
Abstract
The possibility that valency changes due to the Kondo effect induce a charge-density-wave (CDW) transition and lead to zero-thermal-expansion by compensating the accompanying structural changes is appealing from both a fundamental and applied physics perspective. Theoretical studies have predicted CDW-order caused by the Kondo effect, whereby a material would exhibit a temperature-dependent dual Kondo effect comprising of two sublattices with different single-ion Kondo temperatures, but its experimental realization remains elusive. Here, we show direct evidence of a dual Kondo effect providing the electronic energy gain for a CDW accompanied by zero-thermal-expansion, in a strongly correlated f-electron material. YbPd undergoes a cubic to tetragonal transition with an incommensurate-CDW below T1 = 130 K, which becomes commensurate below T2 = 105 K. Bulk-sensitive spectroscopy reveals temperature-independent ytterbium single-site mixed-valence above T1, and a clear temperature-dependent mixed-valence charge-disproportionation of two crystallographic ytterbium sites in the CDW phases. Simplified single-impurity Anderson model calculations prove existence of a dual Kondo mixed-valency coupled to the CDW changes associated with the two ytterbium sites, and quantify site-dependent single-ion Kondo temperatures. The dual Kondo temperatures track the evolution of lattice parameters, resulting in a cell-volume compensated Kondo-CDW phase. The results provide a route to develop room temperature intermetallic zero-thermal-expansion materials.
Similar content being viewed by others

Introduction
Many-body interactions often lead to unusual behavior due to an emergent low-energy scale, which is responsible for the low-temperature (T) physical properties of solids. Symmetry breaking and phase transitions are spectacular effects originating in many body interactions. Superconductivity1 and charge-density-wave (CDW)-order2 are typical examples of collective broken symmetry ground states, which can arise from electron-phonon interactions. Singular physics can also emerge from a single orbital with strong on-site Coulomb correlations hybridized with a conduction band, as in the Kondo effect3,4. The coexistence of CDW-order and superconductivity is known for many systems such as transition-metal dichalcogenides5,6, intermetallics, organic and inorganic compounds7, including the high-Tc cuprates8,9. In an early theoretical study of the 1D (dimensional) Kondo-lattice model with strong on-site Kondo coupling J, a CDW-order was predicted at quarter-filling10. More recently, using single-site dynamical mean-field theory (DMFT)11, a bipartite Kondo lattice at quarter-filling showed a pure ferromagnetic phase at low-T, a coexistence regime of ferromagnetism and CDW-order at intermediate T, and a paramagnetic insulating CDW phase at higher T. In another study12 using cellular (C)-DMFT and variational Monte-Carlo calculations in 2D, it was shown that an antiferromagnetic (AF) insulating CDW phase at T = 0 became a paramagnetic CDW-metal at small T. A CDW phase characterized by charge disproportionation due to partial Kondo screening was also reported for the periodic Anderson model on a 2D triangular lattice with repulsive on-site Coulomb interactions even in the absence of inter-site Coulomb interactions13,14,15. Surprisingly, in spite of these theoretical predictions10,11,12,13,14,15, there is no experimental evidence of a material exhibiting CDW-order due to the Kondo effect. In particular, while f-electron containing CDW systems have been reported, spectroscopic studies showed that the CDW was associated with non-f electrons16,17. A recent study18 on UPt2Si2 suggested interplay between CDW-order and Kondo-lattice behavior before AF-order sets in at TN = 35 K, but spectroscopic evidence of the role of U 5f electrons is awaited. Considering the more general case of Kondo effect modifying lattice properties, a Kondo mixed-valence driven zero-thermal-expansion (ZTE) was proposed19 to explain the properties of YbGaGe because Yb2+ ions exhibit a larger crystal radius than Yb3+ ions. However, later studies showed that it was due to interstitial impurities and not mixed-valence20,21,22. Further, in the anomalous thermal expansion square-net compounds RE4MGe8 (RE = Yb, Gd, M = Cr-Ni, Ag), which showed CDW modulations, the role of f-electron mixed-valence was ruled out by comparing RE properties23.
YbPd is a metal that exhibits two sharp transitions in the specific heat at T1 = 130 K and T2 = 105 K, becoming more metallic with steps in electrical resistivity at T1 and T2, followed by AF-ordering at TN = 1.9 K arising from a competition between Ruderman-Kittel-Kasuya-Yosida (RKKY) interaction and the Kondo effect24,25,26,27,28. The resistivity further reduces below TN, and YbPd remains metallic down to the lowest measured temperature of 40 mK28. YbPd lies between trivalent YbPd3 and divalent Yb3Pd in the Yd-Pd phase diagram24, corroborating the Yb valence instability evidenced by experiments. 170Yb-Mössbauer measurements at T = 0.05 K and 1.4 K confirmed the AF-ordering below TN and mixed-valence indicating two Yb sites with differing charge states, one magnetic and the other non-magnetic25. The Yb3+ site showed a magnetic moment much smaller than the free ion value, inconsistent with a Γ8 symmetry ground state inferred from inelastic neutron scattering26, suggesting a significant 4f-conduction band hybridization25. Further, the other non-magnetic Yb site suggested a stronger hybridization indicative of a dominant Kondo effect25,26.
