
Abstract
Perovskite solar cells continue to attract interest due to their facile preparation and high power conversion efficiencies. However, the highest efficiency perovskite solar cells inevitably contain lead, which raises concerns over contamination of drinking water when a solar module is broken and then flooded. We previously showed that conventional synthetic hydroxyapatite (HAP) nanoparticles could capture some of the lead from broken solar cells, but the amount of lead released was well above the safe drinking water level. Here, we modify the HAP synthesis to prepare new spherical-HAP (s-HAP) nanoparticles with a 60% increase in the Pb absorption capacity. We blend s-HAPs with TiO2 nanoparticles to construct mixed scaffolds and investigate their effect on (FAPbI3)0.97(MAPbBr3)0.03 solar cell performance and lead capture. Replacement of 80% of the TiO2 nanoparticles with s-HAP causes the power conversion efficiency to increase from 18.61% to 20.32% as a result of decreased charge carrier recombination. Lead contamination of water from devices subjected to simulated hail damage followed by flooding is shown to decrease exponentially with increasing s-HAP content. The lead concentration in water after 24 h is below the US safe water drinking limit.
Similar content being viewed by others
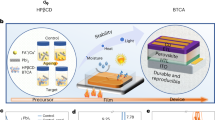


Introduction
Perovskite solar cells (PSCs) have continued to excite the solar cell community due to their outstanding power conversion efficiencies (PCEs), which have reached 25.7%1,2,3,4, and ease of fabrication5,6,7. They are solution processable that provides potentially low preparation costs for manufacture8,9,10,11. Unfortunately, there are two obstacles to their widespread implementation: their operational stability12,13,14 and the presence of Pb9,15,16,17. While the stability challenge is being addressed admirably via several routes13,18,19, the challenge of preventing Pb loss in the event of catastrophic device failure requires more attention. This is because the high PCEs for PSCs appear to be inextricably linked to the presence of Pb20,21, which is toxic to humans20. In a proof-of-concept study, we demonstrated a bioinspired approach using hydroxyapatite (HAP) to partially capture Pb from PSCs in the event of catastrophic failure followed by flood22. That study suggested several new frontiers for developing these materials. Firstly, how can the Pb absorption capacity of HAP be increased? Secondly, does inclusion of HAP nanoparticles with a different morphology (e.g., spheres) in the PSC scaffold also increase the PCE? Finally, is it possible to increase the Pb sequestration efficiency so that the Pb released under conditions of simulated hail damage and flood remains below the safe-drinking water level? We address these important questions in the present study.
The photoactive layer of Pb-based PSCs can contain up to 30 wt.% by mass of this element23. Furthermore, the Pb present in Pb-PSCs is water-soluble. Pb is a toxic, regulated, substance24,25 and has a half-life of 20–30 years in the human skeleton26. It also poisons almost every organ in the body27 and is carcinogenic26. Consequently, there are concerns about the consequences of damaged perovskite solar modules being flooded and Pb leaking into the environment. Hailegnaw et al were the first to study Pb release from damaged PSCs28. They concluded that total destruction of a perovskite solar power plant would not be catastrophic to the environment. Nevertheless, if PSCs are to be accepted by the public and provide a major contribution to next generation renewable energy, low-cost, fail-safe methods for preventing Pb release may be required29.
Methods for preventing (or minimizing) Pb release from damaged PSCs to date have mostly focussed on organic30 and/or polymer-based absorbents31,32,33,34. Chen et al. used a polymer-based cationic exchange resin to capture Pb and reduced the leakage in water to <7 ppb31. Niu et al. used in situ polymerization of acrylamide to limit the release of Pb after immersion of PSCs in water34. An on-device lead-absorbing polymer tape was used by Li et al. which captured 99.9% of the Pb released from their devices35. Epoxy resins and phosphonic-PEO polymers have also been successfully used to reduce Pb leakage33,36. Nonetheless, polymer-based capture systems have limited mechanical toughness and may not be fully reliable if longer-term capture is required after a module is damaged and then flooded (e.g., careless disposal in landfill or in water without recycling). Indeed, the solubility of the final, captured, Pb species should be negligible to prevent slow leaching from used modules in such circumstances. Our unique approach is inorganic-based and provides a stable solution for trapping Pb from PSCs. Moreover, we position our trapping system within the scaffold of the PSCs.
HAP (Ca10(PO4)6(OH)2) is an inorganic calcium-phosphate biomineral that occurs naturally in bone37,38. It can also be chemically synthesized and synthetic HAP is used for bone tissue engineering39. Like bones in humans, HAP has a strong tendency to absorb Pb40,41,42,43,44,45 (exchanging Ca2+ for Pb2+) and transforms to hydroxylpyromorphite (Pb-HAP)46. The latter has a miniscule solubility product of 10−77 over the pH range of 3–10 and is, hence, a very stable form of insoluble Pb under a variety of environmental conditions47. HAP can be prepared by a simple hydrothermal synthesis48,49 and the conventional morphology is nanorods49. The absorption capacity of HAP for Pb is potentially very high and a theoretical value of ~ 2100 mg g−1 can be calculated assuming the product is Pb10(PO4)6(OH)2. Consequently, synthetic HAP is an excellent candidate for scavenging released Pb from damaged PSCs.
In our previous proof-of-concept study we established that conventional HAP nanorods could be used in the scaffold of PSCs to decrease the amount of Pb released when the devices were broken and immersed in water22. The final concentration of Pb in the contaminated water was 0.38 ppm. However, this value is still 25 times higher than the US safe-drinking water Pb threshold of 0.015 ppm31. Furthermore, our previous work showed, surprisingly, that replacing the majority of the electrically conducing TiO2 nanoparticles of the mesoporous scaffold with insulating HAP nanorods increased the PCE. Such an unexpected result warrants further study.
Here, we synthesize new spherical HAP nanoparticles (s-HAP) with very high Pb absorption capacity and investigate their effects on PSC performance and subsequent Pb release from broken devices based on FAPbI3. The work begins with s-HAP characterization and then PSC performance is measured. We investigate the causes for the operational performance benefits due to HAP using a combination of experimental and computational techniques that were not applied in our proof-of-concept study22. The Pb capture is studied and the effect of scaffold composition probed. We also conduct post-mortem investigations to track the Pb absorption by the s-HAP located in both the scaffold and the encapsulation layer. This study improves our understanding of the reasons for the improved performance of the devices containing s-HAP and also demonstrates Pb contamination of water upon simulated hail damage followed by flood that is lower than the US safe water drinking level for Pb. Consequently, our study potentially brings the use of an in device Pb-trapping systems for PSCs a step closer.
