
Abstract
Realizing efficient near-infrared to visible photon upconversion in the solid state is pivotal for commercial applications in various fields. We previously reported a solid-state upconversion device which imitated the photovoltaic conversion mechanisms of organic solar cells. This leads to a significant improvement of up to 2.3% in the external quantum efficiency, which is two orders of magnitude higher than that of conventional devices. Here, we investigate the upconversion mechanism of this device. We examine exciton and charge dynamics using transient absorption spectroscopy and find that approximately 67% of incident photons are utilized owing to fast singlet exciton diffusion in the nonfullerene acceptor layer. Strikingly, triplet excitons are accumulated near the donor/acceptor interface, enabling accelerated triplet–triplet annihilation by a factor of more than 10.
Similar content being viewed by others
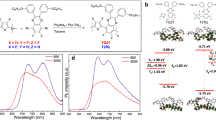

Introduction
Photon upconversion is a phenomenon in which two low-energy photons are converted into a high-energy photon1,2,3,4,5,6,7,8. Upconversion has attracted considerable interest in various research fields, such as solar cells, artificial photosynthesis, and bioimaging, because of its potential applications3,9,10. Conventional upconversion based on triplet–triplet annihilation (TTA) undergoes through a sequence of Dexter energy transfer (Fig. 1a). First, a triplet sensitizer absorbs a low-energy photon and generates a singlet exciton that is rapidly converted into a triplet exciton via intersystem crossing (ISC). The triplet exciton is transferred to an emitter material with low-lying triplet and high-lying singlet state energies. Then, the triplet exciton diffuses randomly, which incidentally can be regarded as a sequence of the Dexter energy transfer between emitter materials, and undergoes TTA when it meets with another emitter triplet, resulting in the formation of a high-energy emitter singlet, followed by rapid deactivation to the ground state by emitting upconverted photon. Solution-based TTA-upconversion has been extensively investigated; for instance, an internal upconversion quantum efficiency ηUC of up to 38% (out of a maximum of 50%)11 and a high external quantum efficiency (EQE), defined as EQE = ηUC × (1−10−A), where A is the absorbance at the excitation wavelength, of 17.4%12 have been reported. In contrast, EQEs of solid-state upconversion have lagged significantly behind those of solution systems: it remained at less than 0.1% until we presented a breakthrough report13,14,15,16,17. This is largely because of the significantly poor light-harvesting ability of conventional solid-state upconversion devices, wherein a very small amount of sensitizers is dispersed in the emitter layer or very thin layer of sensitizers is placed on the emitting layer.

a Upconversion mechanisms of conventional devices. S and E represent triplet sensitizer and emitter, respectively. Abs, TD, and TET refer to photon absorption, triplet diffusion, and triplet energy transfer, respectively. b Upconversion mechanisms of our device. A and D represent electron acceptor and donor, respectively. SD, CT, CD, BR, and BCT refer to singlet diffusion, charge transfer, charge dissociation, bimolecular recombination, and back charge transfer, respectively. c, d Chemical structures of rubrene (electron donor, emitter) and ITIC-Cl (electron acceptor, absorber). e Absorption spectra of pristine rubrene (100 nm) and ITIC-Cl (23 nm) films. Inset photograph shows the upconversion emission from rubrene after photoexcitation at 750 nm with a star-shaped LED. The inset photograph was adapted from ref. 18 with permission. Copyright 2021 Springer Nature.
We previously reported a solid-state upconversion device enabling near-infrared to visible (NIR-vis) light conversion using a completely different strategy from that of conventional upconversion devices, wherein we imitated the photovoltaic conversion mechanisms of organic solar cells (OSCs), as shown in Fig. 1b18. This leads to a significant improvement of up to 2.3% in EQE, which is two orders of magnitude higher than that of conventional devices. The key idea underpinning this concept involves first generating charges at the electron donor/acceptor (D/A) interface. Then, charge recombination leads to the formation of triplets, which then undergo TTA to form high-energy singlets, followed by upconversion emission. One of the greatest advantages of our device is that it can absorb >90% of incident NIR photons owing to the large absorption coefficient of recent novel nonfullerene electron acceptors (NFAs) in the NIR region and a large singlet exciton diffusion constant in the NFA layer (>10−2 cm2 s−1, vide infra). It should also be emphasized that, for practical applications, our devices do not require rare metals, such as Pt and Pd, which are necessary for conventional upconversion devices to increase the spin–orbit coupling of sensitizers.
Herein, we examine the exciton and charge dynamics in our device to provide a comprehensive understanding of the working mechanisms, which has not been addressed in the previous study. OSCs require a D/A interface to dissociate singlets into charges because of the low dielectric constants of organic materials. Singlet excitons diffuse to the D/A interface and undergo charge transfer, forming interfacial charge transfer (CT) states, which are bound electron–hole states trapped at the D/A interface19. If the electrons and holes that constitute CT states dissociate beyond their Coulomb capture radius, they become free carriers (FCs); otherwise, CT states undergo geminate recombination (GR) to the ground state20. On the other hand, all FCs finally undergo bimolecular recombination (BR) in our device as the device is held at open-circuit condition during upconversion measurements, at which no charges are extracted from the device. Charge recombination leads to the formation of triplet excitons, which then diffuse randomly followed by TTA and upconversion emission, as in the case of conventional upconversion devices. We show that GR does not form triplets, and triplets are generated by BR. Strikingly, TTA is accelerated owing to the exciton accumulation effect at the D/A interface. We discuss future perspectives for further improving upconversion efficiency.