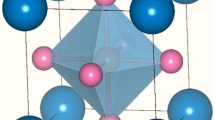
X-ray absorption spectroscopy (XAS) at the Yb-L3 edge28 and low-energy photoemission29 showed mixed-valency with an average Yb valence vav ∼ 2.8 at T = 300 K. While many studies have shown that T-dependent high-energy spectroscopies are powerful to analyze the mixed-valence of Ce and Yb compounds and estimate the Kondo energy30,31,32,33,34, such an analysis of YbPd has not been reported, although T-dependent valency changes were reported28. Recent X-ray diffraction studies revealed an incommensurate (I)-CDW order below T1 = 130 K with a wave vector Q1 = (±0.07 ± 0.07 1/2), which becomes a commensurate (C)-CDW at T2 = 105 K, with wave vector Q2 = (0 0 1/2)27,35. The high-T cubic YbPd with lattice parameters a = b = c and one Yb crystallographic site, deforms into a tetragonal (Tʹ) structure below T1 with aT = bT \(\ne\) cT [see Fig. 1(a), (b)] and a displacement of Pd atoms along the c-axis. This leads to a unit-cell doubling along the c-axis (2cT = cTʹ) in the Tʹ-phase with a trivalent magnetic Yb1-site and a mixed-valent non-magnetic Yb2-site with valency v ~ 2.6, consistent with the Mössbauer study25.

Panels a and b show the schematic crystal structure in the cubic and tetragonal phases of YbPd. The cubic phase has a single crystallographic site, while the tetragonal phase has two distinct crystallographic sites, Yb1 and Yb2, respectively. Panel c shows the experimental Yb-3d5/2 core-level HAXPES of YbPd between 20 K and 200 K. The solid black line is the Shirley background subtracted to estimate the Yb-3d5/2-spectral weight. Below the CDW transition (T1 = 130 K), the spectra exhibit a T-dependent valence change but are T-independent above T1. The experimental and calculated difference spectra for the T = 20 K and T = 115 K obtained by subtracting the T = 200 K spectrum shows consistent change in mixed valence as discussed in the text, while the difference spectra for T = 135 K and T = 160 K show negligible changes.
A CDW transition is governed by a competition of elastic and electronic energies since a structural distortion with positive energy gets compensated by a negative electronic energy. In the classic 1D-Peierls transition, the electronic energy stabilization occurs via electron-phonon coupling with a Kohn anomaly, divergence in the electronic susceptibility, and Fermi surface nesting (FSN), which opens a gap in electronic states2. For some quasi-2D-systems like 2H-NbSe2, a more general momentum-dependent electron-phonon coupling causes an I-CDW with maxima in the electronic susceptibility without FSN5,6,36,37. In an alternative picture for 2H-NbSe2, and for Cr-metal, both of which show continuous pressure-driven quantum-critical points38, the I-CDW/I-SDW (spin-density-wave) transitions were discussed as itinerant density wave instabilities, rooted in long range electron-electron correlations and exchange interactions39. Since YbPd is a 3-D cubic system at high-T, which becomes more metallic in the CDW phase28, it suggests absence of FSN. Band structure calculations in the local density approximation with on-site Coulomb interaction U (LDA + U) have not found FSN, but show that YbPd is a strongly correlated metal with an enhanced quasiparticle effective mass factor of ~9, by comparing the calculated specific heat γ (69.99 mJ/(K2mol)) with the experimental value of γ (~600 mJ/(K2mol))28,40. However, since YbPd shows relatively large CDW ordering temperatures (T1 = 130 K, T2 = 105 K) compared to TN = 1.9 K, it indicates strong charge fluctuations compared to spin fluctuations, thereby ruling out Kondo coherence properties in which charge fluctuations are generally considered unimportant41.
In the following, we show that the strongly correlated f-electrons of YbPd exhibit a dual Kondo effect CDW, which leads to ZTE. From a single-impurity Anderson model (SIAM) analysis of the T-dependent mixed-valence observed in the Yb 3d core level and Yb 4 f valence band spectra, we obtain site- and T-dependent single-ion Kondo temperatures (TK) across the CDW transitions. The site- and T-dependent TK values follow the evolution of lattice parameters in the CDW phase. The results show a direct link of the dual Kondo effect with structural changes, leading to a cell-volume compensated Kondo-CDW phase.
Results and discussions
Temperature-dependent mixed valence
T-dependent measurements of well-characterized YbPd were carried out using bulk-sensitive hard X-ray photoemission spectroscopy (HAXPES)42 to study the Yb valence across the CDW transition. Figure 1(c) shows the HAXPES Yb 3d5/2 core-level spectra for several temperatures between T = 20 K–200 K. The spectra have been normalized to the total integrated intensity. The T = 200 K spectrum exhibits features consisting of a single peak due to 3d4f14 (Yb2+) states at ∼1520 eV binding energy(BE), and a structured feature centered at 1530 eV BE corresponding to the 3d4f13 (Yb3+) multiplet states. The presence of Yb2+ and Yb3+ features arising from a single crystallographic site indicates mixed-valent Yb ions. Indeed, in many Yb compounds30,31,32,34,43,44, the ground state is a hybridized state with a non-integer 4 f number and the spectral weight ratio of Yb2+ and Yb3+ features provides a measure of mixed-valence. A reliable quantitative approach based on the SIAM was developed by Gunnarsson and Schönhammer45 for determining the 4f-valence from core-level PES, which showed that on-site Coulomb energy Uff between f-electrons causes the ~10 eV BE separation between Yb2+ and Yb3+ features. More importantly, Fig. 1(c) shows a clear T-dependent spectral-weight transfer from Yb3+ to Yb2+ upon cooling, consistent with earlier work28 but in contrast to a constant average mixed-valency claimed recently from L-edge XAS measurements35. The T-dependence indicates a decrease in average Yb valence vav with decreasing T, which was also reproducible by T-cycling. The change in valency upon cooling cannot be due to oxidation because oxidation would lead to an increase in Yb3+at low-T, and our results show the opposite with an increase in Yb2+ content at low-T. Two T-ranges can be identified: above T ≥ 135 K, the spectra show negligible changes, while from T = 115 K to 20 K, the spectra are T-dependent. In Fig. 1(c), we also plot the experimental difference spectra for T = 20 K, 115 K, 135 K and 160 K with respect to the 200 K spectrum. Since the I-CDW sets in at T1 = 130 K, it is understood that the spectra show changes only below, and not above T1. We have also plotted the calculated difference spectra for T = 20 K and 115 K with respect to T = 200 K, which show similar mixed-valence changes, and are discussed in detail in the following. Interestingly, the peak energy positions and peak widths of the Yb2+ and Yb3+ features show negligible T-dependence, in contrast to CDW transitions reported for transition-metal dichalcogenides46, hinting at an unusual CDW in YbPd. This is likely due to the fact that for Yb2+ (4f14) states, the 4f shell is fully filled making it effectively insensitive to changes in peak shape and position, while for Yb3+ (4f13) states, the core levels are dominated by multiplet effects spread over much larger energies (~10 eV) than typical CDW-induced changes in peak widths and positions of ~0.2 eV (ref. 46).