Results and discussion
s-HAP and scaffold characterization
We aimed to increase the absorption capacity of HAP for Pb in this study so as to greatly improve the Pb capture efficiency for damaged PSCs. The HAP used here was synthesized over a shorter time period (4 h) and dried at a higher temperature (100 °C) compared to the 16 h and 60 °C used in the previous study22. These synthesis conditions gave spherical HAP (s-HAP) nanoparticles as shown by TEM imaging (Fig. 1a and Supplementary Fig. 1). Higher magnification images (Supplementary Fig. 1a) indicate that s-HAP is hollow. The number-average diameter is 49 ± 19 nm. A spherical morphology is rare for HAP50, which normally forms rod-like nanoparticles49,51. The s-HAP nanoparticles dispersed very well in isopropanol (IPA) and had a z-average diameter measured by dynamic light scattering (DLS) of 55 nm (Supplementary Fig. 2a). The XRD pattern for the dried s-HAP nanoparticles (Supplementary Fig. 2b) has peaks that agree with those reported elsewhere48. The ability of the s-HAP to absorb Pb from solution (depicted in Fig. 1b) was studied by constructing a Langmuir absorption isotherm (Supplementary Fig. 3a). The Langmuir equation gave an absorption capacity of 2165 ± 110 mg g−1. This value is 60% higher than that measured for conventional HAP in our earlier study22. The higher absorption capacity is likely a result of the high surface area-to-volume ratio for the s-HAP nanoparticles which benefited from their tendency to remain colloidally dispersed; whereas, the earlier nanorods aggregated which decreased their effective surface-area-to-volume ratio22.

a Representative s-HAP TEM image. b Depiction of spherical s-HAP absorbing soluble Pb2+ to form a Pb-HAP. c Depiction of the s-HAP:TiO2/Perovskite scaffold. d SEM images of the scaffolds used in this study.
Perovskite solar cells prepared using s-HAP scaffolds
Our n-i-p PSCs contained (FAPbI3)0.97(MAPbBr3)0.03 and were constructed by depositing Spiro-OMeTAD and then Au onto the stack depicted in Fig. 1c. The s-HAP dispersed well, so achieving well mixed scaffolds was straightforward as shown by the SEM images in Fig. 1d. In contrast, earlier work found that conventional HAP nanorods formed aggregates within those scaffolds22. Here, the s-HAP and TiO2 nanoparticles have similar morphology and so are difficult to distinguish by SEM in the mixed scaffolds, although the spherical s-HAP particles are readily apparent in the 100% scaffold (Fig. 1d).
PSCs were constructed using each scaffold (Fig. 1d) and box plots of the measured PSC performance parameters are shown in Fig. 2a. Interestingly, the PCEs of the 60% and 80% systems are greater than that for the 0% s-HAP control. (The 0% scaffold contained 100% TiO2 nanoparticles.) These systems also have the highest average Voc and FF values (see also Supplementary Table 1). The highest PCE (20.32%) was obtained using the 80% s-HAP scaffold (Fig. 2b). We also note that the 100% s-HAP system had a good average PCE of 16.7%. This is 3.2% higher than for the 100% conventional HAP nanorod scaffold system we reported earlier22 and is a remarkably high PCE considering our use of an insulating scaffold.

a Measured short-circuit current densities, open circuit voltages, fill factors and PCEs for devices prepared using scaffolds containing different s-HAP contents. (centre line, average; box limit, standard deviation; whiskers, outliners). b J–V curves of best-performing devices. c EQE data measured for the devices. Bode (d) and Nyquist (e) plots for the impedance data. The circuit model used to fit the data is shown (see text).
The 100% s-HAP system has similarities with the mesoporous Al2O3 layer devices reported by Snaith et al.52. Those devices contained an insulating scaffold and yet provided good PCEs. This is due in large part to the long diffusion lengths for the charge carriers of perovskites53. Representative stabilized power output data are shown in Supplementary Fig. 4. The 80% and 100% s-HAP samples also showed the most stable PCE over the time period of the experiment. EQE data were also measured (Fig. 2c). The integrated Jsc values for 0%, 60%, 80% and 100% are 22.9, 23.4, 23.8 and 22.6 m cm−2. These values are within 7% of the Jsc data obtained from our J–V measurements (Supplementary Table 1).
Electrochemical impedance spectroscopy (EIS) was used to probe charge transport in the devices. The Bode plot (Fig. 2d) shows that there are two charge transport processes present54, which occur at lower and higher frequencies. The relative contribution of the low frequency process increases with s-HAP content and almost matches that for the high frequency process for the 100% s-HAP device. Figure 2e shows the Nyquist plot with the data fitted using the model shown above the figure55. The series resistance (Rs) is constant until the s-HAP content reached 100% and then increases substantially (Supplementary Fig. 5a). This increase is due to the insulating nature of s-HAP and contributes to the low FF for that system (Fig. 2a). We note that the experimental band gap for HAP is 3.95 eV56. The resistance from the low frequency process (RLF) is due to charge recombination57. This value increases strongly for the 80% and 100% systems (Supplementary Fig. 5b) indicating a decreased recombination rate. Hence, s-HAP decreases recombination compared to the TiO2 scaffold. The high frequency resistance (RHF) is ascribed to charge transfer resistance to the hole transport layer58. The RHF value does not significantly change with s-HAP content (Supplementary Fig. 5c). This is expected as the changes to the electron transport layer due to s-HAP are not likely to strongly affect hole transport at the perovskite/Spiro-OMeTAD interface.
Morphology and optoelectronic properties
We examined the perovskite morphology using SEM (Fig. 3a) and 2D grazing angle X-ray diffraction (GIXRD, Fig. 3b). The grain size from SEM analysis (Supplementary Fig. 6a) increases linearly with s-HAP content (Supplementary Fig. 6b). The average grain diameters for the 0%, 60%, 80% and 100% s-HAP systems are 322 ± 90, 552 ± 175, 592 ± 277 and 694 ± 247 nm, respectively. The perovskite grain size more than doubled when meso-TiO2 was replaced by 100% s-HAP. This trend is attributed to increased porosity of the scaffold as the s-HAP content increases. Figure 3a and Supplementary Fig. 6a also show that PbI2 is present (light coloured grains) and occupies less of the surface as the s-HAP content increases, implying greater conversion of PbI2 to perovskite. (We return to this point later.) SEM cross-sections of ITO/scaffold/PVK (Supplementary Fig. 7) show the average thickness of the capping layers for 0%, 60%, 80% and 100% s-HAP films of 338, 417, 379 and 405 nm, respectively. Hence, the average grain diameters for all of the s-HAP-containing systems are greater than the capping layer thicknesses. This implies that single grains extend through the whole of the capping layer depth. This conclusion agrees with the SEM cross-section images of the capping layers in Supplementary Fig. 7.

a SEM images of perovskite films prepared using different scaffolds. b 2D GIXRD scattering patterns for the perovskite films prepared using scaffolds containing different s-HAP concentrations. c Normalized perovskite (100) peaks from the patterns in b. d Azimuthal (β) full width at half maximum height (FWHM) of the perovskite (100) and (200) planes together with their peak intensities. The error bars in d are the uncertainties.
