens. We find that the productivity of CO2-rich fluids is maximum for carbonate-bearing sediments originally containing low to moderate amounts of calcite (10–30 vol%) and metamorphosed at medium- to high-temperature (T) (i.e., T > 600 °C) and at medium pressure (P) (i.e., P > 8 kbar) conditions. We present petrological evidence that most dehydration and decarbonation reactions in these lithologies occur at P–T conditions in the two-phase field of the H2O–CO2–salt ternary fluid systems, generating immiscible CO2-rich vapors and hydrosaline brines. Our results demonstrate a crucial role of fluid immiscibility in driving CO2 transport from the deep crust, explaining how and why significant amounts of CO2 could be effectively degassed at the surface from orogenic belts. This study reconciles geophysical and geochemical observations from active collisional orogens such as the Himalaya, where intense CO2 degassing is currently measured at the surface. Results further highlight the role of hydrosaline brines as metasomatizing and/or granulitizing agents in the lower crust.
Results
Fluid production and evolution in large hot orogens
In large hot orogens (i.e., mountain belts characterized by high crustal temperatures and extreme crustal thickening29,30), CO2 sources are represented mainly by carbonate-bearing sediments ranging from calcareous pelites to marls and impure limestones, important constituents of thick sedimentary sequences deposited along passive margins31. During prograde metamorphism along medium to high dT/dP gradients, these lithologies are transformed into calcic metapelites, calc-silicate rocks, and silicate-bearing marbles and generally show an internally buffered behavior24,31,32,33. Phase equilibria modeling of carbonate-bearing sediments obtained as “synthetic” mixtures of variable proportions of calcite (i.e., 10, 30, 50, 70 vol%, corresponding to initial bulk compositions Cal10, Cal30, Cal50, and Cal70; Table 1) with an average pelite34 allows predicting the fluid evolution and compositions along a dT/dP geothermal gradient typical of large hot orogens (i.e., 750 °C GPa−1, ca. 25 °C km−1 (see ref. 35)). The results of the modeling are illustrated in Figs. 1 and 2.

a, c Mode box changes in calculated mineral proportions (vol%, fluid excluded) during prograde metamorphism of sediments initially containing (a) 10 vol% of calcite (Cal10) and (c) 30 vol% of calcite (Cal30) along the modeled internally buffered P/T–X(CO2) paths. b, d Loss (wt%) of CO2 (black line) and H2O (gray line) relative to the initial CO2 and H2O content in the solids, and X(CO2) of the fluid (blue lines) released during prograde metamorphism of Cal10 (b) and Cal30 (d), along the same internal buffered P/T-X(CO2) path as in (a) and (c). Events I (yellow) and II (red) refer to the main events of CO2 production, as discussed in the text. Dashed lines highlight the main dehydration and decarbonation reactions, corresponding to abrupt consumption and/or complete disappearance of hydrous and CO2-bearing minerals. Reactions corresponding to the event I (R1) and event II (R2a, R2b) are specified.

a, c Mode box changes in calculated mineral proportions (vol%, fluid excluded) during prograde metamorphism of sediments initially containing (a) 50 vol% of calcite (Cal50) and (c) 70 vol% of calcite (Cal70) along the modeled internally buffered P/T–X(CO2) paths. b, d Loss (wt%) of CO2 (black line) and H2O (gray line) relative to the initial CO2 and H2O content in the solids, and X(CO2) of the fluid (blue lines) released during prograde metamorphism of Cal50 (b) and Cal70 (d), along the same internal buffered P/T–X(CO2) path as in (a) and (c). Events I (yellow) and II (red) refer to the main events of CO2 production, as discussed in the text. Dashed lines highlight the main dehydration and decarbonation reactions, corresponding to abrupt consumption and/or complete disappearance of hydrous and CO2-bearing minerals. Reactions corresponding to the event I (R1) and event II (R2b) are specified.
In carbonate-bearing metasediments, volatiles (H2O and CO2) are primarily hosted in phyllosilicates and carbonates; scapolite can represent an additional, often neglected, CO2 reservoir24,25,31,36. Prograde metamorphism generates C–O–H fluids dominated by H2O and CO2 components, the amounts of the other molecular species (CO, CH4, and H2) being negligible (i.e., <0.005 mol%) for all the investigated bulk compositions (Supplementary Data Files S1–S4). The aqueous component of the released C–O–H fluids is generated chiefly by the breakdown of chlorite, muscovite, biotite and/or epidote, and the CO2 component by calcite and/or scapolite consumption. The interplay between formation and consumption of these phases controls the relative amounts of H2O and CO2 that can be released at increasing P–T conditions (i.e., if biotite forms during the muscovite breakdown, part of the H2O is transferred from muscovite to biotite and, similarly, if scapolite formation coincides with calcite consumption, part of the CO2 produced through calcite breakdown is fixed in scapolite). The results of the modeling show that, independently from the original amount of calcite in the sedimentary protolith, the production of C–O–H fluids in internally buffered carbonate-bearing metasediments mainly occurs in pulses (Figs. 1b, d and 2b, d), i.e., through nearly discontinuous reactions operating in narrow P–T intervals (see also refs. 24,25,31).
Two main events of CO2 production are predicted at 575–605 °C (event I) and 630–670 °C (event II), respectively. For any of the modeled initial bulk compositions, event I corresponds to the muscovite-out reaction R1: Cal + Qz + Mu + Pl = Kfs + Scp + F, nearly coinciding with the total consumption of calcite for Cal10 bulk composition. Event II corresponds to the scapolite-out reaction R2a: Bt + Qz + Scp = Kfs + Cpx + Grt + Pl + F for Cal10 bulk composition; for Cal30, Cal50 and Cal70 bulk compositions, event II coincides with the biotite-out reaction R2b: Cal + Bt + Qz + Scp = Kfs + Cpx + Pl + F (Figs. 1 and 2). At each event, the fluid composition (X(CO2) = molar fraction of CO2) varies depending on the initial bulk composition (Figs. 1 and 2). The X(CO2) generally increases at increasing temperatures (i.e., from event I to event II), except for Cal10 where the maximum X(CO2) is reached at event I (Fig. 1b). The X(CO2) of the fluids released at event II is generally higher than 0.5, with maximum values of X(CO2) = 0.64–0.74 (Cal30; Fig. 1d), whereas it is more variable for fluids released at event I, ranging from X(CO2) = 0.62–0.65 (Cal10; Fig. 1b) to X(CO2) = 0.30–0.33 (Cal70; Fig. 2d). The amount of CO2 (i.e., wt% loss of CO2 relative to the initial CO2 content in the solids) produced at each event increases up-temperature, except for Cal10, for which event I releases slightly more CO2 than event II (Fig. 1b). The total CO2 productivity increases from Cal10 (4.7 wt%) to Cal30 (7.0 wt%) and then decreases for Cal50 (3.7 wt%) and Cal70 (1.9 wt%). The same trend is observed when considering the CO2 productivity related to event II only, maximum for Cal30 (3.6 wt%; Fig. 1d) and minimum for Cal70 (0.5 wt%: Fig. 2d).