What could be the physical origin of this T-dependence? The core-level and valence band PES, XAS, and resonant inelastic X-ray scattering of Ce and Yb-based Kondo systems30,31,32,33,34 have shown a T-dependent mixed-valence given by 4f electron occupation number, nf for Ce-ions, or hole number nf = 14 – nf for Yb-ions (the Yb-valence, v = 2 + nf). In the Anderson model, a universal scaling law of (1 – nf) as a function of TK is expected, where TK is the single-ion Kondo temperature3. A recent extensive study47 on many Yb compounds showed a similar scaling law of (1 – nf) with TK. The Kondo ground state is a non-magnetic singlet state composed of mainly 4f13 (nf = 1) and 4f14 (nf = 0) configurations. The first excited states at ε = kBTK (kB is the Boltzmann constant) are essentially composed of magnetic 4f137/2 states corresponding to the singlet breakdown. This low-energy scale explains the crossover from the low-T non-magnetic Fermi liquid regime (mixed-valent state) to the high-T local-moment regime (trivalent states)3,4. A continuous T-dependence of nf through TK is expected, but in the present case, since the spectra are T-independent above T = 130 K, it suggests that the T-dependence is associated only with the CDW phase.
Simplified SIAM calculations of core-level and valence band spectra
In order to understand the T-dependent spectral behavior, we have developed SIAM calculations for the 3d PES and 4f valence band spectra in the simplified zero-bandwidth limit48, but including orbital degeneracy of 4f states. This method simulates the T-dependence of high-energy scale 4f multiplets and also provides the emergent low-energy scale behavior via nf and TK. We use a direct diagonalization in a restricted basis consisting of one f14-state, 14 f13-states (with the two spin-orbit manifolds) and 91 f12-states. The parameters of the SIAM are the energy of a 4f-hole [εf = E(f14) − E(f137/2)], the Coulomb interaction (Uff) between 2f-electrons/holes and the hybridization strength ∆ of 4f states with the conduction band, which couples different Yb configurations. We include the spin-orbit energy εso = 1.27 eV, which has a negligible effect on core-level spectra, but is important for valence band spectra. To simulate the 3d core-level spectra and obtain a good match with a full atomic-multiplet calculation required 4 energy levels, as detailed in Supplementary Note 1 and Supplementary Fig. 1. A systematic evolution of the 3d PES spectral features was confirmed by changing only ∆, with all other appropriate electronic parameters kept fixed (see Supplementary Fig. 2 and Supplementary Note 2). With increasing ∆, a systematic spectral-weight transfer from the 3d4f13 (Yb3+) to the 3d4f14 (Yb2+) feature is obtained with a decrease of Yb vav. Thus, we could simulate the high-T (T = 135 K, 160 K, and 200 K spectra are nearly identical) experimental spectra with the following parameters: εf = −0.2 eV, Uff = 7 eV, εso = 1.27 eV and ∆ = 38 meV. The good match of the experimental spectrum for T = 200 K with the calculation, as shown in Fig. 2(a), indicates the important role of hybridization in the high-T phase, with an average valence vav = 2.84 (nf = 0.84) for the single Yb-site.

a Comparison of experimental spectra (symbols) at T = 200 K and 20 K with simulated spectra (solid lines). The simulated spectrum for T = 20 K is composed of two contributions, one corresponding to the nearly trivalent Yb1 site (dashed lines) and the other one to the mixed-valent Yb2 site (dotted lines) of the tetragonal phase, while for T = 200 K, the two sites become equivalent in the cubic phase. Panels b and c show the variation of the calculated spectra for Yb1 site and Yb2 site at temperatures between 20 K and 200 K consistent with the systematic temperature-dependent evolution of the mixed valency. The individual comparison with experimental spectra for intermediate temperatures are shown in Supplementary Fig. 3.