Results and discussion
Upconversion emission
Our upconversion device consists of a planar heterojunction (PHJ) of rubrene (100 nm, Fig. 1c) and ITIC-Cl (23 nm, Fig. 1d) as the electron donor and acceptor, respectively (the device structure can be found in Supplementary Note 1)21. Rubrene is a well-known material in the research field of upconversion owing to its ideal singlet and triplet energy levels. In this study, rubrene was deposited as an amorphous film. ITIC-Cl is a novel NFA utilized in the research field of OSCs, exhibiting a large absorption band in the NIR region and high transparency in the visible region (Fig. 1e, the absorption spectra of the PHJ film can be found in Supplementary Fig. 2)22,23,24,25. For upconversion measurements, the film surface was covered by Al (70 nm), as in the case of OSCs; thereby, the incident light was reflected by the Al layer and again through the ITIC-Cl layer, which results in ~90% absorption of NIR light owing to the large absorption coefficient and optical interference effect (Supplementary Fig. 2)26,27. When we irradiated the lower-bandgap ITIC-Cl at 750 nm, an orange upconversion emission was observed even with the naked eye (inset photograph in Fig. 1e)18. The maximum EQE of 1.4% was achieved after photoexcitation at 750 nm with a fluence of >150 mW cm−2. EQE of our device was further improved up to 2.3% when a highly emissive dopant tetraphenyldibenzoperiflanthene (DBP) was co-deposited with rubrene owing to the higher photoluminescence (PL) quantum yield of DBP14,28. In this study, however, DBP was not introduced as our aim in this study is to examine triplet generation and decay dynamics. The mechanisms of improvement in the PL quantum yield by adding DBP have been extensively studied by Bossanyi et al.28.
Charge generation
Figure 2a shows the PL spectra of the PHJ film as well as a pristine ITIC-Cl film after photoexcitation at 750 nm to selectively excite ITIC-Cl. The PL intensity attributable to ITIC-Cl fluorescence significantly decreased in the PHJ film compared with that of the pristine ITIC-Cl film, indicating that most of ITIC-Cl singlets diffuse to the D/A interface, at which singlet CT (1CT) states are formed. By analyzing the thickness dependence of the PL quenching yield ηq, the diffusion length LS and diffusion constant DS of ITIC-Cl singlets were determined to be 19.3 ± 3.3 nm and 2.7 ± 0.9 × 10−2 cm2 s−1, respectively (see Supplementary Note 2 for more details). These values are in good agreement with those for other NFAs including ITIC derivatives29,30,31. The diffusion constant of 2.7 × 10−2 cm2 s−1 is considerably larger than that of triplets in typical triplet sensitizers (e.g. the triplet diffusion constant of 4.0 × 10−6 cm2 s−1 was reported for a typical triplet sensitizer PtOEP)32 because singlet diffusion undergoes through the Förster mechanism, which is a long-range resonant energy transfer mediated by dipole–dipole interaction19. The fraction of incident photons that are converted into 1CT states was evaluated as a product of photon absorption efficiency at the excitation wavelength (absorbance of ~0.99 at 750 nm for the PHJ film, Supplementary Fig. 2) and PL quenching efficiency (74.4%, Supplementary Table 1). As a result, we found that ~67% of incident photons were converted into 1CT states. This is in sharp contrast to conventional solid-state upconversion devices, wherein only a few portions of incident photons are utilized.

a PL intensities of the PHJ rubrene/ITIC-Cl film as well as a pristine ITIC-Cl film with the same ITIC-Cl layer thickness. The PL intensities were normalized with the peak PL intensity of the pristine film. Excitation light was irradiated from the ITIC-Cl side. b TA spectra of the PHJ film. The excitation wavelength was 800 nm with a fluence of 2.6 μJ cm−2. The pump and probe pulses were incident from the rubrene side. c, d TA spectra of a BHJ rubrene/ITIC-Cl film in the NIR (c) and visible (d) regions. The excitation wavelength was 800 nm with a fluence of 13 μJ cm−2. e Excitation fluence dependence of ITIC-Cl GSB recovery monitored at 760 nm (ITIC-Cl GSB). f Time evolution of TA signals monitored at 760 nm (ITIC-Cl GSB) and 1000 nm (ITIC-Cl anion).
Figure 2b shows the femtosecond TA spectra of the PHJ film. Here, the excitation wavelength was set to 800 nm to selectively excite the lower-bandgap ITIC-Cl. A sharp photoinduced absorption (PIA) band attributable to ITIC-Cl singlets was observed at 970 nm immediately after photoexcitation (details of assignments can be found in Supplementary Note 3)33,34. Singlets decayed on a time scale of a few tenths of picoseconds, whereas a weak and slightly redshifted PIA band attributable to ITIC-Cl anion remained over 100 ps after photoexcitation33, indicating that 1CT states are generated at the D/A interface. Charges then decayed on a nanosecond time scale.
