
Abstract
New visualization methods can be utilized to resolve structures at resolutions that were previously unachievable. 3D images of porous materials with high resolution have been so far obtained using transmission electron tomography or focused ion beam coupled with scanning electron microscopy. For these methods, ultra-vacuum is required, and only a small volume of the sample is visualized. Here, we demonstrate the application of ptychographic X-ray computed tomography for the visualization of soft matter with a resolution of 26 nm over large fields of view. Thanks to the high-penetration depth of the X-ray beam, we visualize the 3D complex porous structure of polyetherimide hollow fibers in a non-destructive manner and obtain quantitative information about pore size distribution and pore network interconnectivity across the whole membrane wall. The non-destructive nature of this method, coupled with its ability to image samples without requiring modification or a high vacuum environment, makes it valuable in the fields of porous- and nano-material sciences enabling imaging under different environmental conditions.
Similar content being viewed by others


Introduction
Porous materials have essential functions in nature, whether for water and nutrient transport in plants and other biological systems or for the storage of oil and water in rock reservoirs. Synthetic polymeric materials are key for chromatography and membrane separation processes1,2,3. Their application is well-established in water desalination4,5,6,7, hemodialysis8, gas separation9, and is expanding in nanofiltration for chemical separations3,10,11,12. The advantages of membrane technology are low energy requirement, and low carbon footprint combined with a compact design that allows packing a large membrane area within a small volume8. Effective membranes have a complex porous structure to secure selectivity, mechanical stability, and fast transport characteristics. Porosity and interconnectivity determine the membrane performance.
Electron microscopy has enabled a better understanding of transport mechanisms and facilitated the development of membranes for many purposes. However, electron microscopy requires a vacuum. Samples are usually dried and coated with a thin metallic layer for analysis, although the morphology of biological and synthetic polymeric systems can be affected by the environment in which they operate, for instance by swelling. Furthermore, as the pore sizes decrease to the nanoscale, there is a trade-off between resolution and total sampling volume. X-ray tomography could be an advantageous alternative. It is a practical non-destructive visualization method and has been successfully applied for medical and industrial applications13,14,15. The sampling volume is much larger than imaged in typical electron microscopy experiments and the analysis can be done in a regular operation environment without the need for vacuum or additional coating. However, its maximum resolution for soft materials has been limited to a micrometer range or hundreds of nanometers16,17 even for the best laboratory instrumentation and operation conditions17.
This is now changing with the implementation of the latest sources of synchrotron radiation, which can provide nanoscale resolution keeping the advantages of high sampling volume in a diverse environment. Ptychographic X-ray computed tomography (PXCT) is a powerful non-destructive phase-contrast imaging in situ technique that uses a series of 2D projections of the sample in different subsequent angles processed into a 3D high-resolution reconstruction across its volume with quantitative information, for instance on porosity and interconnectivity18,19,20. In this work, these features of PXCT are demonstrated in the example of polymeric membranes.
Membranes are available in different configurations. Among them, hollow fibers have been successful in hemodialysis, seawater desalination, and gas separation due to their effective packing in modules with large surface areas3,8. They are formed by a continuous spinning process in which a polymer dope solution is extruded through an annular die; a coagulating bore fluid is simultaneously injected, and the incipient fiber is collected in a water bath. The exchange between solvent and water or bore fluid induces a liquid–liquid phase separation. The polymer-lean phase forms the pores, while the polymer-rich phase progressively gels and solidifies giving rise to a complex structure21,22. The morphology depends on the polymer dope formulation, bore fluid composition, the gap between the spinneret and the water bath, and the temperature of different parts23.
Porosity gradients and hierarchical structures are frequently intentionally designed to optimize the membrane transport and drive their applications in challenging separations1,24,25. Imaging the membrane structure is, therefore, essential26. The most frequently used techniques for morphological analysis of porous membranes are scanning electron microscopy (SEM), atomic force microscopy (AFM), and transmission electron microscopy (TEM), which mainly provide a 2D image with only restricted information (examples in Supplementary Fig. 1).
3D imaging allows us to better understand the porosity at different layers of the membrane and their interconnectivity26,27. Demonstrated methods are serial block face SEM, focused ion beam (FIB) SEM, and TEM tomography26,28,29. Each of them has advantages and limitations reflected in the already mentioned trade-off between the maximum resolution and the total analyzed volume of the sample. Of the mentioned methods, TEM tomography provides the highest resolution (<1 nm), being the most adequate tool for the characterization of fine structures, such as thin film composite membrane active layer30,31 or block copolymer assemblies28,29. However, the volume that can be effectively analyzed is relatively small: typically, 2–5 μm wide and 100 nm thick. Thinner slices are needed to reduce the excess of scattered electrons and form a clear image resulting from differences in electron density over the analyzed area.