In the CDW phases below 130 K, the displacement of Pd atoms results in two Yb-Pd interatomic distances, with a reduced hybridization for one site and an enhanced hybridization for the other site. As discussed above, at the lowest T, the CDW ordering yields one nearly trivalent magnetic Yb1-site and a strongly mixed-valent non-magnetic Yb2-site (v = 2.6)35. Hence, below T = 130 K, each experimental spectrum is composed of two contributions. The spectrum at T = 20 K for the nearly trivalent site is mainly composed of the 3d4f13 structure and can be simulated with a very small hybridization. The nearly trivalent state with a very small TK (≡TK1 for Yb1-site) is needed for the magnetic-ordering below TN25,27. The contribution of the other strongly mixed-valent site with a large TK (≡TK2 for Yb2-site) is then calculated ensuring the sum of the two calculated contributions matches with the experimental spectrum subject to the condition |vav – v1| = |vav – v2|, i.e., increase in valency for site-1 matches the decrease in valency for site-2, for all T. The results of this procedure are also plotted in Fig. 2(a) where we show the calculated spectra for the Yb1- and Yb2-sites, their sum and the experimental spectrum at T = 20 K. A satisfactory agreement is found with the nearly trivalent (very small mixed valency) Yb1-site spectrum simulated with ∆1 = 11 meV (v = 2.98, nf1 = 0.98) and the strongly mixed-valent Yb2-site spectrum simulated with ∆2 = 260 meV (v = 2.60, nf2 = 0.60), with vav = 2.79. Similarly, we have calculated the spectra for all measured temperatures in the tetragonal CDW phase, as shown in Fig. 2(b), (c). A direct comparison between experimental and calculated spectra for the intermediate-T are shown in Supplementary Fig. 3. The reduction in vav from 2.84 (T = 135–200 K) to 2.79 (at T = 20 K) indicates a charge transfer of 0.05 electrons/Yb. Since the crystal radii of Yb sites depend on its valency (8-co-ordinated Yb2+ has a radius 1.28 Å while 8-co-ordinated Yb3+ has a radius of 1.125 Å, ref. 49), the valency change provides a natural way for compensating the lattice-parameter changes with temperature.
As an independent check of T-dependent changes, we measured the HAXPES valence band of YbPd in the high-T cubic phase (T = 200 K) and in the tetragonal IC-CDW (T = 115 K) and C-CDW (T = 20 K) phases, as shown in Fig. 3(a). We see a small but clear change in the Yb2+ features (at the Fermi level and 1.7 eV binding energy) and Yb3+ features (spread between 6 and 12 eV binding energies) while the Pd 4d states do not change with T. We have also measured the Pd 3d core-level spectra and they also do not change with T, as discussed in Supplementary Note 3 (see Supplementary Fig. 4). Using the same method as that for the core-level spectra with appropriate parameters for the Yb2+ (f13) and Yb3+ (f12) multiplets, and all other parameters fixed, we calculated the valence band spectra for T = 20 K, 115 K, and 200 K (see Supplementary Note 4 for details). We could obtain a good match between calculated and experimental spectra as shown in Fig. 3(a), for the same values of nf as for Yb 3d core-level spectra. The systematics for the calculated spectra for the Yb1- and Yb2-sites are shown in Fig. 3(b), (c), respectively. The comparison of individual calculated spectra with experimental spectra are shown in Supplementary Fig. 5.

Panel a shows T-dependent HAXPES valence band of YbPd after background subtraction in the low-temperature tetragonal commensurate CDW phase (T = 20 K), incommensurate CDW phase (T = 115 K) and in the normal cubic phase (T = 200 K). The experimental spectra are compared with the SIAM calculations for describing the 4f spectral changes in the valence band across the CDW transition. Panels b and c show the variation of the calculated spectra at T = 20 K, 115 K, and 200 K for Yb1 site and Yb2 site, respectively. The individual comparison with experimental spectra are shown in Supplementary Fig. 5.
Dual Kondo effect coupled charge ordering
The obtained valencies for the two sites and vav in the CDW phases, and for the normal phase above T1 = 130 K are plotted in Fig. 4(a), and corresponding nf values are listed in Table 1. The results confirm that all Yb sites are equivalent with the same nf in the high-T cubic phase and undergo a systematic evolution to a dual Kondo mixed-valence of crystallographically distinct Yb1 and Yb2 sites with inequivalent nf1 and nf2 in the low-T tetragonal CDW phase. Thus, since nf scales with TK3,47, the evolution of Kondo mixed-valence corresponds to a gradual transition with charge-disproportionation and results in a bipartite lattice with two distinct single-ion Kondo temperatures TK1 and TK2. Two distinct TK values arising from inequivalent crystallographic sites was originally discussed50 for the cubic heavy fermion system Ce3Pd20Si6. However, it was soon shown that Ce3Pd20Si6 exhibits a quantum-critical transition coupled to destruction of the Kondo effect at very low temperatures51. These experimental results motivated a theoretical study of a Kondo-lattice model consisting of two local-moment sublattices with different Kondo couplings52. While the possibility of CDW order was not investigated, it was shown that depending on the conduction band filling nc, the two Kondo effects compete (for nc < ½) or co-operate (for nc > ½) with each other, where nc = ½ corresponds to the quarter-filled conduction band and is a special point. Subsequently, the quarter-filled Kondo-lattice model was studied using C-DMFT and variational Monte-Carlo calculations12 and it was shown that the ground state is an antiferromagnetic insulating CDW phase at T = 0, which becomes a paramagnetic CDW metal on increasing T. This behavior is reminiscent of the low-temperature properties of YbPd discussed in the introduction. Further, in Fig. 4(a), we have compared the experimental valencies for the two sites with the reported values of the change in the site occupancies n(a) and n(b) as a function of T/t, where T is temperature and t is the hopping integral12. In ref. 12, the authors calculated n(a) and n(b) using a 2D Kondo-lattice model for a two-site (a and b) C-DMFT for the CDW ordered phase12. They could show that charge-order (CO) appears in the intermediate Kondo coupling region with 1.5 ≤ J/t ≤ 3.5 region (J is Kondo coupling strength and t is the hopping), accompanied by a collinear AF-order at the charge-poor sites. In contrast, the charge-rich sites become non-magnetic. It is noted that a 2D Kondo-lattice model does not allow a variation in the localized f-electron charge density, but it allows an increase or decrease in conduction band charge density on the localized f-electron sites due to Kondo coupling. Accordingly, it was shown that the local Kondo singlet at the non-magnetic site is necessary for stabilizing the CO. It is also noted that YbPd is not a quarter-filled system but shows an average valency of vav = 2.84 (T = 135–200 K), which becomes 2.79 at T = 20 K. However, the comparison shows qualitatively similar gradual behavior but the magnitude of the valence change, n(a) – n(b), is roughly half of the experimental results. The small difference in the high-T phase is attributed to the fact that YbPd shows a mixed-valence evolution between high-T non-CDW and low-T CDW phases, while the Kondo-lattice model12 has no mixed-valence in the non-CDW phase. It is also important to discuss the role of inter-site Coulomb interactions as an origin of the CDW transition in YbPd. It was shown53 that in the extended Hubbard model, it is possible to obtain a metallic CDW due to inter-site Coulomb interaction Vʹ with changes in the band density of states near the Fermi level. However, this phase occurs only for small U/W, where U is the on-site Coulomb interaction and W is the bandwidth, while for large values of U/W, one obtains a metal to insulating CDW. However, YbPd exhibits a very large on-site Coulomb energy (Uff ~ 7 eV as estimated from our calculations) and changes in the core levels and valence band occur roughly over the same energy scale. Thus, although we cannot totally rule out effects due to inter-site Coulomb interactions on the experimental spectra, we feel the metallic CDW in YbPd is not caused by inter-site Coulomb interactions.