To ensure a more quantitative discussion, the excitation fluence should be ideally lowered to eliminate the contribution of undesirable bimolecular processes, such as singlet–singlet annihilation (SSA). However, we could not eliminate the contribution of SSA even at the lowest excitation fluence of 2.6 μJ cm−2 because of the relatively fast singlet diffusion. As we measured a PHJ film with a thin ITIC-Cl layer (23 nm), it was very challenging to eliminate SSA altogether while maintaining a reasonable signal-to-noise ratio (SNR). Therefore, we also measured a bulk heterojunction (BHJ) rubrene/ITIC-Cl film, in which the donor and acceptor materials form a fine phase-separated morphology (see Supplementary Note 3 for more details)35. Figure 2c, d shows the TA spectra of the BHJ film in the NIR and visible regions, respectively. The spectral shape is similar to those of the PHJ film. However, singlets decayed within a few picoseconds in the BHJ film, considerably faster than that in the PHJ film owing to the smaller ITIC-Cl domain size; thereby, SSA can be ignored at excitation fluences below 13 μJ cm−2 (Supplementary Note 4). Charges then decayed on nanosecond time scale, as in the case of the PHJ film. To distinguish GR from BR, we measured the excitation fluence dependence of charge decay for the BHJ film (Fig. 2e) because, unlike BR, GR is independent of the excitation fluence (see Supplementary Note 5 for more details). We found that the decay kinetics of charges are independent of the excitation fluence below 24 μJ cm−2, indicating that GR is the dominant decay channel for charges on early times at these conditions. By analyzing the decay kinetics with an exponential function with a constant fraction (fitting results can be found in Supplementary Note 6), the dissociation efficiency of 1CT states into FCs ηCD was determined to be 15% for the BHJ film. This value should be the lower limit of ηCD for the PHJ film, as the morphology of the PHJ film is more suitable for charge dissociation (further discussion can be found in Supplementary Note 6).
As an alternative approach, ηCD of the PHJ film was estimated from EQE for photovoltaic conversion (EQEPV, Supplementary Note 8). EQEPV can be expressed as
where ηabs and ηCC are the efficiencies of photon absorption (~0.90, Supplementary Fig. 2) and charge collection to the respective electrodes, respectively. As we can assume ηCC ~0.9 (Supplementary Note 8), ηCD is estimated to be ~0.3 at short-circuit condition, at which no bias is applied to the device and charges are rapidly extracted from the device. Because ηCD at open-circuit condition (i.e., upconversion and TA measurement conditions) may be lower than that at short-circuit condition because of the absence of a large internal electric field, which is beneficial for charge dissociation36, we consider that ηCD of our upconversion devices is in the range of 15–30%.
Importantly, the decay kinetics of charges coincides with the recovery of GSB (Fig. 2f), and the PIA attributable to ITIC-Cl triplets, which shows PIA at 1200–1300 nm (Supplementary Figs. 7, 8), was not observed on the nanosecond time scale. In addition, the PIA of rubrene, which shows a characteristic PIA at 500 nm28,37, was also not observed. These results indicate that GR does not generate triplets. The spin-mixing rate between 1CT and triplet CT (3CT) is primarily determined by the hyperfine interaction (HFI)38,39,40,41,42, which leads to ISC on timescales of several tenth of nanoseconds in the absence of a large magnetic field43. Therefore, ISC is usually unable to outcompete the nonradiative decay of 1CT states to the ground state (730 ps in this study, Supplementary Note 6)44. This is consistent with the magnetic field dependence of the upconversion emission intensity performed in our previous study, wherein we did not observe a large PL intensity change under magnetic fields of <100 mT18. This means that BR of FCs is the main channel for the generation of rubrene triplets (vide infra).
Notably, SSA in the PHJ film is only a problem in femtosecond TA measurements, and SSA does not limit the upconversion efficiency under a weak CW excitation (further discussion can be found in Supplementary Note 22). The upconversion efficiency of the BHJ film was significantly lower than that of the PHJ film because rubrene singlets generated through TTA can easily reach the interfaces again owing to the fine morphology in the BHJ structure (Supplementary Note 7). Therefore, the PHJ structure is more suitable for the upconversion application, and hence all the measurements in the following sections were again carried out using the PHJ film.
Bimolecular recombination and triplet generation
The lifetime of FCs in the PHJ device was evaluated using the transient photovoltage/photocurrent (TPV/TPC) technique (Fig. 3, see Supplementary Note 9 for details of this technique)45. The charge carrier density and lifetime in the PHJ device at 1 sun (AM1.5G, 100 mW cm−2) open-circuit condition were determined to be 3.2 × 1016 cm−3 and 13 μs, respectively. These are typical values for OSCs45,46. Note that the carrier density under the 1 sun illumination is equivalent to that under monochromatic 750-nm LED illumination at ~32 mW cm−2 (Supplementary Note 10). In other words, the charge density under monochromatic illumination at 750 nm with 150 mW cm−2, at which ηUC reaches the maximum value because it is far beyond the characteristic threshold intensity Ith (39 mW cm−2)18, corresponds to ~4.7 sun illumination. Under this condition, the charge lifetime was extrapolated to be ~5.7 μs (Supplementary Note 10).