Our results show that CO2 is an essential component of C–O–H fluids released at relatively high temperatures (630–670 °C) and medium pressures (8.4–8.9 kbar or 25–30 km depth) during the formation of large hot orogens. The CO2 productivity is maximum for the carbonate-bearing sediments originally containing low to moderate modal amounts of calcite (i.e., Cal10 and Cal30; Fig. 1) (see also refs. 24,25,31), whereas it is considerably lower in impure limestones (Cal70)37. Also, the model predictions are in excellent agreement with those discussed in recent papers focused on thermodynamic modeling of natural rock samples24,25,31,38 and with fluid inclusion studies providing direct measurements of the fluid composition in mixed pelitic–carbonatic metamorphic sequences from collisional orogens39,40,41. As a step forward, determining the physical and chemical properties of the fluids generated at these P–T conditions would be essential for understanding their behavior and, ultimately, the connection between the deep metamorphic production of CO2 in the crust and its release at the surface.
Fluid immiscibility in the H2O–CO2–salt systems
Traditionally, metamorphic fluids have been considered as binary CO2–H2O mixtures in most thermodynamic modeling studies42,43,44,45. However, fluids derived from metasedimentary rocks originally deposited at continental passive margins such as those involved in large hot orogens are generally characterized by variable, relatively high salinity (i.e., from 5 to 50–60 wt% NaCl eq.) even at high metamorphic grades, reflecting the presence of highly saline pore water and/or evaporites in the initial sedimentary sequence37,46,47,48,49. Thick layers of evaporites are well known to occur along present-day passive margins, where they control the formation and storage of oil and gas50,51, but they are also common in ancient sedimentary sequences since the Proterozoic ages52,53. Direct evidence for saline fluids in metamorphosed passive margin sequences is represented by rare halide minerals, fluid inclusions, and/or high Cl contents of key high-grade metamorphic minerals54,55,56,57. Evidence for salinity dilution at high temperatures due to progressive dehydration is feeble, suggesting that even advanced metamorphic dehydration cannot flush away the salinity inherited from the protoliths46,47,52.
In this framework, salts components are particularly relevant because they strongly influence the topology of fluid phase equilibria, expanding the two-phase immiscibility fields37,46,47,58,59,60,61. Fluid(s) produced by decarbonation reactions of calcite-bearing sedimentary protoliths can be reasonably described by the H2O–CO2–NaCl system40,57,62,63,64 and the H2O–CO2–CaCl2 system41,65. These ternary systems have been studied experimentally from 1 to 9 kbar and from 400 to 1000 °C64,66,67,68,69,70, i.e., at P–T conditions compatible with those attained in large hot orogens. In order to describe phase relations of fluids produced during the main events of CO2 production (i.e., event I: 575–605 °C, 7.6–8.0 kbar; event II: 630–670 °C, 8.3–9.0 kbar), we have interpolated the H2O–CO2–salts ternary diagrams obtained experimentally by ref. 69 at 800 °C, 9 kbar and 500 °C, 5 kbar. The phase diagrams resulting from these interpolations (Figs. 3 and 4) are valid at ca. 575 °C, 6 kbar (i.e., for event I) and at ca. 650 °C, 7 kbar (i.e., for event II).

Phase equilibria in the H2O–CO2–NaCl system at ca. 575 °C, 5 kbar and at ca. 650 °C, 7 kbar (interpolated from experimental data by ref. 69), showing phase relations for fluids generated by the modeled sediments (a, b Cal10; c, d Cal30; e, f Cal50) at the event I (a, c, e) and event II (b, d, f), in the hypothesis of salinity of 10 wt% NaCl (corresponding to NaCl ≈ 6–7 mol%). Dashed lines outline the V–L tie-lines within the two-phase fields. Depending on the bulk composition, fluids released during events I and II plot either within the solvus (b, c, e, f) or within the V + NaCl two-phase field (a, d). In the first case, two immiscible fluids coexist: a CO2-rich vapor and a hydrosaline brine (boxes on the solvus), whereas in the second case, a CO2-rich vapor coexists with halite. F one-phase fluid, V vapor, L liquid. Phase relations for Cal70 are reported in Supplementary Fig. S1a, b.

Phase equilibria in the H2O–CO2–CaCl2 system at ca. 575 °C, 5 kbar and at ca. 650 °C, 7 kbar (interpolated from experimental data by ref. 69), showing phase relations for fluids generated by the modeled sediments (a, b Cal10; c, d Cal30; e, f Cal50) at the event I (a, c, e) and event II (b, d, f), in the hypothesis of salinity of 10 wt% CaCl2 (corresponding to CaCl2 ≈ 3–4 mol%). Dashed lines outline the V–L tie-lines within the two-phase fields. Fluids released during events I and II systematically plot within the solvus. Two immiscible fluids thus coexist at every P–T conditions: a CO2-rich vapor and a hydrosaline brine (boxes on the solvus). F one-phase fluid, V vapor, L liquid. Phase relations for Cal70 are reported in Supplementary Fig. S1c, d.
In the H2O–CO2–NaCl system, the evolution of the fluids produced during the most CO2-productive event II proceeds as a function of the initial bulk-rock composition and the initial salinity. Fluids generated during event II have an X(CO2) systematically higher than 0.4 (Cal10: X(CO2) = 0.57–0.64; Cal30: X(CO2) = 0.64–0.74; Cal50: X(CO2) = 0.61–0.68; Cal70: X(CO2) = 0.45–0.51; Figs. 1b, d and 2b, d). In the hypothesis of a bulk salinity of 10 wt% NaCl, these fluids plot in the miscibility gap (Fig. 3b, d, f and Supplementary Fig. S1b), either in the vapor + liquid (V + L) two-phase field (Cal10, Cal50, and Cal70), or in the vapor + salt (V + NaCl) two-phase field (Cal30). In the first case, once liberated, fluids would inevitably split into a CO2-rich phase of a lesser density (i.e., vapor62) (Cal10: CO2 = 60–66 mol%; Cal50: CO2 = 62–66 mol%; Cal70: CO2 = 49–53 mol%; Table 2) and a highly saline aqueous phase (i.e., liquid62) (Cal10: NaCl = 38–42 mol%; Cal50: NaCl = 40–42 mol%; Cal70: NaCl = 23–30 mol %; Table 2), with the CO2-rich vapor being the dominant fluid type (i.e. >85 mol%; Fig. 3b, f and Supplementary Fig. S1b). For Cal10 and Cal50 bulk compositions, the segregation of low amounts of halite from the two immiscible fluids (i.e., three-phase field V + L + NaCl; Fig. 3b, f) is predicted for the highest X(CO2) values of the produced fluid. In the second case, a single vapor-like CO2-rich fluid (Cal30: CO2 = 66–73 mol%; Table 2) would coexist with solid halite. The immiscible nature of the fluids produced at the P–T conditions of event II is further amplified for higher initial fluid salinity, resulting in a more enriched CO2 vapor and a hydrosaline brine with higher salinity; the fraction of the brine increases with initial salinity.