a Below the CDW transition at T1 = 130 K, the two different Yb sites exhibit T-dependent valencies, whereas above T1 the valency of the unique Yb site is constant up to 200 K. The calculated local charge density, n(a) and n(b) (●,■) as a function of T/t, where T is temperature and t is the hopping integral (ref. 12). It shows a gradual CDW formation indicating a second-order transition. b Comparison of T-dependent Kondo temperatures and lattice-parameters (refs. 27,35) across the cubic-to-tetragonal CDW phase transition. The cubic lattice-parameter measured for present sample at T = 300 K is shown as a red circle ( ). c T-dependence of the Kondo energy balance showing CDW order parameter follows the BCS model. d Plot of cubic unit-cell volume (>T1) with cell-volume V = aT \(\times\) bT \(\times\) cT (=half the tetragonal unit-cell volume (<T1), as a function of T. The solid and open symbols represent the tetragonal and cubic phases, respectively. The cell-volume V shows ZTE within error bars in the low-T CDW phase; error bars based on data of lattice-parameters from Mitsuda et al.27 and Takahashi et al.35. The measured cell volume at T = 300 K for present sample is shown as red circle (
). c T-dependence of the Kondo energy balance showing CDW order parameter follows the BCS model. d Plot of cubic unit-cell volume (>T1) with cell-volume V = aT \(\times\) bT \(\times\) cT (=half the tetragonal unit-cell volume (<T1), as a function of T. The solid and open symbols represent the tetragonal and cubic phases, respectively. The cell-volume V shows ZTE within error bars in the low-T CDW phase; error bars based on data of lattice-parameters from Mitsuda et al.27 and Takahashi et al.35. The measured cell volume at T = 300 K for present sample is shown as red circle ( ). The dashed line (----) is the average cell volume below T1. For T > T2 = 105 K, the cell-volume V for data from ref. 35 shows slightly larger deviation compared to ref. 27. Error bars represent standard error (SE).
). The dashed line (----) is the average cell volume below T1. For T > T2 = 105 K, the cell-volume V for data from ref. 35 shows slightly larger deviation compared to ref. 27. Error bars represent standard error (SE).
Kondo mixed-valence, charge-ordering, and structural transition
What is the mechanism linking the mixed valency, CDW and the structural transition in YbPd? Since FSN is unlikely in YbPd, an Ising model with a ferro-coupling of nearest neighbors and an AF-coupling of next-nearest neighbors was proposed, which resulted in a phase diagram with I-CDW and C-CDW regions35. However, the very low Neel temperature, TN = 1.9 K for AF-ordering and the high CDW ordering temperature, T1 = 130 K suggests a different origin. Since our results evidence an interplay between strong mixed-valence and the CDW, and strong mixed-valency is a signature of high TK, it naturally led us to explore an alternative mechanism based on the Yb-site Kondo energies in the two phases. The Kondo energy kBTK represents the electronic energy gained by the formation of a singlet ground state. It was recently shown by Kummer et al.47 that the scaling between TK and (1 − nf) obeys a power law (1 − nf) ≈ ATKα (with α = 2/3 and A = 1/200) over 4 orders of magnitude of TK for many Yb compounds. We used this power law behavior to estimate TK from our experimentally determined nf-values, but a qualitatively similar scaling of (1 – nf) with TK is also known in the non-crossing approximation3. In the cubic phase, TK is estimated to be TKcub = 181 K. Similarly, in the tetragonal phase at T = 20 K, the estimated dual Kondo temperatures for the two sites are TK1 = 8 K and TK2 = 718 K. This is in good agreement with TK values of ∼30 K and ∼800 K estimated from a recent point contact spectroscopy study54. TK1 and TK2 for all measured temperatures are plotted in Fig. 4(b) and listed in Table 1. In Fig. 4(b), we have also overlaid the lattice-parameter values as a function of temperature reported in the literature27,35. The TK1 and TK2 values show a strong similarity with the T-dependent lattice-parameter values reported by Mitsuda et al.27 within error bars, thereby indicating a strong coupling to the lattice. It is also consistent with the data of Takahashi et al.35, if the data are shifted along the y-axis. While we cannot provide a definitive reason for small differences in the data of Takahashi et al., one possibility is that small variations in Yb content can lead to small variations in structural parameters, but nonetheless, the low-temperature CDW phase is correctly observed in both the data sets. If the Kondo coupling to the lattice represents the dominant contribution to the CDW transition, it can be expected to define the CDW order parameter. In order to check the relation of Kondo energies with the CDW order, we plotted the quantity Ψ(T) = TK1 + TK2 − 2TKcub, which equals the electronic energy gain due to the Kondo mechanism in the tetragonal low-temperature phase with respect to the high-temperature cubic phase. The T-dependence of Ψ is plotted in Fig. 4(c) and it is compared with the mean-field Bardeen-Cooper-Schrieffer (BCS)-type gap behavior for superconductivity1,55, which can also be considered as a valid CDW order parameter56,57. The details of the fit to the BCS-type gap function are described in Supplementary Note 5. The T-dependence of Ψ follows the BCS-type gap behavior. The results thus indicate that the dual Kondo mixed valency provides the CDW order parameter of YbPd, consistent with theoretical results reported for the Kondo lattice12, but further theoretical studies are necessary for a better quantitative description of the Kondo-CDW phase in terms of f-electron/hole occupancy. It is noted that our overall results are consistent with the Mössbauer results25, thermodynamic properties with an enhanced effective mass28,40 due to 4f-electrons at the Fermi level as well as the magnetic ordering at low temperatures25,28, as it is expected that TK1 for the nearly trivalent magnetic Yb atoms will decrease further and become comparable to TN = 1.9 K.