a Charge carrier lifetimes plotted against charge density. b BR rate coefficients krec plotted against charge density. The charge density was calculated using the film thickness of the rubrene layer (100 nm). c, d The horizontal axes of (a) and (b) are converted into an equivalent excitation light intensity of a monochromatic 750 nm LED.
Figure 4a shows the microsecond TA spectra of the PHJ film. The excitation wavelength was set to 700 nm to selectively excite ITIC-Cl. Charge PIA and the corresponding GSB of ITIC-Cl were still observed on the microsecond time scale (Supplementary Note 11). The decay kinetics of charges is again coincident with the recovery of GSB (Fig. 4b), and the PIA attributable to ITIC-Cl triplets was not observed (Supplementary Fig. 16).

a TA spectra of the PHJ rubrene/ITIC-Cl film. The excitation wavelength was 700 nm with a fluence of 12.5 μJ cm−2. b Time evolution of TA signals monitored at 800 nm (ITIC-Cl GSB) and 960 nm (ITIC-Cl anion). c Excitation fluence dependence of rubrene triplets decay in the PHJ film. The gray broken line is an exponential function with a time constant of 23 μs, representing the intrinsic lifetime of rubrene triplets, as a guide for the eye. d Triplet excitons decay in the PHJ film (blue, excited at 700 nm with a fluence of 3.8 μJ cm−2) and a pristine rubrene film (orange, excited at 532 nm with a fluence of 0.54 μJ cm−2). The gray lines represent the best fitting curves with Eq. (2).
Strikingly, in contrast to Fig. 2, a characteristic PIA band attributable to rubrene triplets was clearly observed at approximately 500 nm. Emphatically, rubrene triplet PIA was not observed in a pristine rubrene film after photoexcitation at 700 nm (Supplementary Fig. 17), meaning that rubrene triplets observed in the PHJ film were generated through BR.
BR of FCs leads to the formation of both 1CT and 3CT states with a ratio of 25:75 (when considering simple spin statistics, Supplementary Note 12)47,48,49. Then, 3CT states undergo back charge transfer to low-lying local triplet states in either the donor or acceptor material. Upconversion measurements in the solution state revealed that triplet energy transfer from ITIC-Cl to rubrene occurs, indicating that the energy level of rubrene triplets lies below that of ITIC-Cl (Supplementary Note 13). Therefore, rubrene triplets are preferentially generated over ITIC-Cl triplets through BR.
Triplet–triplet annihilation
Figure 4c shows the excitation fluence dependence of rubrene triplet decay in the PHJ film. The triplet decay is independent of the excitation fluence at later times (approximately > 10 μs), indicating the absence of TTA. The intrinsic triplet lifetime τT was determined to be 23 μs by fitting the later time decay with an exponential function, which is in good agreement with that obtained in a previous study50. In contrast, triplets decay faster with increasing excitation fluence at early times, indicating the contribution of TTA. Therefore, we analyzed the decay curves with a rate equation including TTA (Eq. (2))51,52,53,54.
where nT(t), n0, kT, and γ are the triplet exciton density at time t after photoexcitation, initial triplet density, rate constant of monomolecular deactivation given by the inverse of the triplet lifetime τT, and rate constant of TTA, respectively (see Supplementary Note 14 for more details).
Importantly, only the lowest fluence curve (3.8 μJ cm−2, Fig. 4d blue line) can be relatively well fitted with Eq. (2) (details of the fitting results can be found in Supplementary Note 18). Because the widely used TTA model (Eq. (2)) assumes a uniform distribution of excitons in the film, the fact that the fitting with Eq. (2) does not work well in our PHJ film suggests that the density gradients of rubrene triplets across the rubrene layer affect the TTA dynamics, as discussed in detail in the next section. Note that γ was tentatively determined to be 7.0 × 10−13 cm3 s−1 from the fitting (3.8 μJ cm−2).
To gain further insights regarding TTA dynamics, we performed TA measurements on a pristine rubrene film with an excitation wavelength of 532 nm. Upon photoexcitation of rubrene at 532 nm, rubrene singlets were generated, which then underwent singlet fission to form triplets (Supplementary Notes 15–17)28,37. Because the absorption coefficient of rubrene is relatively small (absorbance is ~0.2 at 532 nm for 100 nm thick rubrene, Fig. 1e), we can assume that triplets were uniformly generated across the film. Interestingly, triplet decay dynamics can be well fitted with Eq. (2) irrespective of the excitation fluence (Supplementary Fig. 21b). Most importantly, γ was determined to be 1.1 × 10−13 cm3 s−1, which is considerably smaller than that tentatively determined from the PHJ film, suggesting that TTA is enhanced in the PHJ film (see Supplementary Notes 15–17 for more details).