A similar evolution is predicted for the fluid produced at event I in the H2O-CO2-NaCl system (Fig. 3a, c, e and Supplementary Fig. S1a). At T = 595–605 °C and P = 7–8.0 kbar, Cal10 bulk composition generates a fluid whose CO2-rich composition (X(CO2) = 0.62–0.65; Fig. 1b) plots within the V + NaCl two-phase field or in the nearby V + L + NaCl three-phase field. At these P–T conditions, a CO2-rich vapor (CO2 = 64–66 mol%; Table 2) coexists with halite, ± a very subordinate fraction of a hydrosaline brine (Fig. 3a). On the contrary, fluids produced by Cal30, Cal50, and Cal70 bulk compositions have lower X(CO2) values (Cal30: X(CO2) = 0.49–0.51; Cal50: X(CO2) = 0.37–0.41; Cal70: X(CO2) = 0.30–0.33; Figs. 1d and 2b, d) and thus plot within the V + L two-phase field (Fig. 3c, e and Supplementary Fig. S1a), occasionally close to the boundary of the solvus (Cal50 and Cal70). Two immiscible fluids thus coexist also at these P–T conditions. The composition of conjugate fluids change as a function of the bulk-rock composition: specifically, the amount of CO2 in the CO2-rich vapor progressively decreases from Cal30 (CO2 = 53–57 mol%) to Cal50 (CO2 = 39–42 mol%) and Cal70 (CO2 = 32–38 mol%) and the salinity of the brine decreases from Cal30 (NaCl = 24–27 mol%) to Cal50 (NaCl = 13–15 mol%) and Cal70 (NaCl = 11–12 mol%) (Table 2).
The topology of the ternary diagrams in the H2O–CO2–CaCl2 system shows an even more extensive miscibility gap69 at any P–T conditions (Fig. 4, Supplementary Fig. S1, and Table 3). Thus, fluids produced during events I and II constantly plot within the solvus, independent of the initial bulk-rock composition and even for extremely low C–O–H fluid salinities. Immiscible CO2-rich vapor and a hydrosaline CaCl2-rich brine are predicted for both events.
Therefore, carbonate-bearing sediments undergoing prograde metamorphic decarbonation reactions at relatively high temperatures and medium pressures release a conjugate fluid pair “born this way” (immiscible at the source; i.e., at a crustal depth of ∼25–30 km), even for relatively low-salinity C–O–H initial fluid compositions (i.e., ≥5 wt% NaCl eq). In most cases, the CO2-rich vapor is the dominant phase, coexisting with subordinate hydrosaline brines. Also, in the H2O–CO2–NaCl system, the coexistence of a vapor-like phase strongly enriched in CO2 coexisting with halite is predicted. The immiscible CO2-rich vapors produced during the most productive event II generally consist of more than 60 mol% CO2 (up to 73 and 76 mol% in the NaCl and CaCl2 systems, respectively) and are moderately saline, while hydrosaline brines generally contain > 60 wt% dissolved salt (i.e., close to saline melts; up to 69 wt% NaCl and 85 wt% CaCl2).
Discussion
Implications for the CO2 transport from the deep metamorphic source to the surface
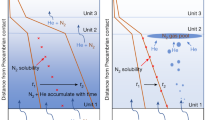
Metamorphic C–O–H–salt fluids involved in dehydration and decarbonation reactions are generally modeled as a single phase. Fluid immiscibility has seldom been considered, although predicted by phase equilibria and documented by fluid inclusions, over different metamorphic environments and P–T–X conditions58,71,72,73,74. The present results illustrate how decarbonation occurs within the solvus, generating immiscible CO2-rich vapors and hydrosaline brines in the deep crust of large hot orogens, at peak metamorphic conditions. Immiscibility favors fluid segregation allowing transport of carbon in the crust. Because of the density and viscosity contrast between coexisting fluids, significantly less dense CO2-rich fluids (∼1.06–1.10 g cm−3 75,76) effectively separate from denser hydrosaline brines (∼1.80 g cm−3), where most solutes concentrate, and migrate upwards. The contrasting wetting behavior and reactivity of CO2 and brines further influence fluid migration from the deep crust. In silicate-dominated rocks, CO2-rich fluids have considerably higher dihedral angles (θ >65°, up to 90°) compared to aqueous fluids, in which lower values are favored by the presence of alkali or alkaline-earth halides (θ < 40 °C)77,78. In carbonate-dominated rocks, the behavior of CO2-rich fluids is less obvious; low solid–fluid dihedral angles (θ < 60°), and therefore wetting properties, are observed for H2O–CO2 fluids with intermediate X(CO2) compositions (0.2 < X(CO2) < 0.6), whereas H2O-rich fluids (X(CO2) < 0.2) and CO2-rich fluids with X(CO2) > 0.6 have high dihedral angles (θ > 65°, up to 90°) and therefore they are non-wetting fluids79. Therefore, CO2-rich fluids generated during events I and II mostly have a non-wetting behavior (except in carbonate-dominated lithologies, i.e., Cal70, whose CO2 productivity is so low that they will not be further considered in the discussion). Thus, it is conceivable that the less dense CO2-rich fluids are highly buoyant and unreactive, whereas the volumetrically minor and denser hydrosaline brines are highly reactive and could migrate by porous flow through thin interconnected films along grain boundaries77 (Fig. 5). Our results also suggest that a solid salt instead of a brine could be segregated from a single CO2-rich vapor-like phase for specific rock and fluid compositions.

Two immiscible fluids are generated in the source rocks: the CO2-rich fluid is shown in yellow-red, with colors changing from yellow to red according to the progressive increase of the fluid pressure within the pores, whereas the hydrosaline brine is reported in blue. The short to long black arrows radiating from the CO2-filled pores indicate the progressive increase of the fluid pressure in the pores. The dihedral angles of the two fluids (CO2-rich fluid: θ > 60°; hydrosaline brine: θ < 60°) imply that the CO2-rich fluid accumulates within isolated fluid pockets, whereas the hydrosaline brine forms thin interconnected films along grain edges. As far as the decarbonation reaction proceeds, fluid pressure within the CO2-rich pockets increases until fluid overpressure is reached, carbo-fracturing the host rocks. The CO2-rich fluid thus rapidly escapes toward the surface along the newly created fracture network, whereas the denser, hydrosaline brine remains trapped in the source rock.
Thus, in a two-fluid flow regime, CO2 and brines migrate separately. CO2-rich fluids generated at depths of 25–30 km (8–10 kbar) would accumulate, eventually forming large reservoirs. With decarbonation reactions proceeding, confined CO2-rich fluids would cause fluid overpressure, inducing carbo-fracturing of the host rocks and migrating upward. Such processes could trigger crustal permeability, possibly resulting in earthquake nucleation and seismicity28,80,81,82,83,84,85). An alternative scenario of a passive ascent of CO2-rich fluids, favored by brittle fracturing of the crust during earthquakes or by ductile deformation, could be equally possible to enhance fluid mobility through channelization along preferential pathways. In both cases, fracturing and faulting would allow fast CO2-rich fluids migration toward the surface28,81,82,85,86,87,88. As CO2-rich fluids are removed from the reacting sites, the remaining hypersaline brines could represent important metasomatic agents in the lower crust48,58,89. Moreover, their low water activity could promote granulitization of the deep crust, delaying its partial melting.
Fluid-rock interaction during ascent is unlikely because the relatively high transport velocities and the nonpolar nature of CO2-rich fluids do not allow chemical equilibration with most metamorphic rock compositions. To our knowledge, pervasive carbonation by fluid-rock interactions at P–T conditions compatible with fluid ascent in large hot orogenic settings is limited to soapstones, listvenites and sagvandites (i.e., talc + magnesite, quartz + magnesite and enstatite + magnesite rocks derived from ultramafic lithologies by reaction with a CO2-rich fluid90,91,92,93,94,95,96,97). However, ultramafic lithologies are rare in large hot orogens and are regarded as exceptions rather than rules. Also, precipitation of epigenetic graphite should be negligible due to the low water activity in CO2-rich fluids coupled with the high temperatures at which decarbonation reactions occur98.