We investigated the films using 2D GIXRD and Debye–Scherrer cones are evident (Fig. 3b). Such data can show orientation effects directly from the 2D diffraction profile. The angle of incidence for these data was 0.5°, which means that experiment tended to probe the bulk of the films59. Inspection of Fig. 3b shows greater intensity along central arc sections of the diffraction rings for 0% and 60% s-HAP systems, indicating greater perovskite crystal domain orientation effects for those systems compared to the 80% and 100% s-HAP systems. Figure 3c shows that preferred orientation was present60 in all films at azimuthal angles (β) of 128°, 180° and 233°. The intensities of the perovskite (100) and (200) peaks decrease by around 10% as the s-HAP content increases (Fig. 3d). Also, the FWHM of (100) and (200) peaks increases, consistent with a decrease in the tendency for preferred orientation. This effect may be due to the larger pores in those scaffolds (Fig. 1d). However, this does not appear to have negatively affected the PCE of the 60% and 80% PSCs. Conversion of PbI2 to perovskite was probed using 1D GIXRD (Supplementary Fig. 8) and increased conversion was apparent with increasing s-HAP content as judged by the intensity ratios of the perovskite (100) to PbI2 peaks (Supplementary Fig. 8b). This supports the qualitative observation concerning PbI2 in the preceding paragraph in relation to Fig. 3a. The coherence length (Lc) was calculated using the Scherrer equation (Supplementary Fig. 8c). The value for Lc increased with increasing % s-HAP used for the scaffold. The latter agrees with the trend for the average grain sizes determined from SEM (Supplementary Fig. 6b). These data indicate the presence of sub-grains that are not visible in the SEM images.
UV-visible spectroscopy data were measured for the films (Supplementary Fig. 9a) and show an increase in absorption with increasing % s-HAP content (Supplementary Fig. 9b). The absorbance increase at 900 nm is an indication of increased light scattering because the perovskite used in this study does not absorb light significantly at 900 nm61. Steady-state PL spectra for the films (Fig. 4a) indicate an 8 nm red-shift and an increase of PL intensity with increasing s-HAP content (Fig. 4b). The red-shift could be due to the increased grain size62. Following Yang et al.63 and Choi et al.64, the increased PL intensity is indicative of decreased quenching. To further probe this suggestion time-resolved photoluminescence (TRPL) data were measured (Fig. 4c). The PL intensity data (IPL) were fitted to a single exponential decay.
The values for the time constant, τ, are plotted in Fig. 4d. The τ values for the films prepared using 0%, 60%, 80% and 100% s-HAP were 108, 113, 123 and 147 ns, respectively. The increase in τ confirms that quenching became less efficient with increasing s-HAP content. These data suggest that the electron extraction efficiency from the perovskite decreases as conducting meso-TiO2 scaffold is progressively replaced by non-conducting s-HAP. The high series resistance for the 100% s-HAP device supports this suggestion (Fig. 5a and Supplementary Information).

a Steady-state PL spectra (λex = 460 nm) and b PL intensity vs. s-HAP content from a. c TRPL data. d TRPL time constants from fits in c. XPS (e) Pb 4f and (f) I 3d core level spectra.

Charge density difference shows Pb-O and Ca-I bond formation. Yellow and aqua charge densities refer to electron accumulation and depletion, respectively.
XPS spectra were obtained for the 0% and 80% s-HAP systems. The Pb 4f peaks (Fig. 4e) show evidence of under-coordinated Pb(0) defects in both systems. Such species have often been reported for perovskite films65 and attributed to Pb metal clusters66. The doublet I 3d peaks (Fig. 4f) for the 0% and 80% systems originate from spin-orbit coupling energy differences67 of the perovskite. These XPS spectra serve as a reference for comparison after the devices are subjected to water soaking (later).
Atomic scale modelling of the perovskite/HAP interface
To complement the experimental work with additional atomic-scale insights, we performed density functional theory (DFT) simulations of the interface between FAPbI3 and HAP (Ca10(PO4)6(OH)2) structures (details of the computational methodology are provided in the Experimental Methods section). We anticipate that the interaction at the perovskite/HAP interface will mainly occur between the predominant FAPbI3 phase (97%) and the contribution from MAPbBr3 (3%) will be insignificant. Figure 5 illustrates the simulated structure and charge density difference of the FAPbI3/HAP interface. (Supplementary Fig. 10 and the Experimental methods section provide additional simulation details.) We focused on the (001) surfaces of the perovskite halide and HAP as they were found to be important low energy surfaces. First, the interface structure was allowed to relax and we found the interlayer separation between the perovskite and HAP structures reduced from the initial configuration by up to 2.8 Å indicating strong interfacial interactions. We analysed the charge density difference at the interface as shown in Fig. 5. We find that there is a localized charge density rearrangement indicating bond formation between Pb and O atoms (of the phosphate groups) and between Ca and I atoms that are in close proximity. Indeed, the final mean Pb-O and Ca-I distances are 2.40 Å and 3.10 Å, respectively, which fall in the typical range of experimental bond lengths of corresponding Pb-phosphate and Ca-iodide type compounds. In particular, such new bond formation between Pb and oxygen of HAP at the interface would assist in the trapping or immobilization of Pb.
In addition, the perovskite Pb/I octahedra are distorted along the x-direction from the compressive strain induced by the incommensurate mismatch between FAPbI3 and HAP structures. This leads to significant Pb/I octahedra tilting along this axial strain. Hence, the passivating effect from Pb-O and Ca-I bond formation combined with the compressive strain could be important factors for the enhanced performance of the HAP-containing devices. Epitaxial compressive strain has been shown previously to stabilize the FAPbI3 perovskite and enhance the performance of PSC devices68. Overall, these DFT simulation results on the perovskite/HAP interface are in line with our experimental findings as well as providing new insights at the atomic level.
The PCE data shown in Fig. 2a can be explained by the competition of opposing effects. Firstly, the perovskite grain size increases as the HAP-content increases (Supplementary Fig. 6b). The number of traps decreases as the grain size increases which increases the PCE69. Also, recombination decreased as the %-s-HAP increased from the EIS data. Secondly, as the s-HAP content increases the electron extraction efficiency decreases based on the PL data (Fig. 4b, d). This will decrease the PCE. FF and Voc (Fig. 2a) are sensitive to traps and strongly contribute to the PCE increase for the devices with 60% and 80% s-HAP. The decrease in electron extraction had not become dominant for those systems. However, the EIS data (Supplementary Fig. 5a) show a relatively high series resistance for the 100% s-HAP device that decreases the FF and hence the PCE. This is why the 100% s-HAP system has the lowest PCE. The good PCE for this system is due to the high electron–hole diffusion length for the perovskite and also the porosity apparent for the scaffold (Fig. 1d). The latter enables contact between the perovskite and the TiO2 hole blocking layer.
In situ lead immobilization studies
We investigated the ability of the devices to release Pb into aqueous solution when subjected to catastrophic fracture followed by immersion in water (simulated hail followed by flood). The devices used in this part of the study had an added s-HAP encapsulation layer (Fig. 6a). A falling rod was used to break the cells from the glass side (Supplementary Fig. 11 and Supplementary Video 1). Figure 6b shows photographs of the broken cells at different times after immersion in water for up to 24 h. The devices change from black to yellow as the perovskite is dissolved to form PbI2. The rate of dissolution decreases as the s-HAP content increases and the Pb concentration found in the aqueous phase also decreases (Fig. 6c). Indeed, the Pb concentration in solution remains remarkably low for both the 80% and 100% samples (0.040 and 0.010 ppm, respectively, after 24 h exposure). Importantly, the latter value is below the US safe drinking water concentration of 0.015 ppm31. Furthermore, the Pb concentration decreased exponentially with s-HAP concentration in the scaffold (Fig. 6d). We note that the Pb concentration reached using the 100% s-HAP scaffold is 38 times lower than achieved with in our previous work using conventional HAP nanorods22.