3D images via FIB-SEM are obtained by the successive removal of thin slices of the sample, by the incidence of an ion beam that exposes a new, deeper cross-section, which is imaged by the SEM electron beam26. For the SEM analysis, to reduce the charging effect, soft matter samples need to be typically sputter-coated with a few-nm layer of gold. To minimize the damages caused by the FIB, an additional protective layer of Pt is deposited at the region of interest prior to FIB milling26,32,33. The serial block face SEM technique is also based on the removal of thin sample slices (typically 10 nm) but using a diamond knife inside the microscope26,34. This is a method frequently applied for biological samples35,36 and in an analogous procedure, the membrane material requires its stabilization within the epoxy resin and staining with heavy atoms like osmium or ruthenium, similar to preparation for TEM analysis26. While FIB-SEM and serial block face SEM offer a higher sampling volume than TEM tomography, their resolution is lower. As mentioned before, a high vacuum is required for all electron microscopy techniques. The only exception is the environmental SEM, where a lower vacuum is used, but this compromise leads to a lower resolution37. Polymeric membranes, like biological materials, can swell in water and solvents, depending on their composition and functional groups. This again affects selectivity and transport performance. The response of porous polymeric materials to stimuli like pH, light, and ionic strength is being used in the development of smart devices as sensors or controlled delivery38,39,40,41. However, the in situ morphological observation with an adequate resolution has been only indirectly possible for instance using cryogenic SEM38.
In contrast to electron microscopy, PXCT could be used for in situ experiments in environments close to the real application allowing the analysis of a large volume. Traditionally, synchrotron-source X-rays have been mostly used in reciprocal space, but the introduction of coherence-based methods at the synchrotron radiation sources in the last decade has enabled diffraction patterns to be phased and inverted to form lensless images42,43. The implementation of 4th generation synchrotron sources provides an enhanced and highly coherent flux of X-rays for coherent diffractive imaging19, with a larger coherent fraction that allows faster data collection, it is possible to achieve nanoscale resolution in 3D with larger and more representative fields of view.
PXCT is a scanning modality of coherent diffractive imaging and has already provided high-resolution images for soft matter and hierarchical structures such as cells44 and bones18. Besides, this is a versatile technique in terms of sample environments allowing for experiments in operando or in situ conditions, via for example humidity control, which would be valuable for highly hydrophilic membranes used in water20. Here, we applied synchrotron source coherent diffractive imaging PXCT to image nanoporous materials and demonstrate the advantages using for example hollow fibers made of polyetherimide (PEI). The obtained diffraction patterns were reconstructed into 3D images with a voxel size of 23 nm × 23 nm × 23 nm. The visualization represents a whole hollow fiber membrane wall from the shell to the lumen side, with volumes of 28,000 and 36,000 µm3 using fields of view of 44 × 84 and 38 × 86 µm2. The obtained spatial resolution of 26 nm corresponds to the highest 3D resolution achieved up to date for a polymer membrane material (or any porous polymer material) using X-ray-based techniques and pushes the limits of the available image technologies for non-crystalline porous- and nanomaterials. The 3D image reconstruction of a representative membrane volume with high resolution, without the preparation steps required for electron microscopy analysis, and therefore under conditions like those observed under operation, enables a much more comprehensive representation of the porous systems with quantitative information to better understand the transport phenomena, the function of the structure and its formation mechanism to guide future developments of the membrane technology. Furthermore, since the method is non-destructive, it can be applied for in situ measurements to study, for example, the changes of nanostructures as a response to different environmental conditions.