Zero thermal expansion in CDW phase
Given that the T-dependence of TK1 and TK2 follows the structural data shown in Fig. 4(b), in order to compare with the cubic unit-cell volume above the CDW transition, we calculated the T-dependent unit-cell volume V = aT \(\times\) bT \(\times\) cT (this is actually half the tetragonal unit-cell volume) as shown in Fig. 4(d), using the lattice-parameter values reported in the literature27,35. The results indicate a ZTE in the CDW phase, followed by a small gradual positive thermal expansion in the cubic phase. This is consistent with the mechanism we propose in terms of Kondo coupling being responsible for generating the ZTE in the low-temperature tetragonal phase, and it is not operative in the cubic phase. The ZTE behavior of YbPd has not been discussed in earlier structural studies of YbPd26,27,35. Since materials normally expand on heating, in order to obtain ZTE, it is necessary to consider a mechanism to obtain negative thermal expansion (NTE). NTE has been observed in several insulating oxides58,59,60 such as CuScO2, Y2W3O12, and ZrW2O8, where it was shown that if there is a M-O-Mʹ (or O-M-O) linkage (M:metal atom; O:oxygen), then a transverse vibration of the central atom O (or M) perpendicular to the M-O-Mʹ (or O-M-O) linkage results in a decrease of the linkage length. Similarly, ZTE reported for the square-net compounds RE4MGe8 was explained on the basis of a square-pyramidal configuration of the M-atom positioned above or below a Ge square net, allowing for transverse vibrations of the M atom perpendicular to the Ge square net23. Since the structure of YbPd involves Yb square-nets with a square-pyramidal arrangement of Pd atoms, which get displaced from the body-centered position in the tetragonal phase along the c-axis (see Fig. 1b), it is considered that a mechanism involving transverse vibrations of Pd atoms along the c-axis (perpendicular to the Yb square-nets) is possible in YbPd. However, it is noted that a recent inelastic X-ray scattering study hinted at the role of a lattice instability in terms of a momentum (k)-dependent softening of phonons around the X (L)-point in the Brillouin zone at room temperature61. However, T-dependent measurements were carried out only at the X (L) k-point and T-dependent phonon softening directly implicating a CDW transition was not found. Further work will shed light on the details of the Yb-Pd distances, Yb-Pd-Yb bond angles and the role of phonons as a function of temperature in YbPd. However, the remarkable similarity of T-dependent variation of TK1 and TK2 values with the T-dependent lattice-parameter values suggests that the Kondo mechanism dominates the structural changes, and electronic energy changes due to electron–phonon coupling are expected to be weak. A very recent study showed a clear NTE in Y-doped SmS based on the change of ionic radii coupled to mixed-valency, albeit in the absence of a CDW62. It is noted that the ZTE or NTE mechanisms discussed above are distinct from that in the first-known Fe-Ni invar alloys, in which the ZTE behavior is derived from the non-collinear alignment of spins63.
Conclusions
The temperature-dependent mixed-valence observed in the Yb 3d core-level and Yb 4f valence band spectra of YbPd could be simulated by SIAM calculations, and provide a site-dependent quantification of the single-ion Kondo temperatures across the CDW transitions. The temperature-dependent dual Kondo temperatures TK1 and TK2 are shown to follow the changes in lattice parameters in the CDW phase, thus linking the structural changes with the Kondo effect. Further, since the crystal radius of Yb2+ is larger than that of Yb3+, it results in an electronic compensation of structural changes leading to ZTE in the CDW phase. The discovery of a dual Kondo mixed-valence coupled charge ordering that leads to ZTE in YbPd will hopefully motivate the search for new intermetallic materials, which exhibit ZTE at room temperature and above for useful applications.
Methods
Sample preparation
The YbPd compound was synthesized from high purity elements with the Yb1.03 Pd starting composition to compensate for the Yb vaporization at elevated temperature25. The mixture of the elements was placed in a Ta crucible, which was sealed by arc-welding under pure Ar atmosphere. The crucible and its content were heated up to about 1550 °C in an induction furnace for several minutes (the melting temperature of YbPd is reported to be 1460 °C). The melted sample was single phasic with the CsCl structure and the unit-cell parameter at room temperature, a = 3.446(3) Å, in good agreement with reported value27.