Fluid immiscibility supports geochemical and geophysical observations from active large hot orogens
Segregation of CO2-rich fluids by phase separation at birth (i.e., directly in the deep crustal source) reconciles geochemical and geophysical evidence from active collisional orogens, e.g., the Himalaya, considered as the archetype of large hot orogens and characterized by present-day intense CO2 surface degassing.
First, immiscibility, with the generation of CO2-dominated fluids (i.e., X(H2O) = 26–38 mol%), could explain the gaseous CO2 emissions measured at the surface. Diffuse degassing occurs over extensive areas in Himalaya, testified by the widespread occurrence of CO2-rich hot springs and gaseous CO2 ground discharges along the main tectonic discontinuities and distributed along the entire length of the orogenic belt9,27,28,85,99,100. Geochemical and isotopic analyses revealed that CO2 released at the surface has a crustal metamorphic signature, produced at >5 km depth9,27,28,85,100. Girault & Perrier84 and Girault et al.88 suggested that CO2 is not dissolved in aqueous fluids but, instead, rapidly outgas along “dry” faults toward the surface. Estimated transport velocities are in the order of 0.1 to 1 m sec−1 to account for the observed radon and CO2 fluxes.
Second, beneath the Himalayan metamorphic core, a major conductive zone is revealed at a depth of ∼20–30 km101, immediately below a zone of intense microseismicity102,103,104. This highly conductive and seismically active zone is located along a mid-crustal ramp below the superficial emergence of the Main Central Thrust (Fig. 6), i.e., the main tectonic discontinuity along which most of the CO2-rich hot springs are concentrated. Lemonnier et al.101 first interpreted the conductivity anomaly as related to highly connected aqueous fluids released by metamorphic dehydration reactions, percolating upward into the brittle portion of the crust, where microseismic activity is observed. In aqueous fluids, conductivity is low but readily increases with salinity105. Thus, this deep conductive zone could result from the stagnation of hydrosaline brines48 left behind by the CO2-rich fluids once they rise toward the surface. Although volumetrically minor, hydrosaline brines have an extremely wetting behavior106. It is, therefore, likely that they are highly connected in the source rocks (Fig. 5). Like Lemonnier et al.101 original interpretation, the zone of intense microseismicity immediately above the conductive anomaly could be related to periodic carbo-fracturing events in the brittle crust induced by the accumulation of CO2-rich fluids at the lower crust–upper crust boundary. Finally, given the striking similarities between actual CO2 fluxes measured at the surface and those predicted by modeling of prograde metamorphism of carbonate-bearing lithologies in Himalaya at depths of 25–30 km (i.e., at 650–750 °C, 8–10 kbar24,25,31,38), it seems likely that CO2-producing processes and transport mechanisms similar to those occurring in Himalaya at present were also active in the past.

The thick red line shows the Main Himalayan Thrust, MHT, which reaches the surface at the front of the Siwalik Hills, coinciding with the Main Frontal Thrust (MFT). The Main Boundary Thrust (MBT) separates the metasediments of the Lesser Himalayan Sequence (LHS) from the molasse deposits of the sub-Himalaya (the Siwaliks Hills). The Main Central Thrust (MCT) places the higher-grade metamorphic rocks of the Greater Himalayan Sequence (GHS) over the LHS metasediments. The GHS is divided into two tectono-metamorphic units (Lower-GHS and Upper-GHS) by the High Himalayan Discontinuity (HHD); geologic cross-section modified from refs. 104,122. The red to yellow area highlights the zone of high conductivity located at depths >15 km beneath the superficial emergence of the MCT, as obtained from the magnetotelluric experiment of ref. 101. The conductivity anomaly is associated with intense microseismic activity, evidenced by the schematic distribution of earthquake hypocentres (small circles). The white diamond indicates the CO2-rich hot springs and gaseous CO2 discharges from the ground, located along the MCT.
In conclusion, we demonstrate that the most productive CO2-source rocks in large hot orogens are calcareous pelites and carbonate-poor marls (i.e., Cal10 and Cal30), which generate immiscible CO2-rich fluids and hydrosaline brines when metamorphosed at T > 590 °C. We suggest that fluid immiscibility provides a physical mechanism to transport carbon liberated during prograde metamorphism. Segregation due to density contrast and different chemical properties between CO2 and brines allows the rapid migration of CO2 from the deep source to the surface. Such a model could explain the CO2 fluxes currently measured at the surface in active collisional orogenic settings such as the Himalaya, which are impossible to explain in the absence of fluid phase separation. Finally, our study contributes to solving the debate on the role of orogenic settings for the global Earth’s CO2 tectonic outgassing. An array of independent observations consistently indicates that active collisional orogens could represent an important (and so far under-considered) source of CO2 on a global scale. Since the Himalayan orogen is a modern analog of ancient large hot orogens, such as the Mesoproterozoic Grenville and Sveconorwegian orogens and the Late Palaeozoic Variscan orogen, we advocate that the same process currently active in the Himalaya should have been repeatedly active at different times, during the main orogenic events, possibly causing periodic perturbations of the atmospheric CO2 levels.
Methods
Model bulk-rock compositions
Model bulk-rock compositions used to calculate the P/T–X(CO2) buffering paths were obtained by adding 10, 30, 50, and 70 vol% of calcite to the average pelite composition of ref. 34 (“shale and slate” group; his Table 2) (Table 1). The modal proportions of the protolith’s minerals have been obtained by applying the least square method (freeware application available on demand107) and using end-member compositions and molar volumes for kaolinite, illite, clinochlore, daphnite, albite, anorthite, quartz, and K-feldspar. The result is considered satisfactory if the residuals (i.e., molar bulk composition of the protolith’s minerals—molar bulk-rock composition) is close to zero. The following assumptions were additionally made: (a) CaO from silicate fraction is equivalent to Na2O108,109 and is incorporated in anorthite; the remaining CaO is incorporated in calcite; (b) chlorite is the only Fe-Mg mineral in the protolith and (c) albite incorporates all the Na2O.
Thermodynamic modeling
Thermodynamic modeling was performed using PerpleX 6.9.0110,111 (version June 2020), the internally consistent thermodynamic dataset (version ds55), and the equation of state for H2O-CO2 binary fluid of ref. 112. P/T–X(CO2) buffering paths were calculated along a P/T geothermal gradient representative of large hot orogens (dT/dP = 750 °C GPa−1 (see ref. 35)). The following solution models were considered: carbonate (i.e., Ca–Mg–Mn–Fe carbonate with calcite structure113), dolomite112, scapolite114, chlorite115, white mica116,117, biotite118, plagioclase119, K-feldspar120, Ca-amphibole (ideal tremolite solution model), clinopyroxene112, garnet112, epidote112, chloritoid112, and staurolite112. A generic hybrid molecular fluid-equation-of-state solution model was used for fluid (COH-Fluid model available in PerpleX), including H2O, CO2, CO, CH4, and H2 molecular species. This model allows considering redox processes, such as the possible reduction of carbon to graphite due to the exchange of oxygen between the C–O–H fluid and the Fe3+-bearing solid phases (white mica, biotite, clinopyroxene, garnet, epidote).