Results and discussion
Membrane formation and characterization
Two hollow fibers were prepared by spinning a dope solution consisting of 20 wt% PEI, 10 wt% diethylene glycol (DEG), and 70 wt% N-methylpyrrolidone (NMP) through a spinneret while injecting bore fluids of two different compositions: 95 wt% NPM/5 wt% water or 90 wt% ethylene glycol (EG)/10 wt% water (Fig. 1). Details of the characterization are available in the Supplementary Methods. The produced fibers are hydrophobic with a water contact angle of 88 ± 3o. The permeance of these hollow fibers to water was evaluated, with that prepared with bore fluid containing NMP being less permeable (285 ± 18 L m−2 h−1 bar−1) than that prepared with EG (341 ± 19 L m−2 h−1 bar−1). As for their selectivity, bore fluids with NMP and EG led to membranes that, respectively, reject 90% of molecules of molecular weight 80 and 55 kg mol−1, or higher. Changes in morphology and dimensions are evident. Visible changes are seen in the total fiber wall thickness (variation from 60 to 80 μm), lengths of internal finger-like cavities (dimensions up to 45 μm), the thickness of a characteristic shell denser layer, and the distribution of sponge-like porous structures (Fig. 1). The morphology is highly affected by the kinetics and the mechanism of phase separation as the solvent from the dope solution is exchanged by the non-solvent components of the bore fluid and coagulation bath during the fiber spinning. The speed of this exchange depends mainly on the fluid viscosities and the mixing enthalpy. Furthermore, a bore fluid containing a large content of solvent reduces the osmotic driving force for the solvent to be transferred to the bore fluid. The polymer/solvent/nonsolvent phase diagram guiding the phase separation is typically delineated by binodal and spinodal curves. A stable homogeneous solution exists in a good solvent (thermodynamic condition outside the area limited by the binodal curve); the separation into two coexisting phases is favored with a large content of nonsolvent (area limited by the spinodal curve); a metastable thermodynamic condition is confined between the binodal and spinodal curves. With a slow solvent–nonsolvent exchange, the system can remain a long time in the metastable condition, and the predominant phase separation mechanism is nucleation and growth with a high probability of leading to a sponge-like pore structure. A fast exchange tends to promote spinodal decomposition, with the formation of a porous structure with more interconnected pores. In addition to this interplay of mechanisms leading to fine pore formation, an ultrafast intrusion of the nonsolvent into a relatively low viscous dope solution leads to the formation of finger-like cavities with fast polymer coagulation around them. With 95 wt% of NMP in the bore fluid, only a small osmotic driving force is built for solvent exchange and a small interfacial tension between the dope solution and bore fluid, resulting in a membrane with a thick sponge layer on the lumen side. The intrusion of pure water from the bath leads to finger-like cavities close to the shell side. No NMP is present in the bore fluid of the second kind of hollow fibers. Both water and EG would have a high osmotic driving force for fast exchange and intrusion with finger-like cavities starting both in the shell and lumen sides23. The SEM 2D images in Fig. 1 provide partial information about the morphology of a fully dry membrane.

a Spinneret and hollow fiber formation. b, e SEM of cross-sections of hollow fibers. c, f SEM of shell surfaces of hollow fibers. d, g SEM of lumen surfaces of hollow fibers prepared bore fluid constituted by b–d 95 wt% NMP/5 wt% water solution and e–g 90 wt% EG/10 wt%. Dope solution: 20 wt% PEI, 10 wt% DEG, and 70 wt% NMP.
Ptychographic x-ray computed tomography of hollow fibers
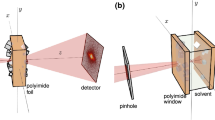
A highly coherent X-ray beam produced by the SIRIUS light source was used at the Cataretê beamline45 for PXCT visualization of the two hollow fibers samples (Fig. 2). PXCT is a lensless coherent X-ray technique where a specimen is raster scanned in two dimensions by a coherent probe, recording the diffraction patterns in the far-field. In such a lensless technique, the resolution is limited by wavelength, detector distance, and detector size. In our setup, a 5 μm pinhole was used to select a portion of the collimated coherent beam to illuminate the sample. The illuminated regions are overlapping (~80%) to reconstruct the image of the sample, retrieving its amplitude and phase. Coupled with the tomography method (collecting diffraction patterns at different projection angles), PXCT enables to obtain three-dimensional high-resolution images. When coupled with a highly coherent X-ray beam from a 4th generation synchrotron, PXCT reveals the three-dimensional structure of nanomaterials over large fields of view, with quantitative electron density contrast. The image of the object is retrieved by an advanced phase retrieval algorithm as the diffracted wave is proportional to the Fourier transform of the object46. The PXCT experimental setup and diffraction patterns with respective 2D electron density projections are presented in Fig. 2a–d.

a Experimental setup, where the coherent X-ray beam is defined by a pinhole and reaches the sample. The latter is scanned two-dimensionally, and an X-ray diffraction pattern is collected at each position. The sample is then rotated and the 2D scans are repeated to obtain a 3D dataset. b Photograph of the experimental setup at the Cateretê beamline. c, d 2D Fourier space coherent X-ray diffraction patterns for hollow fibers prepared with 95 wt% NMP/5 wt% water or 90 wt% EG/10 wt% EG as bore fluids and examples of their respective 2D electron density projections.