a Schematic of the device with a HAP encapsulation layer. b Photographs of failure tests from broken PSCs immersed in water for various times. The black colour turns to yellow showing perovskite degradation to PbI2. c Pb concentration in the water surrounding the broken cells as a function of time. d Pb residual concentration in water after 24 h for each of the devices. At 100% s-HAP the Pb concentration is below the safe level for drinking water in the US (red dashed line).
We used high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDS) spectrum imaging to probe the 80% s-HAP scaffold after the water soaking experiment. A cluster of five s-HAP nanoparticles is evident in Fig. 7a together with a non-spherical TiO2 nanoparticle. The presence of both Ti and Ca in the EDS STEM nanoscale elemental distributions show the intimate mixture of TiO2 and s-HAP nanoparticles in the mixed scaffold (Fig. 7b–d). The Pb elemental distribution (Fig. 7e) is inhomogeneously segregated to the spherical HAP particle surfaces. The XPS Pb 4f and I 3d spectra are shown in Fig. 7g, h, respectively. Interestingly, the under-coordinated Pb(0) species that was present in the as-made 0% and 80% systems (Fig. 4e) is absent. This is strong evidence for a substantial change in Pb bonding. Indeed, there is a major increase of the widths at half-height for both the Pb 4f and I 3d peaks for the 80% sample (compare to Fig. 4e, f). These data indicate that the bonding environment for Pb and I have changed considerably as a result of water soaking. The new environments may be due to Pb bonded to oxygen70 of PO4 and/or I within20 PbI2.

HAADF STEM and STEM-EDS data (a–f) recorded for a fragment of the 80% s-HAP scaffold layer after absorption of Pb for 24 h. f is a composite colour map combining elemental STEM-EDS data for Ca, Ti, P and Pb in b–e. EDS quantification from the whole spectra image shows a Pb:Ca atomic ratio of 1:11.7. XPS spectra showing the g Pb 4f and h I 3d core level signals. i Relative Pb contents in the scaffold and encapsulation layers after 24 h water soaking determined using XPS. These values were calculated using the strongest peaks which were the Pb and I signals for the scaffold or from Pb, I and P for the encapsulation layer.
An aggregate from the s-HAP encapsulation layer was also analysed by HAADF STEM and EDS spectrum imaging (Supplementary Fig. 12). Once again, Pb was found absorbed to the outside of the s-HAP particles (Supplementary Fig. 12e) which is an indication of strong binding. XPS spectra were measured for the encapsulated layer after the water-soaking experiment (Supplementary Fig. 12g, h). The Pb 4f and I 3d peaks had broadened considerably compared to the respective XPS peaks for the as made perovskite (Fig. 4e, f). This indicates a broad range of bonding environments for both Pb and I within the s-HAP-based encapsulation layer. These data provide evidence that HAP can trap both Pb and I, with our theoretical calculations suggesting that I is likely bonded to Ca (see Fig. 5).
The XPS data from the scaffold and encapsulation layer were obtained for all of the PSCs (Supplementary Fig. 13) and the variations of atomic percentages with time are plotted in Fig. 7i. These values are calculated relative to the species with the greatest intensities. The Pb contents for the scaffold are relative to Pb and I; whereas, for the encapsulation layer they are relative to Pb, I and P. After the 24 h leak test, the 0% s-HAP film shows the lowest Pb found in the scaffold indicating that more Pb is leaked/leached away. (This result is consistent with Fig. 6c.) Hence, that scaffold is less efficient in containing the Pb. Detailed analysis of the data is presented in the Supplementary Information using Supplementary Fig. 14, which concludes that the overwhelming majority of the Pb was released into the water phase by the 0% scaffold. We attribute the Pb signal from XPS to a minor proportion of PbI2 residing on the scaffold surface.
On the other hand, Fig. 7i shows that 100% s-HAP scaffold has the highest Pb and hence is best at immobilizing Pb. Moreover, the immobilized Pb concentration in the scaffold increased with s-HAP concentration showing that less Pb is released which concurs with Fig. 6c. The encapsulation layers, by contrast, showed a mostly consistent Pb concentration after leak test. This indicates that the s-HAP encapsulation layer was less efficient at capturing released Pb compared to the scaffold and highlights the value of placing s-HAP within the scaffold. An interesting question concerns whether there was sufficient s-HAP in the 100% scaffold to capture all of the Pb that was originally present in the device. A detailed analysis for this question is presented in the Supplementary Information in which we conclude that there was likely to be sufficient absorption capacity for the 100% s-HAP scaffold to absorb all of the Pb initially present in principle.
It can be speculated whether exchange of Pb2+ with Ca2+ could have occurred in the scaffold layer when PbI2 in DMF:DMSO (95:5) was added in the first step of perovskite formation. Chen et al. 71 measured the solubility of HAP in DMF after 24 h of equilibration and reported a mole fraction solubility of 5 × 10−3. They noted that HAP dissolution was non-spontaneous. In the present work the duration for potential s-HAP dissolution by DMF was only 150 s (see Experimental Methods). The SEM cross-section image for the 100% s-HAP film (Supplementary Fig. 7d) shows that the s-HAP scaffold has a similar thickness to the other systems. Furthermore, the strong decrease in Pb solution concentration with increasing s-HAP concentration from Fig. 6d is due to the presence of the s-HAP scaffold. Accordingly, while HAP dissolution was possible during PbI2 deposition if given enough time (e.g., 24 h), it follows that under the short time scale used here only a small proportion of s-HAP dissolved and reacted with Pb.
In summary, we have established a viable new approach for addressing the urgent problem of Pb leakage from damaged PSCs, which is one of the most important issues that is hindering their application. Previous approaches have focused on polymer31,34,35 and/or organic sequestering agents30, which may have limited ability to retain the captured Pb longer term. Such approaches have located the capturing systems external to the charge collection and transport regions of the device stack. The present study has demonstrated a new and efficient capture system (s-HAP) that is in intimate contact with the photoactive layer within the device stack. Moreover, Pb captured by HAP forms Pb-HAP which has a negligible solubility constant in water over a wide range of pH47 and is also strong mechanically and a biomaterial.
Conclusions
In this study, we synthesized spherical HAP nanoparticles and found they had a greatly improved Pb absorption capacity compared to conventional HAP. The s-HAP nanoparticles were used to replace the TiO2 scaffold at different proportions and in all cases viable solar cells were obtained despite the insulating nature of s-HAP. The highest PCE obtained was 20.32% for a PSC containing 80% s-HAP scaffold. The s-HAP strongly decreases charge recombination. DFT simulations provided atomic-scale insights into the FAPbI3/HAP interface, indicating strong interfacial interactions including Pb-O and Ca-I bond formation, which supports Pb immobilization of the perovskite by the HAP scaffolds. This effect of bond formation combined with compressive strain at the interface could be important factors for the enhanced performance of the s-HAP-containing devices. We investigated the ability of devices with an encapsulation s-HAP layer to prevent Pb release upon simulated hail damage followed by flooding for 24 h. The results showed a dramatic effect of the scaffold type with an exponentially decreasing relationship between Pb water contamination and s-HAP used for the scaffold. The 100% HAP scaffold device restricted the Pb release after 24 h to less than the US safe drinking water value and this scaffold also gave a viable device. Hence, the present study has provided a new approach for a fail-safe solution to the Pb release concern for PSCs, which has excellent potential to effectively lock up Pb within PSCs in the event of catastrophic failure and/or irresponsible discarding of used devices to landfill or in water.