Calculation of P/T–X(CO2) buffering paths requires fixing a P–T–X(CO2) starting condition; this was fixed at 350 °C, 4 kbar, X(CO2) = 0.005, such as the predicted mineral modes are as close as possible to the calculated protolith’s mineral modes (with muscovite replacing illite). The Werami routine of Perple_X was used to calculate the amount of H2O, C, and O2 stored in solid phases at the starting P–T–X(CO2) fluid-saturated conditions (Cal10_0, Cal30_0, Cal50_0, and Cal70_0 compositions in Supplementary Tables S1–4). To start calculating the buffering paths, 1 mol% of the equilibrium fluid with X(CO2) = 0.005 was added to each model bulk-rock composition (Cal10_1, Cal30_1, Cal50_1, Cal70_1 compositions in Supplementary Tables S1–4). The addition of a small amount of fluid is required because, at fluid saturation conditions, the Werami routine returns the amounts of H2O, C, and O2 stored in minerals, but not the amount of free fluid in equilibrium with solid phases, which in turn depends on the porosity of the system (see ref. 121 for further details). P/T–X(CO2) buffering paths were calculated following the constant porosity model of ref. 121. According to this model, the fluid is allowed to accumulate within the rock until it reaches a specified molar proportion threshold; each time this threshold is exceeded, fluid loss occurs through a stepwise process. Starting from the initial fluid mole proportion of 0.01 (1 mol%, relative to the solids + fluid assemblage), we have fixed the threshold at 0.02; this implies that when the fluid proportion reaches this value, its amount is reduced to 0.01. Bulk compositions after each fractionation step are reported in Supplementary Tables S1–4. Supplementary Tables S1–4 also include the modeled amounts of fluid (vol%, relative to the solids + fluid assemblage) immediately before and immediately after each fractionation step, and show that fractionation of fluid allows maintaining a porosity <2 vol%. The two extreme cases of an internally buffered, completely closed system (i.e., no fluid loss is allowed) and of an internally buffered completely open system (i.e., all the fluid that is produced is immediately lost) were further considered, which provide the minimum and maximum X(CO2) values, respectively, for the same starting conditions.
Figures 1 and 2 show the variations in mineral proportions (vol%, fluid excluded), the loss (wt%) of CO2 and H2O relative to the initial CO2 and H2O content in the solids, and the composition (X(CO2) = molar fraction of CO2) of the fluid released along the calculated buffering paths. The complete speciation of the fluid along the modeled buffering paths is reported in Supplementary Data Files S1–4.
Estimate of the amounts of CO2 and H2O released along the modeled prograde P/ T–X (CO2) buffering paths
The Werami routine of Perple_X was used to infer: (i) the abundances (wt%) of both C-bearing minerals (carbonates and scapolite) and hydrous minerals (chlorite, muscovite, biotite, epidote) along the calculated P/T–X(CO2) buffering paths, (ii) the amounts (wt%) of C, O2, and H2O hosted in these minerals at each P–T conditions. Although scapolite can incorporate significant amounts of Cl in compositions close to the marialite end-member, it is to be noted that in the available scapolite solution model114 marialite is treated as a hypothetical CO3 end-member. This would potentially result in an overestimation of the CO2 content in scapolite. However, this should not significantly influence the final estimate of CO2 production because available data on natural scapolite from large hot orogenic contexts suggest that most scapolite contain negligible (if not null) amounts of Cl24,25. The amounts (wt%) of CO2 and H2O released during prograde metamorphism from each model bulk composition were then calculated by adding carbonates’ contribution with that of scapolite and chlorite’s contribution with that of muscovite, biotite, and epidote, respectively. Supplementary Data Files S1–4 summarize the variations in mineral proportions (vol% and wt%) and the amounts (wt%) of C, O2, and H2O released along the calculated buffering paths and used for Figs. 1 and 2.
Data availability
The authors declare that all data supporting the findings of this study are available within the article (Tables 1–3) and its Supplementary Information files (Supplementary Tables S1–S4 and Supplementary Data Files S1–S4). The thermodynamic database and solution models used for thermodynamic modeling are available on the Perple_X website (https://www.perplex.ethz.ch/).
Code availability
No computer code was written in preparing the paper. The Perple_X code used for the thermodynamic modeling (Perple_X 6.9.0, version June 2020) is available on the Perple_X website (https://www.perplex.ethz.ch/).
References
Flesia, C. & Frezzotti, M. L. The dilemma of the dwarf Earth’s CO2 degassing: irrelevant or crucial? J. Geochem. Explor. 152, 118–122 (2015).
Bowen, N. L. Progressive metamorphism of siliceous limestone and dolomite. J. Geol. 48, 225–274 (1940).
Ferry, J. M. Regional metamorphism of the Waits River Formation, eastern Vermont: delineation of a new type of giant metamorphic hydrothermal system. J. Petrol. 33, 45–94 (1992).
Bickle, M. J. Metamorphic decarbonation, silicate weathering and the long-term carbon cycle. Terra Nova 8, 270–276 (1996).
Kerrick, D. M. & Caldeira, K. Metamorphic CO2 degassing from orogenic belts. Chem. Geol. 145, 213–232 (1998).
Ague, J. J. Release of CO2 from carbonate rocks during regional metamorphism of lithologically heterogeneous crust. Geology 28, 1123–1126 (2000).
Stewart, E. M. et al. Carbonation and decarbonation reactions: implications for planetary habitability. Am. Mineral. 104, 1369–1380 (2019).
Kerrick, D. M. & Caldeira, K. Was the Himalayan orogen a climatically significant coupled source and sink for atmospheric CO2 during the Cenozoic? Earth Planet. Sci. Lett. 173, 195–203 (1999).
Becker, J. A., Bickle, M. J., Galy, A. & Holland, T. J. B. Himalayan metamorphic CO2 fluxes: quantitative constraints from hydrothermal springs. Earth Planet. Sci. Lett. 265, 616–629 (2008).
Gaillardet, J. & Galy, A. Himalaya-carbon sink or source? Science 320, 1727–1728 (2008).
Evans, K. A. Metamorphic carbon fluxes: how much and how fast? Geology 39, 95–96 (2011).
Hazen, R. M. & Schiffries, C. M. Why deep carbon? Rev. Mineral. Geochem. 75, 4–6 (2013).
Skelton, A. D. L., Bickle, M. J. & Graham, C. M. Fluid-flux and reaction rate from advective-diffusive carbonation of mafic sill margins in the Dalradian, southwest Scottish Highlands. Earth Planet. Sci. Lett. 146, 527–539 (1997).
Skelton, A. D. L. Flux rates for water and carbon during greenschist facies metamorphism. Geology 39, 43–46 (2011).
Kleine, B. I., Pitcairn, I. K. & Skelton, A. D. The mechanism of infiltration of metamorphic fluids recorded by hydration and carbonation of epidote–amphibolite facies metabasaltic sills in the S.W. Scottish Highlands. Am. Mineral. 100, 2702–2717 (2015).
Kleine, B. I., Pitcairn, I. K. & Skelton, A. D. Mineralogical controls on metamorphic fluid flow in metabasaltic sills from Islay, Scotland. Lithos 248, 22–39 (2016).
Kleine, B. I., Zhao, Z. & Skelton, A. Rapid fluid flow along fractures at greenschist facies conditions on Syros, Greece. Am. J. Sci. 316, 169–201 (2016).
Piccoli, F. et al. Carbonation by fluid–rock interactions at high-pressure conditions: implications for carbon cycling in subduction zones. Earth Planet. Sci. Lett. 445, 146–159 (2016).