To confirm our hypothesis, we also evaluated γ by the excitation fluence dependence of upconversion emission intensity as an alternative approach. The threshold intensity Ith, above which ηUC approaches its maximum value, of our device is expressed as
where α is the absorption coefficient of the upconversion device at the excitation wavelength (see Supplementary Note 20 for more details). The experimental results presented above lead to an effective TTA rate constant γeff of 12.5 × 10−13 cm3 s−1 (we assumed ηCD of 0.2), where the word “effective” is used to denote the apparent TTA rate constant by taking into account the exciton accumulation effect. This value is approximately one order of magnitude larger than that obtained from TA measurements of the rubrene pristine film but close to the roughly estimated γ from the PHJ film. It should be emphasized that this value was obtained under actual upconversion measurement conditions using a CW LED light source; thereby, this reflects the actual situation of the PHJ film most accurately. This result confirms our hypothesis that TTA is enhanced in the PHJ film. As we reported previously, exciton accumulation in a small region has a positive impact on accelerating TTA because TTA is a bimolecular reaction17. As the diffusion length of rubrene triplets in amorphous film was determined to be ~11 nm (Supplementary Note 15), which is considerably shorter than the rubrene layer thickness (100 nm), a large exciton density gradient should exist across the rubrene layer (Supplementary Note 19). Figure 5a shows the probability density function (PDF) of rubrene triplets, wherein rubrene triplets were accumulated within the first 20 nm region even if the contribution of TTA was disregarded. When TTA is taken into account, triplets are further accumulated near the D/A interface because the apparent triplet lifetime (apparent diffusion length) will be shortened in the presence of TTA. If we assume that the triplet lifetime is reduced by 1/3, >90% of triplets are accumulated within the first 15 nm region (blue line). This is in good agreement with an experimentally determined spatial distribution of rubrene triplets reported previously55. This is the origin of the efficient TTA in the PHJ film despite the amorphous nature of the rubrene layer (Supplementary Note 21).

a PDF of rubrene triplets in the PHJ film in the absence or presence of TTA, wherein we assumed that the triplet lifetime was reduced by 1/3 due to TTA, which approximately corresponds to ηTTA of 67%. b Schematic showing the upconversion mechanism of the PHJ rubrene/ITIC-Cl device. ηCD of 0.2 was assumed for the calculation of ηTTA. The lifetime of ITIC-Cl singlets in the PHJ film was determined to be ~36 ps based on the intrinsic lifetime and ηq. The lifetime of 1CT states of the PHJ film was assumed to be the same with that of the BHJ film. The lifetime of FCs was extrapolated to be ~5.7 μs under the 750-nm LED excitation at 150 mW cm−2.
Conclusion
In this study, we comprehensively investigated the upconversion mechanisms in a PHJ film consisting of rubrene and ITIC-Cl. Figure 5b summarizes the upconversion mechanism with the quantum efficiency of each fundamental photophysical process, where the quantum efficiency of TTA ηTTA (out of a maximum of 100%) was calculated with the following equation:
The success of this device is owing to the large absorption coefficient in the NIR region including the optical interference effect (high ηabs), fast singlet diffusion in the NFA layer (high ηq), and efficient TTA by triplet accumulation near the D/A interface (high ηTTA). In contrast, the remaining challenge is the geminate recombination loss of 1CT. ηCD of the PHJ device was estimated in the range of 15–30%, which is the greatest bottleneck in our device. This value is considerably lower than that of the state-of-the-art OSCs, where ηCD is ~100%. Recent studies have pointed out that the energy cascade near the D/A interface is a key driver for efficient charge dissociation56,57,58. Therefore, optimizing the interfacial structure will further improve ηCD, and, hence, ηUC and EQE59,60,61.
Methods
Device fabrication
Rubrene (Tokyo chemical Industry, sublimed grade) and ITIC-Cl (Ossila) were used without further purification. Chloroform solution containing 2.5 mg mL−1 of ITIC-Cl was spin-coated onto quarts substrates in a N2-filled glovebox (UNICO). Rubrene layer was deposited on the top of ITIC-Cl by thermal evaporation (0.1 nm s−1) under high vacuum (~10−5 Pa) in a vacuum evaporation system (ULVAC, VTS-350M), housed inside a N2-filled glovebox (Miwa, DSO-1.5 S MS3-P). The film surface was covered by Al (70 nm) for upconversion emission measurements while it is maintained transparent for other optical measurements. All films were encapsulated by a glass substrate and epoxy resin.
Steady-state absorption and emission spectra
UV–visible absorption spectra were acquired using a UV–visible spectrometer (Jasco, V-570). PL spectra were measured using a fluorescence spectrophotometer (Horiba Jobin Yvon, FluoroLog) equipped with an photomultiplier tube (Hamamatsu, R928P). A single-color LED (Thorlabs, LED750L) with a constant current flow controlled by a source meter (Keithley, 2450) was used as an excitation source. The center wavelength and bandwidth of the LED were 750 nm and 23 nm, respectively. The LED light was focused on the sample by an aspheric lens. The spot size of the incident light was measured by a beam profiler and the beam diameter, defined as 1/e2 width, was 4.01 mm. The excitation intensity was measured by using a power meter (Ophir Photonics, 3A-P). The PL decay kinetics were measured by the time-correlated single-photon-counting (TCSPC) method (Horiba Jobin Yvon, FluoroCube).
