
Abstract
Effective individual wound management, particularly in cases of prolonged healing and increased infection vulnerability, has prompted the development of wound theranostics, combining real-time diagnostic assessment and on-demand treatment. Here, we present a multifunctional conducting polymer-based smart theranostic bandage that integrates pH sensing, pH-compensated uric acid (UA) biosensing, and on-demand antibiotic release using different conducting polymers, each leveraging their advantageous intrinsic properties. Specifically, the polyaniline-based pH sensor operates reversibly across a pH range of 4–10, while the functionalized poly(3,4-ethylenedioxythiophene)-based UA biosensor exhibits a linear response up to 0.9 mM UA. Simultaneous detection of pH and UA allows accurate UA determination via pH compensation. Upon detecting abnormal pH/UA levels, the polypyrrole-based drug carrier releases ciprofloxacin via 0.6 V electrical stimulation, successfully inhibiting bacterial growth in vitro. The array is assembled as a 3D patch, connected to a flexible printed circuit board, and embedded in a wound bandage, offering potential for remote wound monitoring, targeted treatment, and wireless wound management.
Similar content being viewed by others
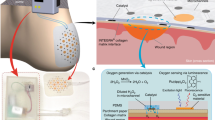


Introduction
Wounds are common skin injuries caused by factors like trauma, burns, injury infections, and underlying medical conditions such as diabetes mellitus, pressure injuries and age-related vascular diseases1. The healing process involves intricate biochemical events, making it highly vulnerable to infection, particularly in cases of deep skin injuries and chronic wounds, thus imposing a substantial burden on both individual patients and the healthcare system2. Conventionally, wound severity and healing status are assessed by clinicians through qualitative visual inspection or time-consuming laboratory testing3. Proper individual wound management is crucial in promoting wound healing, which often involves the usage of wound bandages or dressings. However, there is a significant advantage in incorporating smart systems into these bandages to provide quantitative monitoring of wound conditions and to implement therapy as necessary. Such innovation in wound theranostics reduces the need for frequent dressing changes and visits to medical facilities, thus enhancing wound care for individual patients.
There are many physiological parameters, e.g., biomarkers, present in the wound exudate that are amenable to monitoring using integrated sensors and biosensors, including oxygen, pH, uric acid (UA), and various immune proteins3,4. Continuous monitoring of such biomarkers in real-time and over an extended period can provide information about the healing status and conditions of the wound environment, particularly concerning the risk of bacterial infection. Among these, the pH of wounds is one critical biomarker, which undergoes dynamic change throughout different stages of healing and treatment. It typically maintains a mildly acidic state (5.5–6.5) during normal healing5, but transitions to an alkaline state (7.15–8.9) indicative of either pro-longed healing or the presence of high bacterial levels1,6. Therefore, relying solely on wound fluid pH determination proves insufficient for distinguishing between bacterial infection (bacterial growth) and the severity of the wound (cell damage). UA is another important biomarker associated with wound healing, as it promotes inflammation and correlates with highly reactive superoxide radicals. The UA concentration in chronic wounds fluctuates within the 220–750 μM range and increases with wound severity due to the metabolism of adenosine triphosphate (ATP) released into the extracellular matrix. Conversely, the UA level decreases in the presence of bacterial infection due to the catalysis of microbial uricase1. Consequently, this dynamic fluctuation in UA levels serves as an indicative pattern, reflecting either the severity of the wound or the presence of bacterial infection. Although enzymatic UA biosensors are available, offering good selectivity for detecting UA under relatively stable physiological pH conditions, such as in serum, previously reported amperometric UA biosensors have overlooked the fluctuating pH levels in real wound exudates7,8. This omission is inadequate for dynamic wound monitoring, potentially resulting in inaccurate UA concentration estimation due to variations in enzyme activity under different pH conditions. Therefore, simultaneous detection of pH level and UA concentration not only allows pH compensation of the UA sensor response, but also enables the differentiation between wound infection and severity. Moreover, it is imperative to integrate a therapeutic system into wound bandages to align with the paradigm of personalized therapy based on the assessment of wound status through the sensor and biosensor system. Conventional passive dressings function as physical barriers, while medicated dressings are designed to deliver healing promotion agents (e.g., growth factors and monoterpenes)9 in a passive manner. The integration of a localized and active drug delivery system for administering antimicrobial agents, tailored to specific needs and providing an accurate dosage, is pivotal for enhancing therapeutic efficacy against bacterial infection10,11,12.
To establish a theranostic system within wound bandages, the development of flexible electronics is crucial, necessitating the fabrication of a flexible electrode array and the modification of various materials with distinct functionalities. Currently, the fabrication of flexible electrode arrays typically requires sophisticated equipment and the use of masks for depositing electrical components onto flexible substrates13,14, such as carbonaceous electrodes by screen printing7,15 and metal electrodes by evaporation and photolithography12,16. Beyond these, laser irradiation technology offers a cost-effective and straightforward approach for generating laser-induced graphene (LIG) electrode arrays on flexible substrates, allowing contact-free and mask-free fabrication in a customer-designed pattern17,18. Moreover, it is essential to implement modular modifications to the flexible electrode array to confer specific functionalities to individual electrodes.
Conducting polymers (CPs) are promising functional materials fulfilling all the abovementioned requirements in wound theranostics, including biosensing and chemical sensing19,20, actuation21 and drug delivery22, based on their advantageous intrinsic properties. For instance, polyaniline (PANi) can undergo a reversible protonation and de-protonation process of the imine (=N-) and amine (-NH-) groups in its backbone23. This reversible transition between different states of PANi results in changes in its resistance, which is suitable for potentiometric pH sensing. Poly(3,4-ethylenedioxythiophene) (PEDOT) presents a stable and conductive “doped” state, which is beneficial for serving as an electrically conductive matrix for in-situ embedding of a catalyst, such as the widely used ‘artificial peroxidase’ Prussian blue (PB) in catalytic biosensors24,25. However, conventional methods to fabricate PB-doped PEDOT (PEDOT:PB) typically involve multi-step processes, either through the polymerization of PEDOT with Fe(CN6)3- and subsequent conversion into PB by potentiodynamic cycling in Fe3+, or through the polymerization of PEDOT in the appearance of pre-synthesized PB obtained by wet-chemistry. The exploration of a one-step fabrication method for PEDOT:PB, achieved by simultaneously polymerizing PEDOT and in-situ formation of PB within the PEDOT matrix, is particularly attractive. Polypyrrole (PPy) has been intensively used for actuators and drug delivery on account of its fast actuation speed and high strain26,27 Additionally, distribution of electrode arrays, exemplified by 2D electrode arrays, is limited by the size constraints of specific modules, especially in case of drug delivery carriers that need high drug loading. The utilization of a 3D laminated electrode array presents the potential for enhancing theranostics. This design allows for the placement of a large-size drug delivery carrier adjacent to the sensor and biosensor array, facilitating a more efficient integration of components.