Experimental methods
Materials
Calcium nitrate tetrahydrate (≥97%), NaOH (99%), ammonium hydrogen phosphate (98%), nitric acid (98%), ethyl cellulose (viscosity = 100 cP), lead nitrate (99%), ethanolamine (99.9%), methyl ammonium chloride (MACl, 99.99%), isopropanol (IPA, anhydrous, 99.5%), ethanol (anhydrous, 99%), dimethylsulfoxide (DMSO, anhydrous, 99.9%), N,N-dimethylformamide (DMF, anhydrous, 99.8%), chlorobenzene (CBZ, anhydrous, 99.5%), 4-tert-butylpyridine (96%), lithium bistrifluoromethanesulfonimidate (LiTFSI, 99.95%), titanium isopropoxide (TIP), 2-methoxyethanol (98%), poly(methylmethacrylate) (PMMA, MW = 15,000 g/mol), 2,2’,7,7’-tetrakis[N,N-di(4-methoxyphenyl)amino]−9,9’-spirobifluorene (Spiro-OMeTAD, 99.5%) were all purchased from Aldrich and used as received. PbI2 (Tokyo Chemical Industry, 99.999%), PAE 136 Phosphate Ester (PAE 136, Lakeland Chemical LTD) were used as received. TiO2 paste (30NRD), formaminidium iodide (FAI, 99.5%), methylammonium bromide (MABr, 99.99%), FK 209 Co(III) TFSI salt, were purchased from Greatcell Solar and used as received. Ultrapure deionized water was used in all experiments.
s-HAP synthesis
Ca(NO3)2.4H2O (70.846 g, 0.15 mol) was added to water (400 mL) in a 500 mL Teflon bottle and the pH was adjusted to 7.4 using aqueous NaOH solution (5.0 M). This solution is denoted as solution A. Then NH4.H2PO4 (26.412 g, 0.10 mol) was added to water (400 mL) in another Teflon bottle and the pH was adjusted to 4.0 with aqueous HNO3 solution (5.0 M). This solution is denoted as solution B. Solution A was then added dropwise into solution B at constant rate of 13 mL min−1. The pH of the mixed solution was maintained at 10.8. After the addition was finished, the mixed solution was stirred at room temperature for 4 h. The product was then oven-dried at 100 °C for 4 h and stored in an airtight container.
Pb absorption experiments
Various Pb(NO3)2 concentrations were prepared for the Pb absorption experiments by s-HAP. In brief, to obtain 1000 ppm Pb(II) stock solution, Pb(NO3)2 powder (1.60 g, 4.83 mmol.) was dispersed in water (1000 mL). Dilutions were then made with water to obtain the desired concentrations. The appropriate masses of s-HAP were then added into the Pb solutions. The mixed solutions were stirred for 30 min and Pb concentrations measured using a calibrated Pb ion selective electrode (ISE). The equilibrium absorption capacity (qe) was calculated using the following equation72.
where Ce, Co, V and m are the equilibrium Pb(II) concentration, initial Pb (II) concentration (mg L−1), volume of solution and mass of s-HAP, respectively.
s-HAP dispersion preparation
s-HAP powder (1.20 g) was mixed with PAE136 stabilizer (0.20 wt.%) in ethanol (10.0 mL) in a 50 mL vial and sonicated for 5 min with an ultrasonic probe in an ice bath. Meanwhile, ethyl-cellulose (1.0 g) was added to ethanol (1.0 mL) and stirred for 2 h at 50 °C. After that, the two systems were mixed and terpineol (7.0 g) was added. The s-HAP dispersion was stirred mechanically and briefly sonicated prior to use.
Scaffold preparation
The scaffolds are identified in terms of the nominal mass% of s-HAP in the scaffold. It is assumed that the ratio of TiO2 and s-HAP particles in the final mixed scaffold is the same as that added to the solution used to deposit the scaffold. The following method was used to prepare the scaffolds containing s-HAP. For the 60% and 80% scaffolds, 600 and 800 mg s-HAP paste (5.0 wt.%), were blended with 400 and 200 mg TiO2 paste (1:5 in ethanol, 4.0 wt.%), respectively, and mixed thoroughly with a vortex mixer. The dispersion (70 μL) was spin-coated on the substrate using 5,000 rpm 30 s and annealed as described above. For the 100% film, the as-made s-HAP paste was used directly for spin-coating. The scaffold was sequentially annealed using 125 °C for 5 min, 325 °C for 5 min, 375 °C for 5 min and 450 °C for 30 min.
Perovskite film preparation
Perovskite films ((FAPbI3)0.97(MAPbBr3)0.03) were synthesized by using a 2-step route. Firstly, PbI2 solution (600 mg in 0.95 mL DMF and 0.05 mL DMSO) was prepared and stirred at 70 °C for 2 h. Then, 40 µL of the solution was dropped onto the photoanode, then spin-coated at 1700 rpm for 30 s. The substrate was transferred onto a 70 °C hot plate and dried for 2.0 min. The substrate then left to cool at room temperature. Secondly, a cation solution sample (80 µL) was taken from a solution containing FAI; (60 mg, 0.349 mM), MABr; (6.0 mg, 0.054 mM), MACl (6.0 mg, 0.089 mM) in IPA solution (1.0 mL) and dropped onto the PbI2-impregnated scaffold. The substrate was immediately spun at 1700 rpm for 30 s and transferred onto 150 °C hot plate and annealed for 15 min. It was then stored in a desiccator over P2O5.
Device fabrication
The ITO glass substrates (TEC20, ~16 Ω sq−1, Ossila LTD) were etched using Zn powder and HCl. They were cleaned by ultrasonication in an aqueous Hellmanex solution (2%) and rinsed with water, followed by acetone, ethanol and IPA. The substrates were then dried with N2 stream and were placed in UV/ozone for 20 min. A compact TiO2 blocking layer (bl-TiO2) was then deposited by sol-gel method. Briefly, a 30-nm thick TiO2 blocking layer was obtained by mixing titanium isopropoxide (99.99%) and ethanolamine in 2-methoxy ethanol. A portion of the solution (70 μL) was spun at 3000 rpm for 30 s. The sequential annealing procedure described above was then applied. After cooling, the scaffold was deposited by spin-coating the prepared pastes (70 μL) at 5000 rpm for 30 s followed by the same annealing procedure. The perovskite photoactive layer was then deposited by 2-step deposition method as described above. Subsequently, a solution (55 µL) of Spiro-OMeTAD (0.090 g), tBP (34 µL), LiTFSI solution in acetonitrile (0.52 g mL−1, 19 µL) and FK209 solution in acetonitrile (0.30 g mL−1, 10 µL) dissolved in 1.0 mL CBZ was spin-coated on the perovskite using 4000 rpm for 25 s. The device was then kept in dry air (<25% RH) for about 12 h. A gold layer (80 nm) was deposited on top of the Spiro-OMETAD layer by thermal evaporation under high vacuum.