Reconstruction of ptychographic diffraction patterns
The ptychographic reconstruction procedure consisted in applying a combination of the relaxed averaged alternating reflections and alternating direction method of multipliers methods, as described by Glowinski and Le Tallec47, Luke48, and Chang et al.49. Assembling all 2D reconstructions led to the full 3D dataset. We cropped a region of interest around the sample and unwrapped the phase for each projection. This provided a tomogram that did not satisfy the Ludwig–Helgason conditions, necessary for tomographic reconstructions since the projections were misaligned in both vertical and horizontal directions. Alignment of projections has been proposed in the literature, for instance as an iterative refinement of each projection by the use of the filtered back projection inversion scheme50. In this work, we applied an approach that combines the scheme proposed by Prince et al.51, using a least-squares approach to find shifts and align the frames in the horizontal direction. Subsequently, a momentum-based correction is applied to finish alignment in the vertical direction. This approach is parallelized and is therefore not time-consuming. Based on the corrected and consistent tomogram, reconstructions were obtained using an expectation-maximization-based method52.
3D images of hollow fibers: analysis of porosity and interconnectivity
The high-performance computer reconstruction of the collected 2D projections resulted in detailed 3D images of the hollow fibers, with a voxel size of 23 nm × 23 nm × 23 nm. Such a detailed model contains not only visual but also quantitative information, and it was used to evaluate the porosity gradient across the membrane walls for both hollow fibers membranes, as shown in Fig. 3.

Membranes prepared with the following solutions as bore fluid: a 95 wt% NMP/5 wt% water and b 90 wt% EG/10 wt% water. The depth of 0 μm corresponds to the surface of the shell side.
A dense skin layer is seen on the shell side of the hollow fiber prepared with NMP/water in the bore fluid in a depth range between 0 and 4 μm. A porosity lower than 5% could be quantified, followed by a gradual increase in porosity up to 59% at a depth of 33.8 μm, where a great part of the morphology consists of finger-like cavities. Closer to the lumen (depth of 41–50 μm), a sponge layer is observed, with a decrease of porosity to 20–30%, followed by some increase to 47% at 52.4 μm, due to the distinctive part with increased pore sizes. The sponge layer becomes denser again approximating the lumen side, with a porosity of 28% at 54 μm and 15% of porosity just close to the lumen surface. In comparison to this membrane, the one prepared with EG/water bore fluid has a thinner (1.5 μm) layer with porosity as low as 1–3% within. Then, an open sponge structure is present with porosity up to 46% down to a depth of 6.5 μm. The porosity decreases to 30% just before the first finger-like cavities initiate, corresponding to a gradual growth until reaching the highest porosity of 74% at 46 μm. The porosity drops to 37% before the second layer of finger-like cavities. It increases to 67% at a depth of 55.6–58.7 μm, after which a gradual drop was observed until reaching 2.8% close to the lumen. The total porosity of the two membranes differs, with the double-symmetrical hollow fiber prepared with EG reaching porosities of 74% with two denser layers on the shell and the lumen side, respectively. In comparison, 59% was quantified for the single-symmetrical hollow fiber prepared with NMP, which has a distinguished denser layer only on the shell side of the fiber. The pore sizes in the dense layers with porosity lower than 5% are below the resolution limits of what could be achieved with the method demonstrated here. Pores below 26 nm remain undetected. Quantitative information regarding the pore size above the resolution limits can be easily extracted from the 3D reconstructions. Apart from the porosity, we were able to study the evolution of the diameter size of every detected pore channel across the membrane thickness within the resolution limits (26 nm diameter), to evaluate the pore size distribution and changes across the entire membrane wall, from the shell to the lumen side, as presented in Fig. 4.

Hollow fibers prepared with bore fluids constituted by a 95 wt% NMP/5 wt% water and b 90 wt% EG/10 wt% water. The black points represent the diameter change of every pore channel detected across the membrane that is within the resolution limits (26 nm). The depth of 0 μm corresponds to the surface of the shell side.
For obtaining the pore diameter, the large cavities (represented with the blue color in the 3D visualizations in Fig. 3) were excluded, as their influence on the average pore size will be too high to evaluate the sub-micron pores. The average pore diameter for both hollow fibers was within the range of 50–200 nm, while it must be considered that pores below 26 nm remain undetected, due to resolution limits. For both fibers, the pore diameter did not increase within the parts’ large cavities. The size increase was instead well aligned with the highest porosity regions in the cavity-free volumes. Therefore, the increased porosity in these regions corresponds to the increased pore diameters and reflects (after the cavities region) the second most open parts of the membranes, where the expected mass transfer should be least constrained. Such a conclusion will be valid only if the measured pores are not dead-ended, in other words, the pores should be interconnected across the membrane from the shell to the lumen side. Therefore, further understanding of the complexity and interconnectivity of the pore network is needed. A quantitative analysis was obtained and is shown in Fig. 5. Detailed 3D reconstructions of the porous structures of the two membranes are shown in Supplementary Movies 1–6.