Here, we present a multiplex sensor and drug delivery array based on CPs, and their assembly into a flexible patch and further integration into a smart bandage platform for wound theranostics. As illustrated in Fig. 1, the system leverages the unique properties of the selected CPs modified on the LIG electrode array, including the redox reversibility for selective protonation/de-protonation in PANi, the electrical conductivity of PEDOT, and the volume expansion of PPy. This integrated smart bandage platform enables electrochemical measurements of pH levels (over the range of 4–10) and UA concentrations (up to 0.9 mM) as indicators of wound status, while also facilitating on-demand release of Ciprofloxacin (Cipro) via a 0.6 V electrical stimulation during severe conditions. The pH sensor and UA biosensor array are assembled with a drug carrier into a 3D multiplex patch, which is integrated with a flexible printed circuit board (FPCB) featuring an on-board microcontroller and Bluetooth. Such a smart bandage platform is promising for wound biomarkers detection, controlled drug delivery and wireless connectivity in wound management, thus enabling wound theranostics.

a Schematic illustration of the 3D assembled patch, integrating a PANi-based pH sensor, a PEDOT-based UA biosensor and a PPy-based drug carrier; (b) Schematic illustration of the integration of the patch with an FPCB featuring an on-board microcontroller and Bluetooth, further embedded into a wound bandage for wireless connectivity in individual wound management.
Results and discussion
PANi-based all-solid-state pH sensor
Figure 2a illustrates schematically the all-solid-state pH sensor building on a PANi-based flexible LIG electrode. The design and fabrication of LIG electrode patterns are shown in Supplementary Figs. 1, 2). The pH sensor is comprised of a LIG working electrode modified with a PANi layer (PANi/LIG) as the sensing element to hydrogen ions (H+), and a solid-state pseudo-reference electrode modified with Ag/AgCl and a polyvinyl butyral (PVB)-KCl reservoir (PVB-KCl/Ag/AgCl/LIG). The pH sensing ability of the PANi layer is ascribed to the reversible protonation and de-protonation of the emeraldine state of PANi molecules (Fig. 2a and Supplementary Fig. 3). In acidic solutions, the emeraldine base form of PANi is protonated with H+ and doped with anion (A-), resulting in a highly conductive emeraldine salt form of PANi. While in alkaline solutions, the conductive emeraldine salt form of PANi is deprotonated by the neutralization effect of OH-, leading to the non-conductive emeraldine base form of PANi. As a result, the electromotive force (EMF) at the electrode surface changes due to resistance variations of PANi. The change in the resistance of the PANi-based pH sensor is validated via the gradually suppressed redox peaks and background current with increasing pH (see Supplementary Fig. 3).

a Schematic illustration of the pH sensor and its sensing mechanism; (b) SEM images of PANi/LIG, inset scale bar is 200 nm; (c) SEM images of PVB-KCl on Ag/AgCl/LIG, the inset is a cross-sectional image, scale bar is 10 μm; (d) Potentiometric curve of the EMF response of the pH sensor to stepwise pH increases from 4 to 10 followed by stepwise decreases from 10 to 4; (e) Calibration plots of EMF versus pH (n = 3); Long-term stability (f) and recovery capability (g) of the pH sensor at pH value of 5, 7 and 9.
The electropolymerization of PANi was confirmed via Fourier-transform infrared (FTIR) spectroscopy (see Supplementary Fig. 4) by characteristic bands of PANi23. The surface morphology of PANi/LIG was examined using scanning electron microscopy (SEM). As depicted in Fig. 2b, the PANi layer was deposited on the porous LIG structure (SEM of bare LIG is shown in Supplementary Fig. 1d‒f) in the form of interconnected nanofibers with an average fiber diameter of 54.0 ± 10.4 nm. The reference electrode (see the digital photographs in Supplementary Fig. 5) employs a PVB-KCl reservoir as a solid-state diffusion layer to protect the pseudo-reference to ensure stability and accuracy by maintaining a high and constant concentration of electrolyte at the solution interface28. The top-view SEM image in Fig. 2c displays a compact polymeric film with a few sub-micro pores, resulting from the dissolution of KCl and subsequent formation of a KCl-based microchannel between the Ag/AgCl pseudo-reference and the solution after conditioning in 3 M KCl for 24 h. The cross-sectional SEM image (inset of Fig. 2c) confirms the sequentially layered structure of PVB-KCl, Ag/AgCl and LIG. The PVB top layer is also verified by FTIR with its typical adsorption bands (see Supplementary Fig. 4)29. Conditioning the modified reference electrode is necessary to achieve a stable output voltage30. The effect of conditioning on the output voltage was investigated by cyclic voltammetry using Fe(CN)63/4− as a pair of redox probes. The results indicate (see Supplementary Fig. 6) a stable redox peak position of the Fe(CN)63/4− probes for the conditioned PVB-KCl/Ag/AgCl/LIG, which is consistent with a commercial glass Ag/AgCl (3 M KCl). However, in the case of un-conditioned PVB-KCl/Ag/AgCl/LIG, the redox peak position shifted negatively with an increasing number of cycles, eventually reaching a value as the Ag/AgCl/LIG reference electrode.
The potentiometric sensing performance of the PANi-based pH sensor was evaluated by measuring the EMF values in solutions with varied pH values over the range of 4‒10. The potentiometric curve in Fig. 2d shows that the EMF value gradually decreased with an increasing pH value from 4 to 10, and then increased as the pH value decreased back to 4, eventually reaching approximately the initial EMF value at pH 4. The calibration curve in Fig. 2e demonstrates a linear relationship between the EMF value (average value for each pH for the two measurements of increasing and decreasing pH) and pH, with a regression equation of EMF (mV) = −59.5 × pH + 442.8 (R2 = 0.997, n = 3). The sensitivity of −59.5 mV pH−1 closely aligns with the theoretical Nernstian slope at RT (~59 mV pH−1)31. The stability of the pH sensor was tested by continuously measuring the EMF values while immersing in solutions with pH values of 5, 7, and 9 for 12 h, respectively. The pH sensor exhibited good long-term measurement stability (Fig. 2f) with a small drift of less than 0.6 mV h−1 23. Additionally, Fig. 2g shows the carry-over evaluation of the pH sensor, displaying a reversible EMF response to dynamically changing between pH 5, 7 and 9 solutions. This indicates an excellent recovery capability of the fabricated pH sensor. The sensing performance of the all-solid-state PANi/LIG pH sensor was compared with other PANi-based pH sensors described in the literature (as summarized in Supplementary Table 1), revealing a comparable sensitivity and sensing range.