TA measurements
Femtosecond TA data were collected using a pump and probe femtosecond TA spectroscopy system, which consisted of a TA spectrometer (Ultrafast Systems, Helios) and a regenerative amplified Ti:sapphire laser (Spectra-Physics, Hurricane, and Solstice). A fundamental pulse with a wavelength of 800 nm was used as the excitation source for ITIC-Cl. The TA data were collected over a time range from −5 ps to 3 ns. Femtosecond TA data of the PHJ film were denoised by total variation (TV) regularization method to increase SNR. Details of this method are described elsewhere58.
The microsecond TA data were collected using a sensitive microsecond TA system. A Nd:YAG pumped OPO-laser (EXPLA, NT342) operating at a wavelength of 700 nm with a repetition rate of 20 Hz was used as the excitation source for ITIC-Cl, whereas a higher repetition rate Nd:YAG laser (Elforlight, SPOT-10-200-532) operating at a wavelength of 532 nm with 128 Hz was used as the excitation source for rubrene. White light from a tungsten lamp with a stabilized power source was used as the probe light. Two monochromators and appropriate optical cut-off filters were placed before and after the sample to increase SNR. Further details of our TA setup have been presented elsewhere62.
TPV/TPC measurements
TPV and TPC were measured under bias white light illumination from a white LED with various intensities. A Nd:YAG laser (Elforlight, SPOT-10-200-532) operating at a wavelength of 532 nm with 100 Hz was used as a small perturbation source. The transient photovoltage generated by the laser pulse was monitored with a 200-MHz digital oscilloscope (Tektronix, TDS2022B). The input impedance of the oscilloscope was set to 1 MΩ for TPV measurements to hold the device at open-circuit condition, whereas it was 50 Ω for TPC measurements to hold the device at close to short-circuit condition. Transient current was calculated on the basis of Ohm’s law.
Data availability
The data supporting the results of this work are available from the corresponding author upon reasonable request.
Code availability
The codes used in this work are available from the corresponding author upon reasonable request.
References
Singh-Rachford, T. N. & Castellano, F. N. Photon upconversion based on sensitized triplet–triplet annihilation. Coord. Chem. Rev. 254, 2560–2573 (2010).
Simon, Y. C. & Weder, C. Low-power photon upconversion through triplet–triplet annihilation in polymers. J. Mater. Chem. 22, 20817–20830 (2012).
Schulze, T. F. & Schmidt, T. W. Photochemical upconversion: present status and prospects for its application to solar energy conversion. Energy Environ. Sci. 8, 103–125 (2015).
Yanai, N. & Kimizuka, N. Recent emergence of photon upconversion based on triplet energy migration in molecular assemblies. Chem. Commun. 52, 5354–5370 (2016).
Gray, V., Moth-Poulsen, K., Albinsson, B. & Abrahamsson, M. Towards efficient solid-state triplet–triplet annihilation based photon upconversion: Supramolecular, macromolecular and self-assembled systems. Coord. Chem. Rev. 362, 54–71 (2018).
Joarder, B., Yanai, N. & Kimizuka, N. Solid-state photon upconversion materials: structural integrity and triplet–singlet dual energy migration. J. Phys. Chem. Lett. 9, 4613–4624 (2018).
Bharmoria, P., Bildirir, H. & Moth-Poulsen, K. Triplet–triplet annihilation based near infrared to visible molecular photon upconversion. Chem. Soc. Rev. 49, 6529–6554 (2020).
Zhou, Y., Castellano, F. N., Schmidt, T. W. & Hanson, K. On the quantum yield of photon upconversion via triplet–triplet annihilation. ACS Energy Lett. 5, 2322–2326 (2020).
Zhou, J., Liu, Q., Feng, W., Sun, Y. & Li, F. Upconversion luminescent materials: advances and applications. Chem. Rev. 115, 395–465 (2015).
Zhu, X., Su, Q., Feng, W. & Li, F. Anti-stokes shift luminescent materials for bio-applications. Chem. Soc. Rev. 46, 1025–1039 (2017).
Hoseinkhani, S., Tubino, R., Meinardi, F. & Monguzzi, A. Achieving the photon up-conversion thermodynamic yield upper limit by sensitized triplet–triplet annihilation. Phys. Chem. Chem. Phys. 17, 4020–4024 (2015).
Harada, N., Sasaki, Y., Hosoyamada, M., Kimizuka, N. & Yanai, N. Discovery of key tips-naphthalene for efficient visible-to-UV photon upconversion under sunlight and room light. Angew. Chem. Int. Ed. 60, 142–147 (2021).
Wu, M. et al. Solid-state infrared-to-visible upconversion sensitized by colloidal nanocrystals. Nat. Photonics 10, 31–34 (2016).
Ogawa, T. et al. Donor–acceptor–collector ternary crystalline films for efficient solid-state photon upconversion. J. Am. Chem. Soc. 140, 8788–8796 (2018).
Abulikemu, A. et al. Solid-state, near-infrared to visible photon upconversion via triplet–triplet annihilation of a binary system fabricated by solution casting. ACS Appl. Mater. Interfaces 11, 20812–20819 (2019).
Lin, T.-A., Perkinson, C. F. & Baldo, M. A. Strategies for high-performance solid-state triplet–triplet-annihilation-based photon upconversion. Adv. Mater. 32, 1908175 (2020).