a, b 3D structure of the whole sample volumes and c, d high magnifications of selected parts; e, f 3D visualization of the pore interconnectivity, the blue color representing the main interconnected network, the other colors corresponding to unconnected pores. Hollow fibers prepared with bore fluids constituted by a, c, e NMP/water and b, d, f EG/water.
For both kinds of membranes, most pores were interconnected, however, there are major differences between the distribution within the two samples, as discussed above. The higher porosity across the membrane wall of the membranes prepared with EG/water instead of NMP/water is also visible in the interconnectivity models. While most of the pores of the membranes prepared with EG/water belonged to one interconnected network, there were also smaller networks disconnected from most of the pores. These disconnected pores were distributed across the whole membrane volume, yet particularly pronounced closer to the lumen side of the membrane wall. Even though the membrane prepared with EG/water has a thicker wall and smaller selective pores, it has higher water permeance. This can be explained by the higher porosity across the membrane wall and higher interconnectivity. The selectivity is driven by the smallest selective pores in the membrane. The membranes prepared with EG/water reject smaller molecules than those prepared with NMP/water. Yet, the former has higher permeance, which indicates that the impact of the membrane structure on the mass transport properties is far beyond the selective layer and highlights the importance of understanding the porous structure across the membrane wall.
Conclusions
The highly coherent X-ray beam generated by the 4th generation synchrotron source allows for the non-destructive analysis via PXCT with nanoscale resolution. The reconstruction of the X-ray coherent diffraction patterns from different projection angles, not only facilitated the visualization of complex porous morphology throughout the entire membrane wall volume but also provided a detailed 3D quantitative model of the porous network. The presented PXCT method is not limited to the measurements of porous structures but can be employed for resolving the nanostructure details in large samples within soft matter. Furthermore, thanks to the high-penetration depth of X-rays, PXCT allows for the analysis of the samples where the sectioning for electron microscopy methods is not feasible. The lack of vacuum requirements and the absence of sample modification, such as heavy metal staining, enable in situ analysis of samples in different environmental conditions. These advantages, coupled with the non-destructive nature of this technique, make it highly applicable in material science and biology for the understanding of complex porous- and nanomaterials offering nanometer-scale resolutions for large sample volumes and providing valuable quantitative information.
Methods
Hollow fibers fabrication
Polyetherimide hollow fiber membranes were prepared by non-solvent-induced phase separation using a dry-jet wet-spinning process. The dope solution consisted of 20/10/70 wt% polyetherimide (PEI)/diethylene glycol (DEG)/N-methyl-2-Pyrrolidone (NMP). PEI was obtained from Sabic, Saudi Arabia, and DEG and NMP from Merck, Germany. The mixture was stirred at 70 °C for 3 days until a clear solution was obtained, followed by degassing under vacuum. The solution was then transferred to a spinning tank and left undisturbed overnight prior to spinning. Information about spinning conditions is presented in Supplementary Table 1. The spun fibers were washed with water for three days to remove residual solvent, dried, and stored prior to further analysis.
Ptychographic X-ray computed tomography measurements
The PXCT measurements were carried out at the Cateretê beamline45 at the Sirius synchrotron facility53. Direct illumination from the first undulator harmonic (peak energy = 3.8 keV) was used for the 3D PXCT experiments, with a bandwidth of Δλ/λ = 2.06%. A 5 µm pinhole was used to define the confined coherent illumination impeding on the sample. We recorded the coherent diffraction patterns using a PIMEGA 540D detector (55 μm pixel size and 3072 × 3072 pixels) (PiTec, SP, Brazil) placed in a vacuum, 10 m downstream of the sample. 2D ptychographic scans consisted of 2552 and 2240 diffraction patterns per projection (for hollow fibers prepared with NMP/water and EG/water as bore fluid, respectively) with an exposure time of 150 ms per diffraction pattern. Sampling position trajectories for each 2D scan were set using a grid, with an average step size of 1.25 μm, resulting in an 80% overlap between successive steps. For each step size, we added a random uncertainty of about 50%. The non-periodic steps break the scanning symmetry and avoid the emergence of periodic artifacts in the reconstructed images54. In such a way we obtained a unique grid trajectory for each 2D ptychographic experiment. 645 and 498 and tomography projections were acquired, covering an angular range of 154° and 150°, resulting in a missing wedge of 25° and 30°, for hollow fiber prepared with NMP/water and EG/water, respectively. The field of view covered in a projection across the samples was 44 × 84 and 38 × 86 μm2 (horizontal × vertical) for each of them. Because the tomography algorithm requires regular angle steps between projections, an array of regularly spaced points is created, and each projection is attributed to the array value that best approximates the real angle value. The regular mesh presented steps of 0.24° and 0.30° for hollow fibers prepared with NMP/water and EG/water, respectively. The projections with low reconstruction quality were removed prior to the alignment of the frames. In total, 498 and 169 projections were, respectively, used for the tomographic reconstruction of the membranes. Further details on the sample preparation are available in the Supplementary Methods.