PEDOT-based enzymatic UA biosensor
The structure and corresponding biosensing principle of the UA biosensor building on uricase/PEDOT embedded with Prussian blue composite (PEDOT:PB) on flexible LIG is schematically depicted in Fig. 3a. PEDOT:PB serves as a transducing layer, which can catalyze the reduction of H2O2 generated by the enzymatic catalysis of UA by uricase at a relatively low potential with high catalytic activity and selectivity32,33. PEDOT:PB is fabricated on the porous LIG surface via a facile one-pot electrochemical deposition using EDOT monomer and ferrous iron (II)-based precursors. With increasing potential in potentiodynamic cycling (see Supplementary Fig. 7), the two ferrous iron (II) species, i.e., Fe(CN)64− and Fe2+, were sequentially oxidized into the ferric iron (III) species of Fe(CN)63− and Fe3+, respectively. The newly formed iron (III) species react with the existing iron (II) species forming PB (FeIII [FeIII(CN)6]) nanoparticles. Successive positive scans resulted in the oxidation of the EDOT monomer and its polymerization into PEDOT, while in-situ embedding the PB nanoparticles in the conductive PEDOT matrix. The FTIR results (see Supplementary Fig. 8) validate the successful preparation of PEDOT:PB composites. Figure 3b shows that the deposition of PEDOT:PB preserves the porous structure of the LIG, which is consistent with the PB/LIG and PEDOT/LIG results (see the SEM images in Supplementary Fig. 9). Uricase was deposited onto the PEDOT:PB/LIG, followed by deposition of a chitosan coating to achieve a physical entrapment of the uricase, in order to provide stable immobilization while preserving the enzymatic activity34. As can be seen in Fig. 3c, this procedure did not affect the porous structure of the LIG electrode. The energy-dispersive X-ray (EDX) spectroscopy and calculated elemental composition further verify the successful deposition of PEDOT:PB and effective loading of uricase (see Supplementary Fig. 9) forming the uricase/PEDOT:PB/LIG biosensor.

a Schematic illustration of the UA biosensor and the UA biosensing principle; SEM images of PEDOT:PB/LIG (b) and uricase/PEDOT:PB/LIG (c), inset scale bar is 200 nm; (d) Current-time response curve of PEDOT:PB/LIG to successive addition of H2O2 in 0.1 M PBS (pH 7.5) at −0.2 V; (e) corresponding calibration curve, n = 3; (f) Amperometric response of the uricase/PEDOT:PB/LIG electrode to increasing UA concentrations from 0 to 1.2 mM; (g) corresponding calibration plot, n = 3; (h) Calibration plots of the current responses (absolute value) versus UA concentrations under different pH ranging from 6 to 9, n = 3; (i) Sensitivity correction factor under different pH ranging from 6 to 9 with the sensitivity at pH 7.5 as 1.0; (j) Amperometric current response of the UA biosensor to 0.5 mM UA in the presence of common interferences in wound exudates, insets is the plotting of current response (absolute value).
The sensing performance of H2O2 at the PEDOT:PB/LIG electrode was evaluated before turning to the complete biosensor. The CVs (see Supplementary Fig. 10) display an increased reduction peak current with a reduction peak potential of −0.10 V in 0.1 M PBS (pH = 7.5) containing 5 mM H2O2 compared to that of the blank PBS. The current-time response of H2O2 at the PEDOT:PB/LIG electrode was measured at −0.10 V with successive addition of H2O2. As shown in Fig. 3d, the current response of the PEDOT:PB/LIG electrode increased with increasing of H2O2, delivering a linear current response as a function of H2O2 concentration over the range of 0.01‒6.76 mM (R2 = 0.998) with a high sensitivity of 31.95 μA mM−1 (Fig. 3e).
For UA biosensing, the immobilized amount of uricase was optimized to be 0.4 U/electrode by the evaluation of amperometric responses towards 0.5 mM UA at −0.1 V at the uricase/PEDOT:PB/LIG electrode with varying uricase amount over 0.1–0.5 U (see Supplementary Fig. 11). The evaluation of the current response to 0.5 mM UA at different potentials from 0 to −0.2 V shows high sensitivity at the potential range between −0.2 and −0.1 V (see Supplementary Fig. 12). However, ascorbic acid (AA), an electroactive species that is usually present together with UA in biofluids, is prone to be oxidized at −0.1 V. When shifting the applied potential negatively, the oxidation current of AA is suppressed and it has no significant effect on the UA detection signal at −0.2 V (see Supplementary Fig. 12). Therefore, −0.2 V was chosen as the operational potential for the UA biosensing.
The performance of the uricase/PEDOT:PB/LIG biosensor in response to UA was then investigated and the amperometric responses are shown in Fig. 3f. The amperometric curves exhibit a distinct current response to the dynamic concentration range of UA up to 1.2 mM. The current response was linearly proportional to the UA concentration up to 0.9 mM (R2 = 0.999) with a sensitivity of 1.20 μA mM−1 (Fig. 3g). According to previous reports, the wound healing process shows a varied pH level either in a mildly acidic range for normal healing or a weak alkaline range for prolonged healing and bacterial infection6. Thus, it is imperative to account for pH levels in ensuring accurate measurements of UA concentration due to their effect on enzymatic activities. Amperometric current responses of the UA biosensor towards varied UA concentrations at different pH ranging from 6 to 9 (with 0.5 interval) were evaluated (see Supplementary Fig. 13), and the corresponding current responses versus UA concentration are plotted in Fig. 3h. The responses reveal distinct calibration curves with varying slopes (i.e., sensitivities) at different pH values. To address this, sensitivity correction factors were derived using the slope at pH of 7.5 as a reference factor of 1.0, neglecting the small error due to the slightly different intercepts. As shown in Fig. 3i, the sensitivity correction factors gradually increase with increasing pH value from 6.0 to 8.0, reaching the highest value at pH 8.0, followed by a decreasing trend with further increasing pH value to 9.0. This trend is consistent with that of the relative activity of uricase from Candida sp. under different pH values (www.toyobo-global.com). To evaluate the selectivity of the UA biosensor, the amperometric response toward 0.5 mM UA in 0.1 M PBS (pH 7.5) was tested in the presence of potential interferences at their physiological concentrations from previous reports7,35. The potential interferents, including 10 mM lactate, 1.2 mM cholesterol, 62 μM creatinine, 5 mM glucose, 0.1 mM ascorbic acid, had no significant effect on the UA current signal (Fig. 3j), indicating a good selectivity of the UA biosensor. The biosensing performance of the fabricated uricase/PEDOT:PB/LIG biosensor is comparable, or in some cases, particularly for the linear range, better than other amperometric UA biosensors described in the literature as summarized in Supplementary Table 2.