Hard X-ray photoemission spectroscopy (HAXPES) experiments
HAXPES was used to measure the Yb and Pd core levels and valence band of YbPd. The HAXPES measurements were carried out at the Taiwan beamline BL12XU, SPring-8 in Hyogo, Japan using a linearly polarized incident beam of photon energy hν = 6500 eV. We used a liquid He flow-type cryostat to cool the sample down to T = 20 K. The Fermi-edge (EF) of a gold thin film was measured at T = 20 K to obtain the energy resolution and calibrate the binding energy (BE) scale. The estimated total energy resolution of HAXPES is 0.25 eV, obtained by fitting the Fermi-edge of gold. The YbPd polycrystals were cleaved in an ultrahigh vacuum preparation chamber at 1 × 10–9 mbar, and immediately transferred to the main chamber at 5 × 10–10 mbar for the measurements. Temperature cycling was carried out to confirm the T-dependence of the reported spectra in two different sets of experiments. Thanks to the high photon energies used for the present measurements, the valence band spectra did not show the Yb 4f surface states normally seen in low-energy photoemission29,32. The O1s and C 1s spectral range were used to monitor surface contamination and the surfaces were clean (<1% intensity for O 1s and C 1s signals) and stable for ~16 h after cleaving.
Data availability
The data sets generated/analyzed during the current study are available from the corresponding author on reasonable request.
References
Bardeen, J., Cooper, L. N. & Schrieffer, J. R. Theory of superconductivity. Phys. Rev. 108, 1175–1204 (1957).
Peierls, R. E. Quantum Theory of Solids. 108–112 (Oxford Univ. Press, 1955).
N.E. Bickers, N. E., Cox, D. L. & Wilkins, J. W. Self-consistent large-N expansion for normal-state properties of dilute magnetic alloys. Phys. Rev. B 36, 2036–2079 (1987).
Coleman, P. in Handbook of Magnetism and Advanced Magnetic Materials (eds Kronmüller, H. & Parkin, S.) (John Wiley and Sons, 2007).
Wilson, J. A., Di Salvo, F. J. & Mahajan, S. Charge-density waves and superlattices in the metallic layered transition metal dichalcogenides. Adv. Phys. 24, 117–201 (1975).
Kiss, T. et al. Charge-order-maximized momentum-dependent superconductivity. Nat. Phys. 3, 720–725 (2007).
Gabovich, A. M., Voitenko, A. I. & Ausloos, M. Charge- and spin-density waves in existing superconductors: competition between Cooper pairing and Peierls or excitonic instabilities. Phys. Rep. 367, 583–709 (2002).
Tranquada, J. M., Sternlieb, B. J., Axe, J. D., Nakamura, Y. & Uchida, S. Evidence of stripe correlations of spins and holes in copper oxide superconductors. Nature 375, 561–563 (1995).
Ghiringhelli, G. et al. Long-range incommensurate charge fluctuations in (Y,Nd)Ba2Cu3O6+x. Science 337, 821–825 (2012).
Hirsch, J. E. Strong-coupling expansion for a Kondo-lattice model. Phys. Rev. B 30, 5383–5385 (1984).
Peters, R., Hoshino, S., Kawakami, N., Otsuki, J. & Kuramoto, Y. Charge order in Kondo lattice systems. Phys. Rev. B 87, 165133 (2013).
Misawa, T., Yoshitake, J. & Motome, Y. Charge order in a two-dimensional Kondo lattice model. Phys. Rev. Lett. 110, 246401 (2013).
Hayami, S., Udagawa, M. & Motome, Y. Partial disorder in the periodic Anderson model on a triangular lattice. J. Phys. Soc. Jpn. 80, 073704 (2011).
Hayami, S., Udagawa, M. & Motome, Y. Partial disorder and metal–insulator transition in the periodic Anderson model on a triangular lattice. J. Phys. Soc. Jpn. 81, 1030707 (2011).
Aulbach, M. W., Assaad, F. F. & Potthoff, M. Dynamical mean-field study of partial Kondo screening in the periodic Anderson model on the triangular lattice. Phys. Rev. B 92, 235131 (2015).
Hossain, Z. et al. Coexistence of magnetic order and charge density wave in a Kondo lattice Yb5Ir4Si10. Phys. Rev. B 71, 060406 (2005).
Brouet, V. et al. Angle-resolved photoemission study of the evolution of band structure and charge density wave properties in RTe3 (R=Y, La, Ce, Sm, Gd, Tb, and Dy). Phys. Rev. B 77, 235104 (2008).
Lee, J. et al. Charge density wave with anomalous temperature dependence in UPt2Si2. Phys. Rev. B 102, 041112 (R) (2020).
Salvador, J. R., Guo, F., Hogan, T. & Kanatzidis, M. G. Zero thermal expansion in YbGaGe due to an electronic valence transition. Nature 425, 702–705 (2003).
Bobev, S., Williams, D. J., Thompson, J. D. & Sarrao, J. L. Thermal expansion in YbGaGe. Solid State Commun. 131, 431–433 (2004).
Muro, Y. et al. Divalent state in YbGaGe: magnetic, thermal, transport and structural studies. J. Phys. Soc. Jpn. 73, 1450–1452 (2004).
Doyle, B. P. et al. Temperature-independent ytterbium valence in YbGaGe. Phys. Rev. B 75, 235109 (2007).