Ptychographic reconstruction
The 2D ptychographic reconstructions were performed using a combination of the relaxed averaged alternating reflections and alternating direction method of multipliers methods as described in the main text. The data were binned 4 × 4. Assembling all the 2D ptychographic reconstructions led to the full 3D dataset. The resulting voxel size was 23.1 nm × 23.1 nm × 23.1 nm. The spatial resolutions of the ptychographic projections were determined by the Fourier ring correlation method (Supplementary Fig. 2), using the half-bit threshold criteria55. We applied the splitting strategy presented in the work of Koho et al.56, where a single image is split into even/odd subsets and these are correlated. The obtained resolution was 26 and 29 nm for the hollow fibers prepared with NMP/water and EG/water, respectively, without data binning, to be compared to 29.5 and 30.5 nm in the case of a 4 × 4 binning. Further details on the ptychographic image reconstruction and segmentation and on the porosity and interconnectivity calculation are available in the Supplementary Methods.
Data availability
The raw, reconstructed, and segmented data are available upon request. All datasets are stored at cloud storage of CNPEM and KAUST and multiple back-up drives.
References
Ulbricht, M. in Comprehensive Membrane Science and Engineering (eds Drioli, E., Giorno, L. & Fontananova, E.) 85–119 (2017).
Nunes, S. P. et al. Thinking the future of membranes: perspectives for advanced and new membrane materials and manufacturing processes. J. Membr. Sci. 598, 117761 (2020).
Nunes, S. P. & Peinemann, K.-V. Membrane Technology in Chemical Industry (Wiley-VCH, 2006).
Werber, J. R., Osuji, C. O. & Elimelech, M. Materials for next-generation desalination and water purification membranes. Nat. Rev. Mater. 1, https://doi.org/10.1038/natrevmats.2016.18 (2016).
Shen, J. et al. Fast water transport and molecular sieving through ultrathin ordered conjugated-polymer-framework membranes. Nat. Mater. 21, 1183–1190 (2022).
Lively, R. P. & Sholl, D. S. From water to organics in membrane separations. Nat. Mater. 16, 276–279 (2017).
Culp, T. E. et al. Nanoscale control of internal inhomogeneity enhances water transport in desalination membranes. Science 371, 72–75 (2021).
Baker, R. W. & Membrane Technology and Applications 3rd edn (John Wiley and Sons Ltd., 2012).
Koros, W. J. & Zhang, C. Materials for next-generation molecularly selective synthetic membranes. Nat. Mater. 16, 289–297 (2017).
Chisca, S. et al. Polytriazole membranes with ultrathin tunable selective layer for crude oil fractionation. Science 376, https://doi.org/10.1126/science.abm7686 (2022).
Li, S. et al. Hydrophobic polyamide nanofilms provide rapid transport for crude oil. Science 377, https://doi.org/10.1126/science.abq0598 (2022).
Sholl, D. S. & Lively, R. P. Seven chemical separations to change the world. Nature 532, 435–437 (2016).
Rubin, G. D. Computed tomography: revolutionizing the practice of medicine for 40 years. Radiology 273, S45–S74 (2014).
Villarraga-Gómez, H., Herazo, E. L. & Smith, S. T. X-ray computed tomography: from medical imaging to dimensional metrology. Precis. Eng. 60, 544–569 (2019).
Scharf, J. et al. Bridging nano- and microscale X-ray tomography for battery research by leveraging artificial intelligence. Nat. Nanotechnol. 17, 446–459 (2022).
Cramer, K. et al. Three-dimensional membrane imaging with X-ray ptychography: determination of membrane transport properties for membrane distillation. Transp. Porous Media 138, 265–284 (2021).