PPy-based controlled drug release
The drug delivery was achieved via an electrically triggered drug release of Cipro from a PPy:Cipro/LIG patch as illustrated in Fig. 4a. The PPy:Cipro drug carrier was deposited on the porous LIG surface by electropolymerization of Py monomers in the presence of Cipro using Cl- as the counterion. During the PPy electropolymerization process, Cipro molecules were loaded into the PPy matrix via physical entrapment. Electrical stimulation at a positive potential could trigger oxidation of the PPy with the injection of anions (e.g., Cl-) and water molecules into the PPy film, which results in an expansion of the PPy film22,36. During the expansion, the physically entrapped Cipro inside the bulk polymer was released36. Figure 4b shows a SEM image of the PPy:Cipro/LIG with a well-preserved inter-connected porous LIG structure with a rough surface (insets of Fig. 4b) due to the deposition of PPy:Cipro, while the bare LIG possesses a smooth surface (see Supplementary Fig. 1). The morphology of the PPy:Cipro film is similar to PPy:Cl- and there is no notable change of the PPy:Cipro film after electrical stimulation at 0.6 V for 60 min (see Supplementary Fig. 14). The loading of Cipro was identified by the presence of typical Cipro bands in the FITR spectrum for PPy:Cipro and the enlarged background current in CVs of PPy:Cipro compared to PPy:Cl- (see Supplementary Fig. 15).

a Schematic illustration of the drug carrier as well as Cipro loading and release in the PPy:Cipro/LIG electrode; (b) SEM images of the PPy:Cipro/LIG electrode, inset scale bar is 200 nm; (c) Drug release amount by electrical stimulation at different potentials from −0.6 to 0.8 V and alternating potentials at a frequency of 0.05 Hz for 60 min in 0.1 M KCl compared to passive release, n = 3; (d) Cumulative release profile of Cipro by PPy:Cipro/LIG electrode under 0.6 V (active, red curve) and passive (black curve) condition, respectively; Ct is the cumulative release amount at time t while C0 is the estimated total loaded amount of Cipro (n = 3); (e) Cumulative release of Cipro by a PPy:Cipro/LIG electrode with alternating periods of electrical stimulation at 0.6 V (active, red segments) and passive (black segments) condition (n = 3).
The electrically triggered drug release of Cipro from the PPy:Cipro drug carrier was evaluated by applying either a constant or a pulsed potential for 60 min. The applied potential determines the redox state and thus the volume changes of the PPy:Cipro drug carrier. As shown in Fig. 4c, the released amount of Cipro displays an ascending trend as the applied constant potential increases from 0 V to 0.6 V, which is ascribed to the oxidation/expansion of the PPy:Cipro drug carrier. The released amount of Cipro triggered under 0–0.6 V was significantly higher (i.e., at least 1.99 times at 0 V, 43.5 ± 4.0 μg) than under passive release condition (21.9 ± 1.6 μg), with the highest released Cipro amount of 73.0 ± 2.1 μg (3.33 times higher than passive release) occurring at 0.6 V. Further increase of the potential to 0.8 V resulted in a decreased release amount, which might be ascribed to the electro-oxidation of Cipro under the applied potential37. It is worth noting that the applied potential in the range from −0.2 to −0.6 V caused an even lower released amount compared to that of the passive condition, showing a decreasing trend with increasingly negative potential. This is reflected in the UV/Vis absorption spectra (see Supplementary Figs. 16, 17) as (1) the decay of the maximum absorption band at 277 nm and the two shoulder bands at relatively high wavelengths (317 and 330 nm); (2) the new bands at 235 nm and ~400 nm. Our hypothesis is that the negative potential is not compatible with Cipro in the medium, causing increased decay of Cipro with increasing electrical stimulation time. The suppressed release under negative potential can also be attributed to charge-dependent retention. The reduction of PPy induced by the negative potential diminishes the positive charge of the polymer backbone, thereby facilitating the retention of the partially positively charged Cipro. In addition to applying a constant potential, the effect of alternating potentials of 0/0.6 V, −0.6/0 V and −0.6/0.6 V at a frequency of 0.05 Hz were also tested for Cipro release. The pulsed potential of 0/0.6 V and −0.6/0.6 V triggered a suppressed release of Cipro compared to the active release at 0.6 V, which are around 65% (47.5 ± 2.8 μg for 0/0.6 V) and 62% (45.0 ± 7.0 μg for −0.6/0.6 V) of the released amount at 0.6 V, respectively. A more significantly suppressed release of Cipro was observed for the pulsed potentials of −0.6/0 V (14.4 ± 0.5 μg). These results indicate the compatibility of positive potential on the electrically stimulated release of Cipro. Detailed UV/Vis absorption spectra at different potentials can be seen in Supplementary Figs. 16, 17. As a result, 0.6 V was chosen as a constant potential for the electrically triggered release of Cipro at the PPy:Cipro/LIG electrode.
The total amount of Cipro loaded in the PPy:Cipro/LIG electrode (1 cm2 working electrode area) was determined to be 80.1 ± 4.1 μg. Release profiles of Cipro from the PPy:Cipro/LIG electrode were performed under an active condition at 0.6 V and a passive condition without applied potential. We define a cumulative release ratio (Ct/C0), where Ct is the cumulative release amount at time t while C0 is the estimated total loaded amount of Cipro. As shown in Fig. 4d, a fast-release rate of the Cipro can be observed in the active condition, reaching a cumulative release ratio of around 74.0% after 18 min, and a final cumulative release ratio of around 80.8% after 210 min. In comparison, the passive condition resulted in a slow-release rate with a cumulative release ratio of 25.0% at the first 18 min and finally 31.3% at the 210 min. The final cumulative release ratio at the active condition is around 2.58 times higher than that of passive release. The release profile of Cipro from the PPy:Cipro/LIG electrode was further investigated by applying periodic stimulation for 1 min and passive condition for 9 min. In Fig. 4e, the release triggered by electrical stimulation (red segments) is much more pronounced than that of passive release (black segments) with a higher slope value, which is consistent with previous work on PPy as drug carrier15,38,39. Although active conditions result in a significantly higher release rate, there remains a concern regarding the contribution of the passive release, driven by natural diffusion to the overall drug release40. Future research endeavours should focus on achieving precise drug release at different stages throughout the life cycle of the delivery system. The fabricated PPy-based drug release patch shows a satisfactory performance from the aspect of final cumulative release ratio and active to passive release ratio when compared to the literature as summarized in Supplementary Table 3.
In vitro evaluation of the multiplex sensor and drug delivery patch
To evaluate the analytical reliability of the smart bandage with the integrated pH sensor and UA biosensor array as well as the anti-microbial efficacy of the drug delivery patch for real applications, the sensor array and drug delivery patch were attached to the exposed dermis layer of porcine skin (Fig. 5a). For the sensor array, artificial wound exudates with various pH values (7.0, 8.0 and 9.0) and UA concentrations (0.1, 0.2, 0.4 and 0.6 mM) were applied on the dermis layer. The preparation of the artificial wound exudate samples is listed in Supplementary Table 4. The EMF value of the pH sensor and the current response of the UA biosensor were measured simultaneously by potentiometry and amperometry, respectively, for 100 s. Figure 5b shows a plot of the pH sensing and UA biosensing of four different artificial wound exudate samples with different UA concentrations at a constant pH value of 8.0. It can be observed that the EMF value remains constant at around −29 mV, which is calculated to be a pH value of 7.92 based on the equation from Fig. 2. The current response of the UA biosensor increased as the UA concentration increased.