Physical measurements
The surface morphologies were examined using a Tescan Mira scanning electron microscope (SEM). Cross-sectional images were obtained using a Ultra 55 Carl Zeiss Sigma field emission gun (FEG)-SEM. The morphology and chemical composition of the scaffold were characterized by scanning transmission electron microscopy (HR-STEM) and energy dispersive X-ray spectroscopy (EDS) using a Thermo Fisher Titan STEM (G2 80–200) equipped with a Cs probe corrector (CEOS) and a high-angle annual dark-field (HAADF) detector, operating at 200 kV. The TEM samples were prepared by dispersing the samples in methanol, with subsequent sonication of the mixture for 5 min and drop-cast onto a copper grid coated with an amorphous holey carbon film. UV-visible spectra were recorded using an Agilent Cary 60 UV−vis spectrophotometer. Photoluminescence (PL) spectra were obtained using an Edinburgh Instruments FLS980 spectrometer. The films were prepared under a nitrogen atmosphere and film was coated with a thin layer of PMMA. Measurements were conducted from the ITO side of the film. Film thicknesses were measured by SEM. The 2D grazing angle X-ray diffraction (GIXRD) data were measured using a Rigaku SmartLab with 2D hybrid semiconductor detector Hypix-3000 with incident beam collimation. All the samples were measured at a 0.5° grazing incident angle. 1D GIXRD data were collected using a Bruker D8 Discover and the grazing incident angle was 0.3°. Dynamic light scattering (DLS) measurements were performed using a Malvern Nano ZS instrument.
X-ray photoelectron spectroscopy (XPS) measurements were performed with a monochromated Al Kα X-ray source and Argus electron energy analyser (1486.6 eV, 300 W, ESCA2SR, ScientaOmicron GmbH). Binding energies were calibrated to C 1s 248.80 eV from adventitious carbon for all samples and quoted to accuracies of ±0.05 eV for high resolution scans measured using 20 eV pass energies. Relative atomic ratios were calculated from HRXPS spectra where a Shirley background and GL(30) function (70% Gaussian and 30% Lorentzian) were applied to fit the core level spectra using CasaXPS software. Scofield sensitivity factors were used for calculating the stoichiometry of the samples. The films were freshly prepared in a nitrogen-filled glovebox and immediately transferred to the XPS vacuum chamber for measurement.
Failure simulation
s-HAP paste (100 μL) was spin-coated at 3,000 rpm for 60 s onto glass and annealed at 500 °C for 30 min to obtain an HAP layer with a thickness of 4.0 μm. The film was then combined with a completed device in such a manner that the s-HAP surface was in contact with the gold contact at the top of the device. The edges were sealed by epoxy and cured. A consistent breakage of the PSC was created by impact from a metal rod (300 g), dropped from a height of 5.0 cm from the outer encapsulation surface (see Supplementary Video 1 and Supplementary Fig. 11). The substrates were coated with 1.5 cm×2.0 cm of perovskite as shown in Fig. 6b. They were then soaked in water (50 mL) for 24 h. Accordingly, the ratio of the water volume-to-substrate area was 16.7 cm3 cm−2. The latter value is similar to that used in related studies33,34. The water was tested for Pb leakage by inductively coupled plasma atomic emission spectroscopy (ICP-AES).
Device measurements
The current density–voltage (J–V) characteristics were measured using a high precision sourcemeter (Xtralien X200) with a calibrated 100 mW cm−2 illumination (AM 1.5 G) sourcing from an ABET 10500 solar simulator in ~40% relative humidity. For this, a Newport 91150 V calibrated reference cell was used and the data were corrected for spectral mismatch. Forward and reverse direction sweeps were measured with a sweep rate of 200 mV s−1. The averages presented in the text are for reverse scans. EQE measurements were performed using a Quantx 300 (Oriel Instrument PLC). Electrochemical impedance spectroscopy (EIS) was measured using phase-sensitive detection of the current through the device. The measurements were made in the dark at the open circuit voltage with a superimposed AC voltage (20 mV) supplied using an Agilent 33210 A function generator. The device area for J–V and EQE measurements was 0.079 cm2. The area for the EIS measurements used was also 0.079 cm2.
Computational simulations
The analysis of the atomic-scale structure and charge density at the interface between FAPbI3 and Hydroxyapatite (HAP) was carried out using DFT methods as implemented in the Quantum ESPRESSO code73,74. Such DFT methods have been applied successfully to other studies of perovskite halides75,76,77. The Kohn–Sham wave-functions and energies were calculated with the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) formalism78,79. A plane-wave basis was used, with energy and charge density cutoffs of 30 and 300 Ry, respectively. The Grimme dispersion correction DFT-D380,81 was used, as well as ultrasoft pseudopotentials82. For geometry optimization, k-point sampling of 1 × 3 × 1 was used for the Brillouin zone integration and a 2 × 4 × 2 k-mesh for the electronic structure analysis following the Monkhorst–Pack scheme83. To avoid the interaction between the repeated images, a vacuum layer of 22 Å was used. For the construction of the interface model, a slab of FAPbI3 supercell structure along (001) with Pb/I surface termination was used. To maintain the bulk electronic properties, the slab thickness is chosen to be three times the unit cell of the bulk cubic phase of FAPbI3 in the out-of-plane direction. The HAP slab was also cut along (001), as it was identified to be the lowest energy surface84, with a thickness of two unit cells of the bulk structure. Since HAP crystallizes in a hexagonal phase and in order to reduce the mismatch between MAPbI3 and HAP slabs, a rectangular box along the in-plane directions was used (as shown in Supplementary Fig. 10). The two slabs were commensurate along the y direction, with a mismatch of 4.8% along the x direction. To reduce this mismatch and fulfil the periodic boundary conditions, the perovskite slab was compressed by 2.4% and the HAP slab was elongated by 2.4%. The simulation box included 664 atoms in total with initial lattice parameters of a = 33.83 Å, b = 9.533 Å, and c = 59.20 Å. The initial distance between the FAPbI3 and HAP slabs was fixed to 4 Å. After relaxation the lattice parameter of the interface shortened by about 2% due to the compressive strain caused by the mismatch.
Data availability
Data that support the findings within this work are available from the corresponding author upon reasonable request.
References
NREL. Best research-cell efficiencies. www.nrel.gov/pv/cell-efficiency.html (2022).
Yoo, J. J. et al. Efficient perovskite solar cells via improved carrier management. Nature 590, 587–593 (2021).
Wu, W.-Q., Rudd, P. N., Wang, Q., Yang, Z. & Huang, J. Blading phase-pure formamidinium-alloyed perovskites for high-efficiency solar cells with low photovoltage deficit and improved stability. Adv. Mater. 32, 2000995 (2020).
Zhang, F. et al. Metastable Dion-Jacobson 2D structure enables efficient and stable perovskite solar cells. Science 375, 71–76 (2022).
Wu, T. et al. The main progress of perovskite solar cells in 2020–2021. Nano-Micro Lett. 13, 152 (2021).
Assadi, M. K., Bakhoda, S., Saidur, R. & Hanaei, H. Recent progress in perovskite solar cells. Renew. Sustain. Energy Rev. 81, 2812–2822 (2018).
Gao, Y. et al. Highly efficient, solution-processed CsPbI2Br planar heterojunction perovskite solar cells via flash annealing. ACS Photonics 5, 4104–4110 (2018).