Menzel, M. D. et al. Carbonation of mantle peridotite by CO2-rich fluids: the formation of listvenites in the Advocate ophiolite complex (Newfoundland, Canada). Lithos 323, 238–261 (2018).
Peng, W. et al. Multistage CO2 sequestration in the subduction zone: Insights from exhumed carbonated serpentinites, SW Tianshan UHP belt, China. Geochim. Cosmochim. Acta 270, 218–243 (2020).
Hu, H. et al. Retrograde carbon sequestration in orogenic complexes: a case study from the Chinese southwestern Tianshan. Lithos 392-393, 106151 (2021).
Groppo, C., Rolfo, F., Castelli, D. & Connolly, J. A. D. Metamorphic CO2 production from calc-silicate rocks via garnet-forming reactions in the CFAS–H2O–CO2 system. Contrib. Mineral. Petrol. 166, 1655–1675 (2013).
Craw, D. & Upton, P. Graphite reaction weakening of fault rocks, and uplift of the Annapurna Himal, central Nepal. Geosphere 10, 720–731 (2014).
Groppo, C., Rolfo, F., Castelli, D. & Mosca, P. Metamorphic CO2 production in collisional orogens: petrologic constraints from phase diagram modeling of Himalayan, scapolite-bearing, calc-silicate rocks in the NKC(F)MAS(T)-HC system. J. Petrol. 58, 53–83 (2017).
Rapa, G. et al. Titanite-bearing calc-silicate rocks constrain timing, duration and magnitude of metamorphic CO2 degassing in the Himalayan belt. Lithos 292–293, 364–378 (2017).
Stewart, E. M. & Ague, J. J. Infiltration-driven metamorphism, New England, USA: regional CO2 fluxes and implications for Devonian climate and extinctions. Earth Planet. Sci. Lett. 489, 123–134 (2018).
Perrier, F. et al. A direct evidence for high carbon dioxide and radon-222 discharge in Central Nepal. Earth Planet. Sci. Lett. 278, 198–207 (2009).
Girault, F. et al. The Syabru-Bensi hydrothermal system in central Nepal: 1. characterization of carbon dioxide and radon fluxes. Geophys. Res, Solid Earth 119, 4017–4055 (2014).
Beaumont, C., Nguyen, M. H., Jamieson, R. A. & Ellis, S. Crustal flow modes in large hot orogens. In Channel Flows, Ductile Extrusion and Exhumation in Continental Collision Zones (eds Law, R. D., Searle, M. P. & Godin, L.) Vol. 268, 91–145 (Geological Society Special Publication, 2006).
Beaumont, C., Jamieson, R. & Nguyen, M. Models of large, hot orogens containing a collage of reworked and accreted terranes. Can. J. Earth Sci. 47, 485–515 (2010).
Groppo, C., Rapa, G., Frezzotti, M. L. & Rolfo, F. The fate of calcareous pelites in collisional orogens. J. Metam. Geol. 39, 181–207 (2021).
Greenwood, H. J. Buffering of pore fluids by metamorphic reactions. Am. J. Sci. 275, 573–593 (1975).
Baker, J., Holland, T. J. B. & Powell, R. Isograds in internally buffered systems without solid solutions: principles and examples. Contrib. Mineral. Petrol. 106, 170–182 (1991).
Ague, J. J. Evidence for major mass transfer and volume change during regional metamorphism of pelites. Geology 19, 855–858 (1991).
Brown, M. Duality of thermal regimes is the distinctive characteristic of plate tectonics since the Neoarchean. Geology 34, 961–964 (2016).
Morissey, L. J. & Tomkins, A. G. Evaporite-bearing orogenic belts produce ligand-rich and diverse metamorphic fluids. Geochim. Cosmochim. Acta 275, 163–187 (2020).
Yardley, B. W. D. The Evolution of Fluids through the metamorphic cycle. In Fluid Flow and Transport in Rocks (eds Jamtveit, B. & Yardley, B. W. D.) 99–121 (Springer, 1997).
Rolfo, F., Groppo, C. & Mosca, P. Metamorphic CO2 production in calc-silicate rocks from the eastern Himalaya. It. J. Geosci. 136, 28–38 (2017).
Pêcher, A. Les inclusions fluides des quartz d’exsudation de la zone du MCT himalayen au Népal central: données sur la phase fluide dans une grande zone de cisaillement crustal. Bull. Minéral. 102, 537–554 (1979).
Sisson, V. B. & Hollister, L. S. A fluid-inclusion study of metamorphosed pelitic and carbonate rocks, south central Maine. Am. Mineral. 75, 59–70 (1990).
Sisson, V. B., Crawford, M. L. & Thompson, P. H. CO2-brine immiscibility at high temperatures: evidence from calcareous metasedimentary rocks. Contrib. Mineral. Petrol. 78, 371–378 (1981).
Carmichael, D. M. Univariant mixed-volatile reactions: pressure–temperature phase diagrams and reaction isograds. Can. Mineral. 29, 741–754 (1991).
Connolly, J. A. D. & Trommsdorff, V. Petrogenetic grids for metacarbonate rocks: pressure-temperature phase-diagram projection for mixed-volatile systems. Contrib. Mineral. Petrol. 108, 93–105 (1991).
Yardley, B. W. D. & Shmulovich, K. I. An introduction to crustal fluids. In Fluids in The Crust: Equilibrium and Transport Properties (eds Shmulovich, K. I., Yardley, B. W. D. & Gonchar, G. G.) 1–12 (Chapman & Hall, 1995).
Castelli, D., Rolfo, F., Groppo, C. & Compagnoni, R. Impure marbles from the UHP Brossasco–Isasca Unit (Dora–Maira Massif, Western Alps): evidence for Alpine equilibration in the diamond stability field and evaluation of the X(CO2) fluid evolution. J. Metam. Geol. 25, 587–603 (2007).
Yardley, B. W. D. & Graham, J. T. The origins of salinity in metamorphic fluids. Geofluids 2, 249–256 (2002).
Yardley, B. W. D. & Bodnar, R. J. Fluids in the continental crust. Geochem. Perspect. 3, 1–123 (2014).
Manning, C. E. Fluids of the lower crust: deep is different. Annu. Rev. Earth Planet. Sci. 46, 67–97 (2018).
Evans, K. A. & Tomkins, A. G. Metamorphic fluids in orogenic settings. Elements 16, 381–388 (2020).
Rowan, M. G. Passive-margin salt basins: hyperextension, evaporite deposition, and salt tectonics. Basin Res. 26, 154–182 (2014).
Xie, F. et al. Passive continental margin basins and the controls on the formation of evaporites: a case study of the Gulf of Mexico Basin. Carbonates Evaporites 34, 405–418 (2019).
Warren, J. K. Evaporites, brines and base metals: fluids, flow and “the evaporite that was”. Australian J. Earth Sci. 44, 149–183 (1997).
Warren, J. K. Evaporites: Sediments, Resources and Hydrocarbons (Springer-Verlag, 2006).
Markl, G. & Bucher, K. Composition of fluids in the lower crust inferred from metamorphic salt in lower crustal rocks. Nature 391, 781–783 (1998).
Newton, R. C., Aranovich, L. Y., Hansen, E. C. & Vandenheuvel, B. A. Hypersaline fluids in Precambrian deepcrustal metamorphism. Precambrian Res. 91, 41–63 (1998).