a Schematic illustration of the attachment of the pH sensor and UA biosensor array as well as the drug delivery patch onto a porcine skin, from which the epidermis layer was removed and an artificial wound exudate was applied; (b) Simultaneous recording of the EMF value and the current response to an artificial wound exudate at pH 8.0 and with different UA concentrations, inset is the pH sensor and UA biosensor array attached to the porcine skin; (c) Comparison plot of the measured pH value and the reference pH value for the artificial wound exudate with various pH values and UA concentrations (n = 3); (d) Comparison plot of the measured UA concentration and the reference UA concentration for the artificial wound exudate with various pH values and UA concentrations (n = 3); (e) Bacterial growth inhibition test from (i) blank, (ii) PPy:Cl- release, (iii) PPy:Cipro passive release, and (iv) PPy:Cipro active release against E. coli cultured in a Petri dish with 6 mm discs of Whatman filter paper (the white circles) with artificial wound exudate samples, inset is the drug delivery patch attached to the porcine skin; (f) Simplified block diagram of the FPCB; (g) 3D assembly of the pH & UA sensor layer and the drug carrier layer; (h) Integration of the 3D assembled patch with a FPCB (top) and their further embedding into a wound bandage (bottom); (i) FPCB user interface for simultaneous pH and UA sensing as well as controlled drug delivery.
For comparison, standard pH (commercial pH meter using a glass electrode) and UA (spectrophotometry based on a colorimetric assay) measurements were used as reference methods to measure the pH values and the UA concentrations in the artificial wound exudate, respectively. The correlation of the pH value and the UA concentration measured by the integrated pH sensor and UA biosensor array and by standard methods is shown in Fig. 5c, d based on the potentiometric and amperometric curves in Supplementary Fig. 18. The pH values measured from the pH sensor for all artificial wound exudate samples containing different concentrations of UA were closely comparable to those obtained from the commercial pH meter as shown in Fig. 5c. In contrast, the UA biosensor showed a varying sensitivity towards UA at different pH values of the wound exudate (Supplementary Fig. 18), which is consistent with the sensitivity correction factor from Fig. 3. The simultaneous measurement of the pH and UA parameters facilitated an accurate detection of the UA level by pH compensation, avoiding over/under-estimation of the UA level owing to pH-affected enzymatic activity change of the uricase/PEDOT:PB LIG biosensors. Therefore, the measured UA concentration was adjusted based on the sensitivity correction factors (Fig. 3i) and compared to that of the colorimetric assay. As shown in Fig. 5d, the adjusted UA concentration shows a good correlation with the standard colorimetric assay. For each spiked UA concentration, the recovery was calculated to be in the range of 94–102% (see Supplementary Table 4). These results suggest the analytical reliability of the fabricated pH sensor and UA biosensor array. The approach also ensures precise measurements of the UA concentration in wound exudates, even when there are dynamic pH fluctuations. Unlike conventional biosensors that typically analyze biological samples under relatively stable physiological conditions, such as serum with a consistent pH, or rely solely on temperature compensation, wounds exhibit significant pH variations that can greatly impact the enzymatic UA biosensor’s sensitivity. Consequently, the demonstrated simultaneous monitoring of pH levels and pH-compensated UA concentrations presents a powerful and comprehensive method for accurately assessing wound severity and detecting infection. Moreover, this innovation holds promise for artificial intelligence-assisted deconvolution of sensor array responses, leveraging advanced machine learning algorithms, further enhancing the potential applications of our concept.
To investigate the in vitro antimicrobial efficacy of the Cipro released from the drug delivery patch (inset of Fig. 5e), the formation of a zone of inhibition (ZOI) was measured by a disc diffusion assay of the released reagents in the supporting medium on an E. coli lawn. As shown in Fig. 5e, there was no obvious formation of a ZOI for the blank artificial wound exudate (i) and the PPy:Cl- released sample (ii). A clear ZOI can be seen for the PPy:Cipro passive released sample (iii) and PPy:Cipro active released sample (iv) against E. coli. The mean diameter of the ZOI for the active release is 3.28 ± 0.03 cm, which is 1.23 times larger than that of the passive release (2.65 ± 0.01 cm). The results confirm not only the retention of the antimicrobial efficacy of Cipro via electropolymerization and subsequent electrically triggered drug release, but the preserved antimicrobial performance of the active Cipro release from the drug delivery patch.
A FPCB was designed and fabricated to serve as an electrochemical analyzer for signal acquisition from the sensor array and electrical stimulation of the drug release patch. The main circuit and electronic components are illustrated in the simplified block diagram in Fig. 5f, including a 2-electrode circuit for potentiometric detection of pH and two 3-electrode circuits for amperometric UA biosensing and electrically triggered drug release, respectively. Figure 5g illustrates the assembly of the sensors and drug delivery patch into a flexible 3D array with multiple layers, including a double-sided medical adhesive tape layer (ViTri Wound film) at the bottom to facilitate secure attachment to a bandage, a pH sensor and UA biosensor array, another medical adhesive tape layer underneath the drug carrier layer with rectangular openings by xurographic cutting to constitute the boundary of the sensing chambers, and a top insulating Kapton layer. The inset of Fig. 5g shows a digital photograph of a 3D assembled patch. The assembled patch was integrated with a FPCB (top of Fig. 5h), which was further prototyped as a smart bandage (bottom of Fig. 5h) by embedding them into a wound bandage (a sterile universal bandage, 4-in-1 mini blood stopper, Cederroth). The patch was seamlessly attached to the absorbent in the bandage and the FPCB was fixed at the back of the bandage. The prototyped smart bandage could achieve conformal contact with the skin surface due to its flexibility, and wireless communication with an electronic module for in-situ signal processing and on-demand drug release that can be controlled by a user-interface. The prototyped smart bandage was employed for in-vitro measurement of pH and UA levels in artificial wound exudate. Figure 5i displays the user-interface for the simultaneous detection of pH and UA levels. The average measured pH value of 7.35 ± 0.06 (n = 3) and UA level of 0.88 ± 0.04 mM (n = 3) are close to the values of our standard artificial wound exudate prepared by adjusting the pH value to 7.40 and spiking the UA concentration to 0.90 mM according to reference methods. The prototyped smart bandage also incorporates a feature for drug administration through electrically triggered release, utilizing a time-control panel, to be activated upon the detection of anomalous pH and UA values. The smart bandage holds the promise of evolving into a closed-loop drug release system, contingent upon additional investigation and clinical validation of the correlation between bacterial infection severity and UA/pH levels. The unit cost considering the chemicals and materials used to produce the prototyped smart bandage was estimated to be ~0.5 € for the disposable 3D sensor array & drug carrier platform, and ~49 € for the reusable FPCB (as summarized in Supplementary Table 5).