Sakamoto, Y., Tamai, Y. & Ohkita, H. Sensitizer-host-annihilator ternary-cascaded triplet energy landscape for efficient photon upconversion in the solid state. J. Chem. Phys. 153, 161102 (2020).
Izawa, S. & Hiramoto, M. Efficient solid-state photon upconversion enabled by triplet formation at an organic semiconductor interface. Nat. Photonics 15, 895–900 (2021).
Tamai, Y., Ohkita, H., Benten, H. & Ito, S. Exciton diffusion in conjugated polymers: from fundamental understanding to improvement in photovoltaic conversion efficiency. J. Phys. Chem. Lett. 6, 3417–3428 (2015).
Tamai, Y. Delocalization boosts charge separation in organic solar cells. Polym. J. 52, 691–700 (2020).
Nakano, K. & Tajima, K. Organic planar heterojunctions: From models for interfaces in bulk heterojunctions to high-performance solar cells. Adv. Mater. 29, 1603269 (2017).
Hou, J., Inganäs, O., Friend, R. H. & Gao, F. Organic solar cells based on non-fullerene acceptors. Nat. Mater. 17, 119–128 (2018).
Kini, G. P., Jeon, S. J. & Moon, D. K. Design principles and synergistic effects of chlorination on a conjugated backbone for efficient organic photovoltaics: a critical review. Adv. Mater. 32, 1906175 (2020).
Moser, M., Wadsworth, A., Gasparini, N. & Mcculloch, I. Challenges to the success of commercial organic photovoltaic products. Adv. Energy Mater. 11, 2100056 (2021).
Armin, A. et al. A history and perspective of non-fullerene electron acceptors for organic solar cells. Adv. Energy Mater. 11, 2003570 (2021).
Sievers, D. W., Shrotriya, V. & Yang, Y. Modeling optical effects and thickness dependent current in polymer bulk-heterojunction solar cells. J. Appl. Phys. 100, 114509 (2006).
Burkhard, G. F., Hoke, E. T. & Mcgehee, M. D. Accounting for interference, scattering, and electrode absorption to make accurate internal quantum efficiency measurements in organic and other thin solar cells. Adv. Mater. 22, 3293–3297 (2010).
Bossanyi, D. G. et al. In optimized rubrene-based nanoparticle blends for photon upconversion, singlet energy collection outcompetes triplet-pair separation, not singlet fission. J. Mater. Chem. C 10, 4684–4696 (2022).
Chandrabose, S. et al. High exciton diffusion coefficients in fused ring electron acceptor films. J. Am. Chem. Soc. 141, 6922–6929 (2019).
Firdaus, Y. et al. Long-range exciton diffusion in molecular non-fullerene acceptors. Nat. Commun. 11, 5220 (2020).
Natsuda, S. et al. Singlet and triplet excited-state dynamics of a nonfullerene electron acceptor Y6. J. Phys. Chem C 125, 20806–20813 (2021).
Lunt, R. R., Giebink, N. C., Belak, A. A., Benziger, J. B. & Forrest, S. R. Exciton diffusion lengths of organic semiconductor thin films measured by spectrally resolved photoluminescence quenching. J. Appl. Phys. 105, 053711 (2009).
Umeyama, T. et al. Efficient light-harvesting, energy migration, and charge transfer by nanographene-based nonfullerene small-molecule acceptors exhibiting unusually long excited-state lifetime in the film state. Chem. Sci. 11, 3250–3257 (2020).
Saito, T., Natsuda, S., Imakita, K., Tamai, Y. & Ohkita, H. Role of energy offset in nonradiative voltage loss in organic solar cells. Sol. RRL 4, 2000255 (2020).
Heeger, A. J. 25th anniversary article: bulk heterojunction solar cells: understanding the mechanism of operation. Adv. Mater. 26, 10–28 (2014).
Clarke, T. M. & Durrant, J. R. Charge photogeneration in organic solar cells. Chem. Rev. 110, 6736–6767 (2010).
Ma, L. et al. Singlet fission in rubrene single crystal: Direct observation by femtosecond pump–probe spectroscopy. Phys. Chem. Chem. Phys. 14, 8307–8312 (2012).
Groff, R. P., Merrifield, R. E., Suna, A. & Avakian, P. Magnetic hyperfine modulation of dye-sensitized delayed fluorescence in an organic crystal. Phys. Rev. Lett. 29, 429–431 (1972).
Groff, R. P., Suna, A., Avakian, P. & Merrifield, R. E. Magnetic hyperfine modulation of dye-sensitized delayed fluorescence in organic crystals. Phys. Rev. B 9, 2655–2660 (1974).
Devir-Wolfman, A. H. et al. Short-lived charge-transfer excitons in organic photovoltaic cells studied by high-field magneto-photocurrent. Nat. Commun. 5, 4529 (2014).
Thomson, Sa. J. et al. Charge separation and triplet exciton formation pathways in small-molecule solar cells as studied by time-resolved EPR spectroscopy. J. Phys. Chem. C 121, 22707–22719 (2017).
Kim, T. et al. Magnetic-field-induced modulation of charge-recombination dynamics in a rosarin-fullerene complex. Angew. Chem. Int. Ed. 60, 9379–9383 (2021).