Howard, I. A. et al. Coated and printed perovskites for photovoltaic applications. Adv. Mater. 31, 1806702 (2019).
Li, D. et al. A review on scaling up perovskite solar cells. Adv. Funct. Mater. 31, 2008621 (2021).
Li, N., Niu, X., Chen, Q. & Zhou, H. Towards commercialization: the operational stability of perovskite solar cells. Chem. Soc. Rev. 49, 8235–8286 (2020).
Du, T. et al. Aerosol assisted solvent treatment: a universal method for performance and stability enhancements in perovskite solar cells. Adv. Energy Mater. 11, 2101420 (2021).
Chi, W. & Banerjee, S. K. Stability improvement of perovskite solar cells by compositional and interfacial engineering. Chem. Mater. 33, 1540–1570 (2021).
Lin, Y.-H. et al. A piperidinium salt stabilizes efficient metal-halide perovskite solar cells. Science 369, 96–102 (2020).
Wilks, R. G. & Bär, M. Perovskite solar cells: danger from within. Nat. Energy 2, 16204 (2017).
Bae, S.-Y. et al. Hazard potential of perovskite solar cell technology for potential implementation of “safe-by-design” approach. Sci. Rep. 9, 4242 (2019).
Ravi, V. K., Mondal, B., Nawale, V. V. & Nag, A. Don’t let the lead out: new material chemistry approaches for sustainable lead halide perovskite solar cells. ACS Omega 5, 29631–29641 (2020).
Wan, J. et al. Lead contamination analysis of perovskite modules under simulated working conditions. Solar Energy 226, 85–91 (2021).
Grancini, G. et al. One-year stable perovskite solar cells by 2D/3D interface engineering. Nat. Commun. 8, 15684 (2017).
Bai, S. et al. Planar perovskite solar cells with long-term stability using ionic liquid additives. Nature 571, 245–250 (2019).
Babayigit, A. et al. Assessing the toxicity of Pb- and Sn-based perovskite solar cells in model organism Danio rerio. Sci. Rep. 6, 18721 (2016).
Ke, W. & Kanatzidis, M. G. Prospects for low-toxicity lead-free perovskite solar cells. Nat. Commun. 10, 965 (2019).
Mokhtar, M. Z. et al. Bioinspired scaffolds that sequester lead ions in physically damaged high efficiency perovskite solar cells. Chem. Commun. 57, 994–997 (2021).
Yan, J. & Saunders, B. R. Third-generation solar cells: a review and comparison of polymer:fullerene, hybrid polymer and perovskite solar cells. RSC Adv. 4, 43286–43314 (2014).
Kajari-Schröder, S. Contain the lead. Nat. Energy 4, 534–535 (2019).
Yang, L., Barrows, A. T., Lidzey, D. G. & Wang, T. Recent progress and challenges of organometal halide perovskite solar cells. Rep. Progr. Phys. 79, 026501 (2016).
Babayigit, A., Ethirajan, A., Muller, M. & Conings, B. Toxicity of organometal halide perovskite solar cells. Nat. Mater. 15, 247–251 (2016).
Horváth, E. et al. Fighting health hazards in lead halide perovskite optoelectronic devices with transparent phosphate salts. ACS Appl. Mater. Interfaces 13, 33995–34002 (2021).
Hailegnaw, B., Kirmayer, S., Edri, E., Hodes, G. & Cahen, D. Rain on methylammonium lead iodide based perovskites: possible environmental effects of perovskite solar cells. J. Phys. Chem. Lett. 6, 1543–1547 (2015).
Goetz, K. P., Taylor, A. D., Hofstetter, Y. J. & Vaynzof, Y. Sustainability in perovskite solar cells. ACS Appl. Mater. Interfaces 13, 1–17 (2021).
Yan, P. et al. Chemical encapsulation of perovskite film by tetra-thiol copper(II) porphyrin for stable and clean photovoltaics. Org. Electron. 93, 106158 (2021).
Chen, S. et al. Trapping lead in perovskite solar modules with abundant and low-cost cation-exchange resins. Nat. Energy 5, 1003–1011 (2020).
Chen, S. et al. Preventing lead leakage with built-in resin layers for sustainable perovskite solar cells. Nat. Sustain. 4, 636–643 (2021).
Jiang, Y. et al. Reduction of lead leakage from damaged lead halide perovskite solar modules using self-healing polymer-based encapsulation. Nat. Energy 4, 585–593 (2019).
Niu, B. et al. Mitigating the lead leakage of high-performance perovskite solar cells via in situ polymerized networks. ACS Energy Lett. 6, 3443–3449 (2021).
Li, X. et al. On-device lead-absorbing tapes for sustainable perovskite solar cells. Nat. Sustain. 4, 1038–1041 (2021).
Li, X. et al. On-device lead sequestration for perovskite solar cells. Nature 578, 555–558 (2020).
Ibrahim, M., Labaki, M., Giraudon, J.-M. & Lamonier, J.-F. Hydroxyapatite, a multifunctional material for air, water and soil pollution control: a review. J. Hazard. Mater. 383, 121139 (2020).
Sadat-Shojai, M., Khorasani, M.-T., Dinpanah-Khoshdargi, E. & Jamshidi, A. Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater 9, 7591–7621 (2013).
Yoshikawa, H., Tamai, N., Murase, Y. & Myoui, A. Interconnected porous hydroxyapatite ceramics for bone tissue engineering. J. R. Soc. Interface 6, S341–S348 (2009).
Ma, Q. Y., Traina, S. J., Logan, T. J. & Ryan, J. A. In situ lead immobilization by apatite. Environ. Sci. Technol. 27, 1803–1810 (1993).
Hasret, E., Ipekoglu, M., Altintas, S. & Ipekoglu, N. A. Microwave-assisted synthesis of hydroxyapatite for the removal of lead(II) from aqueous solutions. Environ. Sci. Pollut. Res. 19, 2766–2775 (2012).
Pham Minh, D., Tran, N. D., Nzihou, A. & Sharrock, P. Hydroxyapatite gel for the improved removal of Pb2+ ions from aqueous solution. Chem. Eng. J. 232, 128–138 (2013).
Park, S. Y. et al. Sustainable lead management in halide perovskite solar cells. Nat. Sustain. 3, 1044–1051 (2020).
Bailliez, S., Nzihou, A., Bèche, E. & Flamant, G. Removal of lead (Pb) by hydroxyapatite sorbent. Process Saf. Environ. Prot. 82, 175–180 (2004).
Meski, S., Ziani, S. & Khireddine, H. Removal of lead ions by hydroxyapatite prepared from the egg shell. J. Chem. Eng. Data 55, 3923–3928 (2010).
Sugiyama, S., Shimizu, Y., Manabe, T., Nakagawa, K. & Sotowa, K.-I. Preparation of a hydroxyapatite film and its application in the removal and regeneration of aqueous cations. J. Coll. Interf. Sci. 332, 439–443 (2009).
Zhu, Y., Zhu, Z., Zhao, X., Liang, Y. & Huang, Y. Characterization, dissolution, and solubility of lead hydroxypyromorphite [Pb5(PO4)3OH] at 25–45°C. J. Chem. 2015, 269387 (2015).