Conclusion
In summary, we have successfully demonstrated a multiplex sensor and drug delivery array based on CPs. This innovation involves functionalizing a flexible LIG electrode with different CPs, namely PANi, PEDOT, and PPy, capitalizing on their unique intrinsic properties: redox reversibility, electrical conductivity, and volume expansion, respectively. The PANi-based all-solid-state pH sensor and the PEDOT-based enzymatic UA biosensor showed good analytical performance for pH and UA determination, respectively. Importantly, we achieved simultaneous pH and UA detection, thereby enhancing the accuracy of UA concentration measurement through the application of a pH-dependent sensitivity correction factor. This simultaneous detection capability holds promise for early bacterial infection detection in wounds. Furthermore, the PPy-based drug release system demonstrated electrically triggered antibiotic release, exhibiting enhanced antimicrobial efficacy compared to passive release mechanisms, which is beneficial for on-demand wound treatment. The assembly of the sensor array and drug release carrier into a 3D patch, along with the design and manufacturing of a FPCB, followed by embedding into a wound bandage, showcases the potential for wound biomarker detection and drug delivery in wireless wound management. In the current version, the drug release depends on the patient’s active decision-making. However, for specific applications, such as chronic wound management, it is straightforward to implement a closed-loop system for sensing and therapeutics. Overall, our study highlights the versatility of CPs in creating a smart bandage system capable of accurate wound monitoring, targeted treatment, and wireless connectivity for individual wound management.
Methods
Materials and instruments
Polyimide thermal insulating film (thickness of 75 µm) and Kapton tape were purchased from RS Components AB (Sweden). Aniline, 3, 4-ethylenedioxythiophene (EDOT), pyrrole, polyvinyl butyral (PVB), uric acid (UA), hydrogen peroxide (H2O2), potassium ferricyanide (K3[Fe(CN)6]), potassium ferrocyanide (K4[Fe(CN)6]), lithium perchlorate (LiClO4), iron(II) chloride (FeCl2), hydrogen peroxide (H2O2), bovine serum albumin (BSA), human serum (from human male AB plasma), uric acid assay kit were purchased from Sigma-Aldrich (USA). Uricase from Candida sp. (4 U mg−1) was purchased from Toyobo (Japan). Ciprofloxacin hydrochloride monohydrate (Cipro) was bought from VWR (Sweden). Silver/silver chloride (Ag/AgCl) was purchased from Dupont (USA). E. coli (ATCC-25922) was obtained from LGC Standards GmbH (Germany). All chemicals were of analytical grade and used without any further treatment. Deionised water from a Milli-Q System was used throughout.
Computer-controlled CO2 Laser platform (HL40-5g, 10.6 μm, Full Spectrum Laser LLC, USA) were used to produce LIG. CompactStat potentiostat (Ivium, Netherlands) was used for electrochemical performance measurement. A robotic cutting device (ScanNut CM900, Brother International) was used for xerographic cutting for assembly of the 3D array. SEM images were recorded using a Zeiss-Sigma 500 Gemini electron microscope (Zeiss, Germany). The chemical composition of LIG and modified LIG were determined by EDX (Oxford Instruments). FTIR spectroscopy was performed using a VERTEX spectrometer (Bruker, USA) equipped with an attenuated total reflection (ATR) measuring cell. Raman spectra were acquired with a LabRAM HR 800 Raman spectrometer (Horiba Jobin Yvon, France) using a 660 nm 5 mW laser. UV/vis spectroscopy was collected using a UV/vis spectrophotometer (UV-2450, Shimadzu, Japan). Commercial pH meter (S20, Mettler Toledo, Sweden) was used to as a standard for pH measurement.
Fabrication of the multiplex sensor and drug delivery array
LIG
LIG was prepared in different patterns (Supplementary Figs. 1, 2) by a CO2 laser operating with a resolution of 1000 ppi in a raster mode under ambient conditions. The laser parameters were optimized to be 50% of the full laser power (full power is 40 W) and 55% of the full scan speed (full speed is 80 inches s−1).
PANi-based pH sensor
Electropolymerization of PANi on LIG was conducted by dynamic cyclic voltammetry between −0.2 and 1.0 V for 30 cycles in 1 M HCl containing 0.1 M aniline at a scan rate of 50 mV s−1. The reference electrode surface was stencil-printed with Ag/AgCl ink and dried under 120 °C on a hotplate for 2 min. Then 9 µL of PVB polymer (10% wt in methanol) containing KCl fine powder (30% w/v) was drop-casted on top of the Ag/AgCl using a mask, followed by conditioning by immersing the reference electrode in 3 M KCl for 24 h.
PEDOT-based UA biosensor
PEDOT:PB/LIG was prepared by cyclic voltammetric scanning from −0.5 to 1.2 V in 0.1 M HCl containing 10 mM EDOT monomer and a 10 mM mixture of K4[Fe(CN)6] and FeCl2 for 10 cycles at the scan rate of 50 mV s−1. An aliquot of uricase solution (4 μL) containing 25 mg mL−1 uricase (100 U mL−1) and 10 mg mL−1 BSA was deposited onto the PEDOT:PB/LIG and dried at 4 °C for 2 h, followed by the addition of 2 μL of a chitosan solution (1 wt%, in 1% acetic acid). The final enzyme electrode (Uricase/PEDOT:PB/LIG) was stored at 4 °C in a sealed petri dish when not in use and was hydrated with either PBS or artificial wound exudate on the working electrode area. The reference electrode surface was stencil-printed with Ag/AgCl and dried under 120 °C on a hotplate for 2 min. The counter LIG electrode was used without further modification.
PPy-based drug carrier
PPy:Cipro/LIG was deposited by potentiostatic polymerization under 0.7 V in 0.1 M HCl solution containing 0.2 M pyrrole and 50 mM Cipro until the charge reached 1.5 Coulomb. The PPy:Cipro/LIG was then vigorously rinsed with water to remove excess monomer on the surface, followed by soaking the electrode in 0.1 M KCl for 1 min to dissolve the attached Cipro on the surface. The reference electrode and counter electrode were prepared using the same procedures as those described for the UA biosensor above.
Performance measurement
Multiplex sensor and drug delivery
The performance of the pH sensor, UA biosensor and drug delivery electrode systems was characterized in a cell containing supporting electrolytes. The response of the pH sensor was characterized by measuring the EMF between the working and the reference electrode in Britton-Robinson buffer with 0.1 M KCl with varied pH of 4‒10. The response of the PEDOT:PB/LIG electrode to H2O2 was conducted by cyclic voltammetry or amperometry with/without 5 mM H2O2 or successive addition of H2O2 in 0.1 M PBS with 0.1 M KCl (pH = 7.5). Amperometric UA biosensing was performed under a static condition in 0.1 M PBS. For the drug delivery electrode system, the electrically triggered release of Cipro was conducted at various potentials at either a fixed value or alternating values with a frequency of 0.05 Hz. Passive release evaluation was also performed by immersing the electrode in a medium without electrical stimulation. The amount of released Cipro was determined via UV/vis spectroscopy at an absorption of 277 nm. The total amount of Cipro loaded in the PPy:Cipro/LIG was estimated by UV/Vis spectroscopy of the medium from an electrically triggered release at 0.6 V for 24 h followed by vigorous bath sonication for 1 h.