Cohen, A. E. Nanomagnetic control of intersystem crossing. J. Phys. Chem. A 113, 11084–11092 (2009).
Gillett, A. J. et al. The role of charge recombination to triplet excitons in organic solar cells. Nature 597, 666–671 (2021).
Maurano, A. et al. Transient optoelectronic analysis of charge carrier losses in a selenophene/fullerene blend solar cell. J. Phys. Chem. C 115, 5947–5957 (2011).
Credgington, D. & Durrant, J. R. Insights from transient optoelectronic analyses on the open-circuit voltage of organic solar cells. J. Phys. Chem. Lett. 3, 1465–1478 (2012).
Rao, A. et al. The role of spin in the kinetic control of recombination in organic photovoltaics. Nature 500, 435–439 (2013).
Lakhwani, G., Rao, A. & Friend, R. H. Bimolecular recombination in organic photovoltaics. Annu. Rev. Phys. Chem. 65, 557–581 (2014).
Chow, P. C., Gelinas, S., Rao, A. & Friend, R. H. Quantitative bimolecular recombination in organic photovoltaics through triplet exciton formation. J. Am. Chem. Soc. 136, 3424–3429 (2014).
Di, D. et al. Efficient triplet exciton fusion in molecularly doped polymer light-emitting diodes. Adv. Mater. 29, 1605987 (2017).
Tamai, Y., Ohkita, H., Benten, H. & Ito, S. Triplet exciton dynamics in fluorene-amine copolymer films. Chem. Mater. 26, 2733–2742 (2014).
Tamai, Y., Matsuura, Y., Ohkita, H., Benten, H. & Ito, S. One-dimensional singlet exciton diffusion in poly(3-hexylthiophene) crystalline domains. J. Phys. Chem. Lett. 5, 399–403 (2014).
Murata, Y. et al. Two-dimensional exciton diffusion in an HJ-aggregate of naphthobisoxadiazole-based copolymer films. J. Phys. Chem. C 124, 13063–13070 (2020).
Murata, Y. & Tamai, Y. Intrachain exciton motion can compete with interchain hopping in conjugated polymer films with a strong J-aggregate property. J. Phys. Chem. Lett. 13, 2078–2083 (2022).
Izawa, S., Morimoto, M., Naka, S. & Hiramoto, M. Spatial distribution of triplet excitons formed from charge transfer states at the donor/acceptor interface. J. Mater. Chem. A 10, 19935–19940 (2022).
Perdigón-Toro, L. et al. Barrierless free charge generation in the high-performance PM6:Y6 bulk heterojunction non-fullerene solar cell. Adv. Mater. 32, 1906763 (2020).
Karuthedath, S. et al. Intrinsic efficiency limits in low-bandgap non-fullerene acceptor organic solar cells. Nat. Mater. 20, 378–384 (2021).
Natsuda, S. et al. Cascaded energy landscape as a key driver for slow yet efficient charge separation with small energy offset in organic solar cells. Energy Environ. Sci. 15, 1545–1555 (2022).
Zhong, Y., Tada, A., Izawa, S., Hashimoto, K. & Tajima, K. Enhancement of voc without loss of jsc in organic solar cells by modification of donor/acceptor interfaces. Adv. Energy Mater. 4, 1301332 (2014).
Izawa, S., Nakano, K., Suzuki, K., Hashimoto, K. & Tajima, K. Dominant effects of first monolayer energetics at donor/acceptor interfaces on organic photovoltaics. Adv. Mater. 27, 3025–3031 (2015).
Natsuda, S., Saito, T., Shirouchi, R., Imakita, K. & Tamai, Y. Delocalization suppresses nonradiative charge recombination in polymer solar cells. Polym. J. 54, 1345–1353 (2022).
Ohkita, H., Tamai, Y., Benten, H. & Ito, S. Transient absorption spectroscopy for polymer solar cells. IEEE J. Sel. Top. Quantum Electron. 22, 100–111 (2016).
Acknowledgements
This study was partly supported by JST PRESTO program Grant Numbers JPMJPR1874 and JPMJPR2101, and JSPS KAKENHI Grant Numbers 18K14115, 21H02012, 21H05394, and 21H05411.
Author information
Authors and Affiliations
Contributions
S.I. and Y.T. conceived and directed the project. S.I. fabricated all devices and performed steady-state optical measurements under the supervision of M.H. Y.T. performed femtosecond TA measurements and numerical simulations. Y.S. performed time-resolved PL, microsecond TA, and TPV/TPC measurements under the supervision of H.O. and Y.T. Y.S., S.I., and Y.T. wrote the manuscript and all authors contributed to revising the manuscript. All authors have approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Ardalan Armin and John Plummer. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sakamoto, Y., Izawa, S., Ohkita, H. et al. Triplet sensitization via charge recombination at organic heterojunction for efficient near-infrared to visible solid-state photon upconversion. Commun Mater 3, 76 (2022). https://doi.org/10.1038/s43246-022-00300-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-022-00300-z
This article is cited by
-
Blue organic light-emitting diode with a turn-on voltage of 1.47 V
Nature Communications (2023)