Batey, D. J. et al. X-ray ptychography with a laboratory source. Phys. Rev. Lett. 126, 193902 (2021).
Dierolf, M. et al. Ptychographic X-ray computed tomography at the nanoscale. Nature 467, 436–439 (2010).
Pfeiffer, F. X-ray ptychography. Nat. Photonics 12, 9–17 (2018).
Esmaeili, M. et al. Ptychographic X-ray tomography of silk fiber hydration. Macromolecules 46, 434–439 (2013).
Lipscomb, G. G. The melt hollow fiber spinning process: steady-state behavior, sensitivity and stability. Polym. Adv. Technol. 5, 745–758 (1994).
Ahmad, A. L., Otitoju, T. A. & Ooi, B. S. Hollow fiber (HF) membrane fabrication: a review on the effects of solution spinning conditions on morphology and performance. J. Ind. Eng. Chem. 70, 35–50 (2019).
Le, N. L. & Nunes, S. P. Ethylene glycol as bore fluid for hollow fiber membrane preparation. J. Membr. Sci. 533, 171–178 (2017).
Sorci, M. et al. Linking microstructure of membranes and performance. J. Membr. Sci. 594, https://doi.org/10.1016/j.memsci.2019.117419 (2020).
Hao, P., Wijmans, J. G., He, Z. & White, L. S. Effect of pore location and pore size of the support membrane on the permeance of composite membranes. J. Membr. Sci. 594, https://doi.org/10.1016/j.memsci.2019.117465 (2020).
Sundaramoorthi, G. et al. 3D membrane imaging and porosity visualization. Ind. Eng. Chem. Res. 55, 3689–3695 (2016).
Alvarez, J. et al. 3D analysis of ordered porous polymeric particles using complementary electron microscopy methods. Sci. Rep. 9, 13987 (2019).
Madhavan, P., Sougrat, R., Behzad, A. R., Peinemann, K.-V. & Nunes, S. P. Ionic liquids as self-assembly guide for the formation of nanostructured block copolymer membranes. J. Membr. Sci. 492, 568–577 (2015).
Chisca, S., Musteata, V.-E., Sougrat, R., Behzad, A. R. & Nunes, S. P. Artificial 3D hierarchical and isotropic porous polymeric materials. Sci. Adv. 4, https://doi.org/10.1126/sciadv.aat0713 (2018).
Pacheco, F., Sougrat, R., Reinhard, M., Leckie, J. O. & Pinnau, I. 3D visualization of the internal nanostructure of polyamide thin films in RO membranes. J. Membr. Sci. 501, 33–44 (2016).
Pacheco, F. A., Pinnau, I., Reinhard, M. & Leckie, J. O. Characterization of isolated polyamide thin films of RO and NF membranes using novel TEM techniques. J. Membr. Sci. 358, 51–59 (2010).
Nunes, S. P. et al. Ultraporous films with uniform nanochannels by block copolymer micelles assembly. Macromolecules 43, 8079–8085 (2010).
Zils, S. et al. 3D Visualisation of PEMFC electrode structures using FIB nanotomography. Fuel Cells 10, 966–972 (2010).
Reingruber, H., Zankel, A., Mayrhofer, C. & Poelt, P. Quantitative characterization of microfiltration membranes by 3D reconstruction. J. Membr. Sci. 372, 66–74 (2011).
Moller, M., Midtgaard, J., Qvortrup, K. & Rath, M. F. An ultrastructural study of the deep pineal gland of the Sprague Dawley rat using transmission and serial block face scanning electron microscopy: cell types, barriers, and innervation. Cell Tissue Res. 389, 531–546 (2022).
He, Q., Hsueh, M., Zhang, G., Joy, D. C. & Leapman, R. D. Biological serial block face scanning electron microscopy at improved z-resolution based on Monte Carlo model. Sci. Rep. 8 https://doi.org/10.1038/s41598-018-31231-w (2018).
Donald, A. M. The use of environmental scanning electron microscopy for imaging wet and insulating materials. Nat. Mater. 2, 511–516 (2003).
Nunes, S. P. et al. Switchable pH-responsive polymeric membranes prepared via block copolymer micelle assembly. ACS Nano 5, 3516–3522 (2011).
Liu, Z., Wang, W., Xie, R., Ju, X.-J. & Chu, L.-Y. Stimuli-responsive smart gating membranes. Chem. Soc. Rev. 45, 460–475 (2016).
Park, Y., Gutierrez, M. P. & Lee, L. P. Reversible self-actuated thermo-responsive pore membrane. Sci. Rep. 6, 39402 (2016).