In-vitro evaluation
The evaluation was performed in an artificial wound exudate on porcine skin41,42. The pH sensor and the UA biosensor were integrated into one sensor array. Porcine skin samples obtained from a local retail store were cleaned and disinfected with ethanol (70% v/v) and PBS solution (with 0.1 M KCl, pH = 7.5). The epidermis layer was removed by a sterile scalpel creating an exposed dermis layer measuring 1.8 × 1.2 cm2 to simulate a wound. The artificial wound exudate was prepared by diluting human serum with a PBS solution (with 0.1 M KCl, pH = 7.5) with a dilution factor of 5, which resulted in an artificial wound exudate with a pH value of 7.23 and a UA level of 0.10 mM. The pH value of the artificial wound exudate was adjusted to the desired values (7, 8, and 9) by adding 5 M KOH or 5 M H3PO4. The UA level in the artificial wound exudate was spiked to be 0.10, 0.20, 0.40 and 0.60 mM. The pH value and the UA concentration were then simultaneously measured using the fabricated pH sensor and UA biosensor attached to the dermis area of the skin that was drop-casted with 100 μl of artificial wound exudate. The measured results were compared to the values obtained by a commercial pH meter and colorimetric assay using a UA assay kit, respectively.
Evaluation of the drug delivery was performed by attaching the drug delivery electrode system to the dermis area of the skin that was drop-casted with 100 μl of artificial wound exudate. Cipro was released into the artificial wound exudate by applying 0.6 V electrical stimulation for 5 min. The artificial wound exudate was collected for the antibacterial activity test against Gram-negative E. coli using a disc diffusion assay. All utensils were sterilized by autoclaving at 121 °C for 15 min prior to each microbiological experiment. The culture medium was prepared by dispensing 15 mL of LB Broth medium with agar (25 g L−1 for LB Broth and 15 g L−1 for agar, at around 50 °C) into a petri dish (90 mm), which was cooled to room temperature to form a solid agar. Then 100 μL of E. coli suspension was uniformly spread over the agar plate, and the resulting E. coli lawns were air-dried. The collected artificial wound exudate samples from different patches (20 μL) on the porcine skin were loaded on sterile discs (6 mm) of Whatman filter paper, followed by placing the loaded disc on the E. coli lawns. After incubating the plate at 37 °C for 12 h, the zone of inhibition (ZOI) around the disc on the E. coli lawns was measured to evaluate the antibacterial activity.
FPCB integration
A FPCB, consisting of three analog front ends (AFE), a microcontroller, a Bluetooth module, and a lithium-ion battery, was designed and fabricated as an electrochemical analyzer for signal acquisition from the sensor array and electrically triggered drug release. The circuit diagram (Supplementary Fig. 19) was designed using Altium Designer. The customized PCBs based on the two-layer printed PCB design were manufactured on conventional rigid FR-4 (fiberglass-reinforced epoxy resin) and flexible polyimide substrate, followed by the surface-mounting of all the components on the PCB by manual soldering (Supplementary Fig. 20). Two different low-power and configurable AFEs, LMP91000 and LMP91200 (Texas Instruments, USA), were accommodated on the PCB for chronoamperometry applied for the three-electrode system in UA biosensing and for the electrically triggered drug release, and potentiometry using a two-electrode system in pH sensing, respectively. A microcontroller (STM32F, STMicroelectronics) was used for potential digitization, signal processing, wireless data communication and peripheral controls. This is facilitated through a Bluetooth Low Energy (BLE, JDY-23A) module to an electronic device (e.g., laptop, phone or tablet). The firmware was written and loaded into the microcontroller via Keil Uvision5. The PCB was interfaced with the multiplex sensor and drug delivery patch using an eight-pin connector. The PCB was powered by a rechargeable lithium-ion polymer battery (170 mAh, 3.7 V, Renata SA), and the battery output was regulated via a low dropout regulator (LDR, MIC5205, Microchip) to supply a precise 3.3 V digital and analog power supply. The assembly of the 3D array was achieved by a xurographic technique according to our earlier report43 using a robotic cutting device.
Data availability
The authors declare that all the relevant data are available within the paper and its Supplementary Information file or from the corresponding author upon reasonable request.
References
Sun, X. et al. A review of recent advances in flexible wearable sensors for wound detection based on optical and electrical sensing. Biosensors 12, 10 (2021).
Scalamandré A., Bogie K. M. Smart technologies in wound prevention and care. In: Innovations and Emerging Technologies in Wound Care. Elsevier (2020).
O’Callaghan, S., Galvin, P., O’Mahony, C., Moore, Z. & Derwin, R. ‘Smart’wound dressings for advanced wound care: a review. J. Wound Care 29, 394–406 (2020).
McLister, A., Mathur, A. & Davis, J. Wound diagnostics: deploying electroanalytical strategies for point of care sensors and smart dressings. Curr. Opin. Electrochem. 3, 40–5 (2017).
Mostafalu, P. et al. Smart bandage for monitoring and treatment of chronic wounds. Small 14, 1703509 (2018).
Gethin, G. The significance of surface pH in chronic wounds. Wounds UK 3, 52 (2007).
Kassal, P. et al. Smart bandage with wireless connectivity for uric acid biosensing as an indicator of wound status. Electrochem. Commun. 56, 6–10 (2015).
Pal, A. et al. Early detection and monitoring of chronic wounds using low-cost, omniphobic paper-based smart bandages. Biosens. Bioelectron. 117, 696–705 (2018).
Saghazadeh, S. et al. Drug delivery systems and materials for wound healing applications. Adv. Drug. Deliv. Rev. 127, 138–166 (2018).
Derakhshandeh, H., Kashaf, S. S., Aghabaglou, F., Ghanavati, I. O. & Tamayol, A. Smart bandages: the future of wound care. Trends Biotechnol. 36, 1259–1274 (2018).
Mostafalu P. et al. Smart flexible wound dressing with wireless drug delivery. In: 2015 IEEE biomedical circuits and systems conference (BioCAS). IEEE (2015).
Bagherifard, S. et al. Dermal patch with integrated flexible heater for on demand drug delivery. Adv. Healthc Mater. 5, 175–184 (2016).
Gao, W., Ota, H., Kiriya, D., Takei, K. & Javey, A. Flexible electronics toward wearable sensing. Acc. Chem. Res. 52, 523–533 (2019).
Li, Q. et al. Review of printed electrodes for flexible devices. Front. Mater 5, 77 (2019).