Bertoni, E., Bigi, A., Falini, G., Panzavolta, S. & Roveri, N. Hydroxyapatite/polyacrylic acid nanocrystals. J. Mater. Chem. 9, 779–782 (1999).
Kandori, K., Fujiwara, A., Yasukawa, A. & Ishikawa, T. Preparation and characterization of hydrophobic calcium hydroxyapatite particles grafting oleylphosphate groups. Coll. Surf. (A) 150, 161–170 (1999).
Zhang, Y. & Lu, J. A simple method to tailor spherical nanocrystal hydroxyapatite at low temperature. J. Nanopart. Res. 9, 589–594 (2007).
Santos, C., Franke, R. P., Almeida, M. M. & Costa, M. E. V. Nanoscale characterization of hydroxyapatite particles by electron microscopy. Microsc. Microanal. 14, 67–70 (2008).
Ramirez, D. et al. Meso-superstructured perovskite solar cells: revealing the role of the mesoporous layer. J. Phys. Chem. C 122, 21239–21247 (2018).
Stranks, S. D. et al. Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 342, 341–344 (2013).
Nikbakht, H. et al. Polymer amplification to improve performance and stability toward semitransparent perovskite solar cells fabrication. Energy Technol. 8, 1900728 (2020).
Li, Z. et al. Laminated carbon nanotube networks for metal electrode-free efficient perovskite solar cells. ACS Nano 8, 6797–6804 (2014).
Rosenman, G. et al. Photoluminescence and surface photovoltage spectroscopy studies of hydroxyapatite nano-Bio-ceramics. J. Lumin. 122-123, 936–938 (2007).
Li, Z. et al. Carbon nanotubes as an efficient hole collector for high voltage methylammonium lead bromide perovskite solar cells. Nanoscale 8, 6352–6360 (2016).
Zhang, S. et al. PVDF-HFP additive for visible-light-semitransparent perovskite films yielding enhanced photovoltaic performance. Sol. Energy Mater. Sol. Cells 170, 178–186 (2017).
Wu, G. et al. Surface passivation using 2D perovskites toward efficient and stable perovskite solar cells. Adv. Mater. 34, 2105635 (2022).
McMeekin, D. P. et al. Crystallization kinetics and morphology control of formamidinium–cesium mixed-cation lead mixed-halide perovskite via tunability of the colloidal precursor solution. Adv. Mater. 29, 1607039 (2017).
Jiang, Q. et al. Planar-structure perovskite solar cells with efficiency beyond 21%. Adv. Mater. 29, 1703852 (2017).
Han, L. et al. Environmental-friendly urea additive induced large perovskite grains for high performance inverted solar cells. Solar RRL 2, 1800054 (2018).
Yang, D. et al. High efficiency planar-type perovskite solar cells with negligible hysteresis using EDTA-complexed SnO2. Nat. Commun. 9, 3239 (2018).
Choi, K. et al. Thermally stable, planar hybrid perovskite solar cells with high efficiency. Energy Environ. Sci. 11, 3238–3247 (2018).
Wu, T. et al. Efficient defect passivation for perovskite solar cells by controlling the electron density distribution of donor-π-acceptor molecules. Adv. Energy Mater. 9, 1803766 (2019).
Wei, D. et al. Ion-migration inhibition by the cation–π interaction in perovskite materials for efficient and stable perovskite solar cells. Adv. Mater. 30, 1707583 (2018).
Men, S., Lovelock, K. R. J. & Licence, P. X-ray photoelectron spectroscopy of trihalide ionic liquids: comparison to halide-based analogues, anion basicity and beam damage. Chem. Phys. Lett. 679, 207–211 (2017).
Chen, Y. et al. Strain engineering and epitaxial stabilization of halide perovskites. Nature 577, 209–215 (2020).
Lei, Y., Xu, Y., Wang, M., Zhu, G. & Jin, Z. Origin, influence, and countermeasures of defects in perovskite solar cells. Small 17, 2005495 (2021).
Eighmy, T. T., Kinner, A. E., Shaw, E. L., Eusden, J. D. Jr & Francis, C. A. Plumbogummite (PbAl3(PO4)2(OH)5⋅H2O) characterization by XPS: an environmentally important secondary mineral. Surf. Sci. Spectra 6, 202–209 (1999).
Chen, Y., Xu, X. & Xie, L. Thermodynamic parameters on corresponding solid-liquid equilibrium of hydroxyapatite in pure and mixture organic solvents. J. Mol. Liq. 229, 189–197 (2017).
Madadrang, C. J. et al. Adsorption behavior of EDTA-graphene oxide for Pb (II) removal. ACS Appl. Mater. Interfaces 4, 1186–1193 (2012).
Giannozzi, P. et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 29, 465901 (2017).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 21, 395502 (2009).
Ghosh, D., Smith, A. R., Walker, A. B. & Islam, M. S. Mixed A-cation perovskites for solar cells: atomic-scale insights into structural distortion, hydrogen bonding, and electronic properties. Chem. Mater. 30, 5194–5204 (2018).
Lanzetta, L. et al. Degradation mechanism of hybrid tin-based perovskite solar cells and the critical role of tin (IV) iodide. Nat. Commun. 12, 2853 (2021).
Senocrate, A. et al. Tuning ionic and electronic conductivities in the “hollow” perovskite {en}MAPbI3. Chem. Mater. 33, 719–726 (2021).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Perdew, J. P. et al. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (2008).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892–7895 (1990).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Slepko, A. & Demkov, A. A. First principles study of hydroxyapatite surface. J. Chem. Phys. 139, 044714 (2013).
Acknowledgements
We would like to thank the EPSRC for funding (EP/R020590/1). This work including TEM access was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1 and EP/P025498/1. S.J.H. acknowledges the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant ERC-2016-STG-EvoluTEM-715502). R.C. and S.J.H. would like to thank the UK Catalysis Hub for resources and support provided via the membership of the UK Catalysis Hub Consortium and funded by EPSRC grant: EP/R027129/1. We gratefully acknowledge supercomputer resources via the MCC/Archer HPC consortium (EP/R029431/1).
Author information
Authors and Affiliations
Contributions
M.Z.M. conducted most of the synthesis, experiments, and data collection. A.A. performed the SEM and helped with device preparation. B.W. conducted the GIXRD/GIWAXS and D.H. assisted with data interpretation. R.S. helped with STEM/HAADF and S.J.H. helped with data interpretation and manuscript writing. B.F.S. conducted the XPS measurements, J.C.-R.K. and A.T. assisted with data interpretation. Q.C. assisted with the cross-sectional SEM measurements. J.J. conducted the IS measurements. R.C. and D.J.L. assisted with writing the manuscript and data interpretation. N.Z. conducted computational modelling and M.S.I. helped with simulation interpretation. B.R.S. wrote much of the manuscript and led the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks Tao Xu and the other anonymous reviewers for their contribution to the peer review of this work. Primary handling editor: John Plummer.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mokhtar, M.Z., Altujjar, A., Wang, B. et al. Spherical hydroxyapatite nanoparticle scaffolds for reduced lead release from damaged perovskite solar cells. Commun Mater 3, 77 (2022). https://doi.org/10.1038/s43246-022-00299-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-022-00299-3