Huang, T. et al. Advanced stimuli-responsive membranes for smart separation. Chem. Soc. Rev. https://doi.org/10.1039/D2CS00911K (2023).
Robinson, I. & Harder, R. Coherent X-ray diffraction imaging of strain at the nanoscale. Nat. Mater. 8, 291–298 (2009).
Miao, J., Charalambous, P., Kirz, J. & Sayre, D. Extending the methodology of X-ray crystallography to allow imaging of micrometre-sized non-crystalline specimens. Nature 400, 342–344 (1999).
Deng, J. et al. Correlative 3D x-ray fluorescence and ptychographic tomography of frozen-hydrated green algae. Sci. Adv. 4, https://doi.org/10.1126/sciadv.aau4548 (2018).
Meneau, F. et al. Cateretê: the coherent X-ray scattering beamline at the 4th generation synchrotron facility SIRIUS. Acta Crystallogr. Sect. A A77, https://doi.org/10.1107/S0108767321093995 (2021).
Miao, J., Ishikawa, T., Roinson, I. K. & Murnane, M. M. Beyond crystallography: diffractive imaging using coherent x-ray light sources. Science 348, https://doi.org/10.1126/science.aaa1394 (2015).
Glowinski, R. & Le Tallec, P. in Studies in Applied and Numerical Mathematics (Society for Industrial and Applied Mathematics, 1989).
Luke, D. R. Relaxed averaged alternating reflections for diffraction imaging. Inverse Probl. 21, 37–50 (2005).
Chang, H. et al. Advanced denoising for X-ray ptychography. Opt. Express 27, 10395–10418 (2019).
Guizar-Sicairos, M. et al. Quantitative interior x-ray nanotomography by a hybrid imaging technique. Optica 2, 259 (2015).
Prince, J. L. & Willsky, A. S. Hierarchical reconstruction using geometry and sinogram restoration. IEEE Trans. Image Process. 2, 401–416 (1993).
Fessler, J. in Handbook of Medical Imaging Vol. 2. In Medical Image Processing and Analysis (eds Michael Fitzpatrick, J. & Sonka, M.) Ch. 1, 1–70 (SPIE, 2000).
Liu, L., Milas, N., Mukai, A. H. C., Resende, X. R. & De Sá, F. H. The Sirius project. J. Synchrotron Radiat. 21, 904–911 (2014).
Thibault, P., Dierolf, M., Bunk, O., Menzel, A. & Pfeiffer, F. Probe retrieval in ptychographic coherent diffractive imaging. Ultramicroscopy 109, 338–343 (2009).
van Heel, M. & Schatz, M. Fourier shell correlation threshold criteria. J. Struct. Biol. 151, 250–262 (2005).
Koho, S. et al. Fourier ring correlation simplifies image restoration in fluorescence microscopy. Nat. Commun. 10 https://doi.org/10.1038/s41467-019-11024-z (2019).
Acknowledgements
The authors acknowledge King Abdullah University of Science and Technology (KAUST) and thank for the funding of the project 3D Image of porous polymeric membranes by in situ advanced methods, award number OSR-2019-CRG8-4040.2. The authors also thank the Brazilian Synchrotron Light Laboratory (LNLS) for the provision of the beamtime for experiment number 20221482. The authors also thank KAUST Imaging Core Lab, particularly Dr. Valentina Musteata for discussions in the starting phase of this work.
Author information
Authors and Affiliations
Contributions
R.G.: conceptualization, manuscript writing, sample preparation, ptychographic data processing, membrane characterization, scanning, and transmission electron microscopy analysis. C.C.P.: ptychographic data processing, reconstruction, and segmentation of the 3D data, manuscript writing. T.A.K.: beamline operation, code development, collection, and processing of ptychographic data, and reconstruction. E.X.S.M.: code development, ptychographic data processing, and reconstruction, manuscript writing. Y.R.T.: code development, ptychographic data processing, and reconstruction. L.U.: hollow fiber preparation and performance characterization, scanning electron microscopy. F.M.: conceptualization, manuscript writing, beamline operation, collection and processing of ptychographic data, and reconstruction. S.P.N.: conceptualization, manuscript writing, critical review, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Simone Sala and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Jet-Sing Lee. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Górecki, R., Polo, C.C., Kalile, T.A. et al. Ptychographic X-ray computed tomography of porous membranes with nanoscale resolution. Commun Mater 4, 68 (2023). https://doi.org/10.1038/s43246-023-00396-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-023-00396-x