Touret, J. L. R. Fluid regime in southern Norway: the record of fluid inclusions. In The Deep Proterozoic Crust in the North Atlantic Provinces (eds Tobi, A. C. & Touret, J. L. R.) 517–549 (Reidel, 1985).
Trommsdorff, V., Skippen, G. & Ulmer, P. Halite and sylvite as solid inclusions in high-grade metamorphic rocks. Contrib. Mineral. Petrol. 89, 24–29 (1985).
Heinrich, W. Fluid immiscibility in metamorphic rocks. Rev. Mineral. Geochem. 65, 389–430 (2007).
Liebscher, A. Aqueous fluids at elevated pressure and temperature. Geofluids 10, 3–19 (2010).
Manning, C. E. & Aranovich, L. Y. Brines at high pressure and temperature: thermodynamic, petrologic and geochemical effects. Precambrian Res. 253, 6–16 (2014).
Manning, C. E. & Frezzotti, M. L. Subduction-Zone fluids. Elements 16, 395–400 (2020).
Trommsdorff, V. & Skippen, G. Vapour loss (“boiling”) as a mechanism for fluid evolution in metamorphic rocks. Contrib. Mineral. Petrol. 94, 317–322 (1986).
Skippen, G. & Trommsdorff, V. The influence of NaCl and KCl on phase relations in metamorphosed carbonate rocks. Amer. J. Sci. 286, 81–104 (1986).
Heinrich, W., Churakov, S. S. & Gottschalk, M. Mineral-fluid equilibria in the system CaO-MgO-SiO2-H2O-CO2-NaCl and the record of reactive fluid flow in contact metamorphic aureoles. Contrib. Mineral. Petrol. 148, 131–149 (2004).
Crawford, M. L., Kraus, D. W. & Hollister, L. S. Petrologic and fluid inclusion study of calc-silicate rocks, Prince Rupert, British Columbia. Amer. J. Sci. 279, 1135–1159 (1979).
Zhang, Y. G. & Frantz, J. D. Experimental determination of the compositional limits of immiscibility in the system CaCl2-H2O-CO2 at high temperatures and pressures using fluid inclusions. Chem. Geol. 74, 289–308 (1989).
Shmulovich, K. I. & Plyasunova, N. V. Phase equilibria in ternary systems formed by H2O and CO2 with CaCl2 or NaCl at high T and P. Geochem. Internat. 30, 53–71 (1993).
Shmulovich, K. I., Tkachenko, S. I. & Plyasunova, N. V. Phase equilibria in fluid systems at high pressures and temperatures. In Fluids in the Crust (eds Shmulovich, K. I., Yardley, B. W. D. & Gonchar, G. G.) 193–214 (Chapman and Hall, 1995).
Shmulovich, K. I. & Graham, C. M. An experimental study of phase equilibria in the systems H2O-CO2-CaCl2 and H2O-CO2-NaCl at high pressures and temperatures (500-800 °C, 0.5-0.9 GPa): geological and geophysical applications. Contrib. Mineral. Petrol. 146, 450–462 (2004).
Duan, Z., Møller, N. & Weare, J. H. Equation of state for the NaCl-H2O-CO2 system: prediction of phase equilibria and volumetric properties. Geochim. Cosmochim. Acta 59, 2869–2882 (1995).
Yardley, B. W. D. & Bottrell, S. H. Immiscible fluids in metamorphism: implications of two-phase fluid flow for reaction history. Geology 16, 199–202 (1988).
Frezzotti, M. L. & Touret, J. L. R. CO2, carbonate-rich melts, and brines in the mantle. Geosci. Front. 5, 697–710 (2014).
Frezzotti, M. L. & Ferrando, S. The chemical behavior of fluids released during deep subduction based on fluid inclusions. Am. Mineral. 100, 352–377 (2015).
Li, Y. Immiscible C-O-H fluids formed at subduction zone conditions. Geochem. Perspect. Lett. 3, 12–21 (2017).
Bower, T. S. & Hegelson, H. C. Calculation of the thermodynamic and geochemical consequences of non-ideal mixing in the system H2O–CO2–NaCl on phase relations in geological systems: equation of state for H2O–CO2–NaCl fluids at high pressures and temperatures. Geochim. Cosmochim. Acta 47, 1247–1275 (1983).
Bakker, R. J. Package FLUIDS 1. Computer programs for analysis of fluid inclusion data and for modelling bulk fluid properties. Chem. Geol. 194, 3–23 (2003).
Watson, E. B. & Brenan, J. M. Fluid in the lithosphere. I. experimentally determined wetting characteristics of CO2-H2O fluids and their implications for fluid transport, host-rock physical properties, and fluid inclusion formation. Earth Planet. Sci. Lett. 85, 497–515 (1987).
Holness, M. B. Equilibrium dihedral angles in the system quartz-CO2-H2O-NaCl at 800 °C and 1-15 kbar: the effects of pressure and fluid composition on the permeability of quartzites. Earth Planet. Sci. Lett. 114, 171–184 (1992).
Holness, M. B. & Graham, C. M. Equilibrium dihedral angles in the system CO2-H2O-NaCl-calcite, and implications for fluid flow during metamorphism. Contrib. Mineral. Petrol. 108, 368–383 (1991).
Irwing, W. P. & Barnes, I. Tectonic relations of carbon dioxide discharges and earthquakes. J. Geophys. Res. 85, 3115 (1980).
Chiodini, G. et al. Carbon dioxide Earth degassing and seismogenesis in central and southern Italy. Geophys. Res. Lett. 31, 2–5 (2004).
Chiodini, G. et al. Correlation between tectonic CO2 Earth degassing and seismicity is revealed by a 10-year record in the Apennines, Italy. Sci. Adv. 6, eabc2938 (2020).
Frezzotti, M. L., Peccerillo, A. & Panza, G. Carbonate metasomatism and CO2 lithosphere-asthenosphere degassing beneath the Western Mediterranean: An integrated model arising from petrological and geophysical data. Chem. Geol. 262, 108–120 (2009).
Girault, F. & Perrier, F. The Syabru-Bensi hydrothermal system in central Nepal: 2. modeling and significance of the radon signature. J. Geophys. Res, Solid Earth 119, 4056–4089 (2014).
Girault, F. et al. Large-scale organization of carbon dioxide discharge in the Nepal Himalayas. Geophys. Res. Lett. 41, 6358–6366 (2014).
Miller, S. A. et al. Aftershocks driven by a high-pressure CO2 source at depth. Nature 427, 724–727 (2004).
Sibson, R. H. Earthquake rupturing in fluid-overpressured crust: how common? Pure Appl. Geophys. 171, 2867–2885 (2014).
Girault, F. et al. Persistent CO2 emissions and hydrothermal unrest following the 2015 earthquake in Nepal. Nat. Comm. 9, 2956 (2018).
Newton, R. C. & Manning, C. E. Role of saline fluids in deep‐crustal and upper‐mantle metasomatism: insights from experimental studies. Geofluids 10, 58–72 (2010).
Halls, C. & Zhao, R. Listvenite and related rocks: perspectives on terminology and mineralogy with reference to an occurrence at Cregganbaun, Co., Mayo, Republic of Ireland. Mineral Deposita 30, 303–313 (1995).
Johannes, W. An experimental investigation of the system MgO-SiO2-H2O-CO2. Am. J. Sci. 267, 1083–1104 (1969).