Xu, G. et al. Battery‐free and wireless smart wound dressing for wound infection monitoring and electrically controlled on‐demand drug delivery. Adv. Funct. Mater 31, 2100852 (2021).
Gao, Y. et al. A flexible multiplexed immunosensor for point-of-care in situ wound monitoring. Sci. Adv. 7, eabg9614 (2021).
El-Kady, M. F., Strong, V., Dubin, S. & Kaner, R. B. Laser scribing of high-performance and flexible graphene-based electrochemical capacitors. Science 335, 1326–1330 (2012).
Lin, J. et al. Laser-induced porous graphene films from commercial polymers. Nat. Commun. 5, 8 (2014).
Meng, L., Turner, A. P. & Mak, W. C. Modulating electrode kinetics for discrimination of dopamine by a PEDOT: COOH interface doped with negatively charged tricarboxylate. ACS Appl. Mater Inter. 11, 34497–34506 (2019).
Meng, L., Turner, A. P. F. & Mak, W. C. Tunable 3D nanofibrous and bio-functionalised PEDOT network explored as a conducting polymer-based biosensor. Biosens. Bioelectron. 159, 112181 (2020).
Cao, D., Martinez, J. G., Hara, E. S. & Jager, E. W. Biohybrid variable‐stiffness soft actuators that self‐create bone. Adv. Mater 34, 2107345 (2022).
Alshammary, B., Walsh, F. C., Herrasti, P. & Ponce de Leon, C. Electrodeposited conductive polymers for controlled drug release: polypyrrole. J. Solid State Electrochem. 20, 839–859 (2016).
Yoon, J. H. et al. High performance flexible pH sensor based on polyaniline nanopillar array electrode. J. Colloid Interface Sci. 490, 53–58 (2017).
Hui, Y., Bian, C., Xia, S., Tong, J. & Wang, J. Synthesis and electrochemical sensing application of poly (3, 4-ethylenedioxythiophene)-based materials: a review. Anal. Chim. Acta 1022, 1–19 (2018).
Meng, L., Turner, A. P. F. & Mak, W. C. Conducting polymer-reinforced laser-irradiated graphene as a heterostructured 3D transducer for flexible skin patch biosensors. ACS Appl. Mater Inter. 13, 54456–54465 (2021).
Ting, M. S., Narasimhan, B. N., Travas-Sejdic, J. & Malmström, J. Soft conducting polymer polypyrrole actuation based on poly (N-isopropylacrylamide) hydrogels. Sensor Actuat. B-Chem. 343, 130167 (2021).
Puiggalí-Jou, A., Del Valle, L. J. & Alemán, C. Drug delivery systems based on intrinsically conducting polymers. J. Control Release 309, 244–264 (2019).
Moya, A. et al. Stable full-inkjet-printed solid-state Ag/AgCl reference electrode. Anal. Chem. 91, 15539–15546 (2019).
Chen, J., Cheng, Z., Yuan, Y., Zhang, J. & Cao, J. J. R. A. Shape-controllable nanofibrous membranes with well-aligned fibers and robust mechanical properties for PM 2.5 capture. RSC Adv. 9, 17473–17478 (2019).
Mousavi, Z., Granholm, K., Sokalski, T. & Lewenstam, A. An analytical quality solid-state composite reference electrode. Analyst 138, 5216–5220 (2013).
Rahimi, R. et al. A low-cost flexible pH sensor array for wound assessment. Sensor Actuat B-Chem 229, 609–617 (2016).
Ricci, F. & Palleschi, G. Sensor and biosensor preparation, optimisation and applications of Prussian Blue modified electrodes. Biosens. Bioelectron. 21, 389–407 (2005).
Karyakin, A. A. Prussian blue and its analogues: electrochemistry and analytical applications. Electroanalysis 13, 813–819 (2001).
Sassolas, A., Blum, L. J. & Leca-Bouvier, B. D. Immobilization strategies to develop enzymatic biosensors. Biotechnol. Adv. 30, 489–511 (2012).
Meng, L., Chirtes, S., Liu, X., Eriksson, M. & Mak, W. C. A green route for lignin-derived graphene electrodes: a disposable platform for electrochemical biosensors. Biosens. Bioelectron. 218, 114742 (2022).
Esrafilzadeh, D., Razal, J. M., Moulton, S. E., Stewart, E. M. & Wallace, G. G. Multifunctional conducting fibres with electrically controlled release of ciprofloxacin. J Control Release 169, 313–320 (2013).
Hosseini, A., Sohouli, E., Gholami, M., Sobhani-Nasab, A. & Mirhosseini, S. A. Electrochemical determination of ciprofloxacin using glassy carbon electrode modified with CoFe2O4-MWCNT. Anal. Bioanal. Electrochem. 11, 996–1008 (2019).
Feiner, R. et al. A stretchable and flexible cardiac tissue–electronics hybrid enabling multiple drug release, sensing, and stimulation. Small 15, 1805526 (2019).
Qu, J., Zhao, X., Ma, P. X. & Guo, B. Injectable antibacterial conductive hydrogels with dual response to an electric field and pH for localized “smart” drug release. Acta Biomaterialia 72, 55–69 (2018).
Seyfoddin, A. et al. Electro-responsive macroporous polypyrrole scaffolds for triggered dexamethasone delivery. Eur. J. Pharm. Biopharm. 94, 419–426 (2015).
Mirani, B. et al. An advanced multifunctional hydrogel‐based dressing for wound monitoring and drug delivery. Adv. Healthc. Mater 6, 1700718 (2017).
Zhu, Y. et al. A multifunctional pro‐healing zwitterionic hydrogel for simultaneous optical monitoring of pH and glucose in diabetic wound treatment. Adv. Funct. Mater 30, 1905493 (2020).
Kongkaew, S. et al. Craft-and-Stick Xurographic manufacturing of integrated microfluidic electrochemical sensing platform. Biosensors 13, 446 (2023).
Acknowledgements
We acknowledge the Sweden strategic research area Security Link for financial support. W.C. Mak acknowledges the financial support from the startup grant of The Chinese University of Hong Kong.
Funding
Open access funding provided by Linköping University.
Author information
Authors and Affiliations
Contributions
L.M., M.E., and W.C.M. conceived the concept. L.M. performed the experiments and analyzed all data for pH sensor, UA biosensor, drug delivery, in vitro measurements, and device integration. S.L. performed the design, printing, and testing of the FPCB. B.A.B. supplied bacterial culture protocol. L.M. wrote the original draft. L.M., M.E., and W.C.M. revised the manuscript. M.E. and W.C.M. supervised the project jointly.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Sina Naficy and Sahika Inal for their contribution to the peer review of this work. Primary Handling Editors: Rona Chandrawati and John Plummer. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meng, L., Liu, S., Borsa, B.A. et al. A conducting polymer-based array with multiplex sensing and drug delivery capabilities for smart bandages. Commun Mater 5, 28 (2024). https://doi.org/10.1038/s43246-024-00469-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00469-5