Falk, E. S. & Kelemen, P. B. Geochemistry and petrology of listvenite in the Semail ophiolite, Sultanate of Oman: complete carbonation of peridotite during ophiolite emplacement. Geochim. Cosmochim. Acta 160, 70–90 (2015).
Beinlich, A., Plümper, O., Hövelmann, J., Austrheim, H. & Jamtveit, B. Massive serpentinite carbonation at Linnajavri, N-Norway. Terra Nova 24, 446–455 (2012).
Menzel, M. D. et al. Carbonation of mantle peridotite by CO2-rich fluids: formation of listvenites in the Advacate ophiolite complex Newfoundland, Canada. Lithos 323, 238–261 (2018).
Schreyer, W., Ohnmacht, W. & Mannchen, J. Carbonate-orthopyroxenites (sagvandites) from Troms, Northern Norway. Lithos 5, 345–364 (1972).
Ohnmacht, W. Petrogenesis of carbonate-orthopyroxenites (sagvandites) and related rocks from Troms, Northern Norway. J. Petrol. 15, 303–324 (1974).
Bucher, K. & Stober, I. Interaction of mantle rocks with crustal fluids: sagvandites of the Scandinavian caledonides. J. Earth Sci. 30, 1084–1094 (2019).
Huizenga, J. M. & Touret, J. Granulites, CO2 and graphite. Gondw. Res. 22, 799–809 (2012).
Evans, M. J., Derry, L. A. & France-Lanord, C. Geothermal fluxes of alkalinity in the Narayani river system of central Nepal. Geoch. Geophys. Geos. 5, Q08011 (2004).
Evans, M. J., Derry, L. A. & France-Lanord, C. Degassing of metamorphic carbon dioxide from the Nepal Himalaya. Geoch. Geophys. Geos. 9, Q04021 (2008).
Lemonnier, C. et al. Electrical structure of the Himalaya of Central Nepal: High conductivity around the mid-crustal ramp along the M.H.T. Geophys. Res. Lett. 26, 3261–3264 (1999).
Cattin, R. & Avouac, J. P. Modeling mountain building and the seismic cycle in the Himalaya of Nepal. J. Geophys. Res, Solid Earth 105, 13389–13407 (2000). B6.
Bollinger, L., Avouac, J. P., Cattin, R. & Pandey, M. R. Stress buildup in the Himalaya. J. Geophys. Res. 109, B11405 (2004).
Avouac J.-P. Mountain Building: From Earthquakes to Geologic Deformation. In Treatise on Geophysics (ed. Schubert, G.) 2nd edition, Vol. 6, 381–432 (Elsevier, 2015).
Wannamaker, P. E., Caldwell, T. G., Doerner, W. M. & Jiracek, G. R. Fault zone fluids and seismicity in compressional and extensional environments inferred from electrical conductivity: the New Zealand Southern Alps and US Great Basin. Earth, Planets and Space 56, 1171–1176 (2004).
Nesbit, B. E. Electrical resistivities of crustal fluids. J. Geophys. Res. 98, 4301–4310 (1993).
Godard, G. Two orogenic cycles in eclogite-facies gneisses of the Southern Armorican Massif (France). Eur. J. Mineral. 21, 1173–1190 (2009).
Hofer, G., Wagreich, M. & Neuhuber, S. Geochemistry of fine grained sediments of the upper Cretaceous to Paleogene Gosau Group (Austria, Slovakia): Implications for paleoenvironmental and provenance studies. Geosci. Front. 4, 449–468 (2013).
McLennan, S. M., Hemming, S., McDaniel, D. K. & Hanson, G. N. Geochemical approaches to sedimentation, provenance, and tectonics. GSA Special Paper 284, 21–40 (1993).
Connolly, J. A. D. Multivariable phase diagrams: an algorithm based on generalized thermodynamics. Am. J. Sci. 290, 666–718 (1990).
Connolly, J. A. D. The geodynamic equation of state: what and how. Geoch. Geophys. Geos. 10, Q10014 (2009).
Holland, T. J. B. & Powell, R. An internally consistent thermodynamic data set for phases of petrologic interest. J. Metam. Geol. 16, 309–343 (1998).
Massonne, H. J. Phase relations and dehydration behaviour of calcareous sediments at very-low to low grade metamorphic conditions. Period. Mineral. 79, 21–43 (2010).
Kuhn, B. K., Reusser, E., Powell, R. & Günther, D. Metamorphic evolution of calc-schists in the Central Alps, Switzerland. Schweiz. Mineral. Petrogr. Mitt. 85, 175–190 (2005).
Holland, T., Baker, J. & Powell, R. Mixing properties and activity‐composition relationships of chlorites in the system MgO‐FeO‐Al2O3‐SiO2‐H2O. Eur. J. Mineral. 10, 395–406 (1998).
Coggon, R. & Holland, T. J. B. Mixing properties of phengitic micas and revised garnet–phengite thermobarometers. J. Metam. Geol. 20, 683–696 (2002).
Auzanneau, E., Schmidt, M. W., Vielzeuf, D. & Connolly, J. A. D. Titanium in phengite: a geobarometer for high temperature eclogites. Contrib. Mineral. Petrol. 159, 1–24 (2010).
White, R. W., Powell, R. & Holland, T. J. B. Progress relating to calculation of partial melting equilibria for metapelites. J. Metam. Geol. 25, 511–527 (2007).
Newton, R. C., Charlu, T. V. & Kleppa, O. J. Thermochemistry of the high structural state plagioclases. Geochim. Cosmochim. Acta 44, 933–941 (1980).
Thompson, J. B. & Hovis, G. L. Entropy of mixing in sanidine. Am. Mineral. 64, 57–65 (1979).
Elmer, F. L., White, R. W. & Powell, R. Devolatilization of metabasic rocks during greenschist–amphibolite facies metamorphism. J. Metam. Geol. 24, 497–513 (2006).
Rapa, G., Groppo, C., Mosca, P. & Rolfo, F. Petrological constraints on the tectonic setting of the Kathmandu Nappe in the Langtang-Gosainkund-Helambu regions, Central Nepal Himalaya. J. Metam. Geol. 34, 999–1023 (2016).
Acknowledgements
This study was funded by the Italian Ministry of University and Research (PRIN 2017, Project no. 2017LMNLAW). M.L.F. acknowledges funding by MUR project Dipartimenti di Eccelllenza 2018–2022.
Author information
Authors and Affiliations
Contributions
All authors jointly conceived the study. C.G. performed the thermodynamic modeling and interpreted the results. M.L.F. interpreted the consequence of fluid immiscibility in terms of transport dynamics. F.R. and C.G. extended the results to the Himalayan orogeny and put them in a broader perspective. All authors discussed the results and their implications and participated in writing the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests. Maria Luce Frezzotti is an Editorial Board Member for Communications Earth & Environment, but was neither involved in the editorial review of nor the decision to publish this article.
Peer review
Peer review information
Communications Earth & Environment thanks Manuel Menzel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Joe Aslin, Heike Langenberg. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Groppo, C., Rolfo, F. & Frezzotti, M.L. CO2 outgassing during collisional orogeny is facilitated by the generation of immiscible fluids. Commun Earth Environ 3, 13 (2022). https://doi.org/10.1038/s43247-022-00340-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-022-00340-w
This article is cited by
-
Methane-hydrogen-rich fluid migration may trigger seismic failure in subduction zones at forearc depths
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