Peter, S. C., Chondroudi, M., Malliakas, C. D., Balasubramanian, M. & Kanatzidis, M. G. Anomalous thermal expansion in the square-net compounds RE4TGe8 (RE = Yb, Gd; T = CrNi, Ag). J. Amer. Chem. Soc. 133, 13840–13843 (2011).
Iandelli, A. & Palenzona, A. The crystal structure and lattice constants of RE3Pd4, Y3Pd4 and Th3Pd4 compounds. Rev. Chim. Miner. 10, 303 (1973).
Bonville, P., Hammann, J., Hodges, J. A., Imbert, P. & Jéhanno, G. J. Magnetic and nonmagnetic charge states in YbPd. Phys. Rev. Lett. 57, 2733–2766 (1986).
Walter, U. & Wohlleben, D. Unusual magnetic response of intermediate-valent YbPd and Yb3Pd4 as studied by inelastic neutron scattering. Phys. Rev. B 35, 3576–3584 (1987).
Mitsuda, A. et al. Origins of phase transitions in valence fluctuating YbPd. J. Phys. Soc. Jpn. 82, 084712 (2013).
Pott, R. et al. Magnetic order and other phase transitions in mixed-valent YbPd. Phys. Rev. Lett 54, 481–484 (1985).
Domke, M. et al. Bulk and surface valence in YbPdx compounds. Phys. Rev. B 32, 8002–8006 (1985).
Malterre, D., Grioni, M. & Baer, Y. Recent developments in high energy spectroscopies of Kondo systems. Adv. Phys. 45, 299–348 (1996).
Allen, J. W. The Kondo resonance in electron Spectroscopy. J. Phys. Soc. Jpn 74, 34–48 (2005).
Tjeng, L. H. et al. Temperature dependence of the Kondo resonance in YbAl3. Phys. Rev. Lett. 71, 1419–1422 (1993).
Reinert, F. et al. Temperature dependence of the Kondo resonance and its satellites in CeCu2Si2. Phys. Rev. Lett. 87, 106401 (2001).
Moreschini, L. et al. Comparison of bulk-sensitive spectroscopic probes of Yb valence in Kondo systems. Phys. Rev. B 75, 035113 (2007).
Takahashi, R. et al. Valence ordering in the intermediate-valence magnet YbPd. Phys. Rev. B 88, 054109 (2013).
Johannes, M. D. & Mazin, I. I. Fermi surface nesting and the origin of charge density waves in metals. Phys. Rev. B 77, 165135 (2008).
Zhu, X., Guo, J., Plummer, E. W. & Zhang, J. Classification of charge density waves based on their nature. Proc. Natl Acad. Sci. USA 112, 2367–2371 (2015).
Feng, Y. et al. Itinerant density wave instabilities at classical and quantum critical points. Nat. Phys. 11, 865–872 (2015).
Overhauser, A. W. Exchange and correlation instabilities of simple metals. Phys. Rev. 167, 691–698 (1968).
Jeong, T. & Kwon, Y. Electronic and magnetic properties of a heavy fermion compound YbPd. Phys. Lett. A 362, 500–504 (2007).
Yang, Y. F., Fisk, Z., Lee, H. O., Thompson, J. D. & Pines, D. Scaling the Kondo lattice. Nature 454, 611 (2008).
Woicik, J. C. Hard X-ray Photoelectron Spectroscopy (HAXPES), Springer Series in Surface Sciences. Vol. 59, (ed. Woicik, J. C.) (Springer International, 2016).
Okawa, M. et al. Strong valence fluctuation in the quantum critical heavy fermion superconductor β-YbAlB4: a hard X-ray photoemission study. Phys. Rev. Lett. 104, 247201 (2010).
Kuga, K. et al. Quantum valence criticality in a correlated metal. Sci. Adv. 4, eaao3547 (2018).
Gunnarsson, O. & Schönhammer, K. Electron spectroscopies for Ce compounds in the impurity model. Phys. Rev. B 28, 4315–4341 (1983).
Hughes, H. P. & Pollak, R. A. Charge density waves in layered materials observed by X-ray photoemission. Philos. Mag. 34, 1025–1046 (1076).
Kummer, K. et al. Similar temperature scale for valence changes in Kondo lattices with different Kondo temperatures. Nat. Commun. 9, 2011 (2018).
Imer, J. M. & Wuilloud, E. A simple model calculation for XPS, BIS and EELS 4f-excitations in Ce and La compounds. Zeit. Phys. B Condens. Matter 66, 153–160 (1987).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A32, 751–767 (1976).
Winkler, H. et al. Chemical pressure, dilution and disorder in the heavy fermion compounds Ce3−xLaxPd20Si6 (x = 1/3, 2/3). J. Phys.: Condens. Matter 23, 094208 (2011).
Custers, J. et al. Destruction of the Kondo effect in the cubic heavy-fermion compound Ce3Pd20Si6. Nat. Mat. 11, 189 (2012).
Benlagra, A., Fritz, L. & Vojta, M. Kondo lattices with inequivalent local moments: Competitive versus cooperative Kondo screening. Phys. Rev. B 84, 075126 (2011).
Camjayi, A., Haule, K., Dobrosavljevic, V. & Kotliar, G. Nature Phy. 4, 932–935 (2008).
Shiga, M. et al. Observation of Kondo resonance in valence-ordered YbPd. Phys. Rev. B 100, 245117 (2019).
Scalapino, D. J. in Superconductivity. Vol. I, Chap. 10 (ed. Parks, R. D.) (Dekker, 1969).
Grüner, G. Density Waves in Solids, p. 68 (Perseus Publishing, 1994).
Solyom, J. The Fermi gas model of one-dimensional conductors. Adv. Phys. 28, 201–303 (1979).
Forster, P. M. & Sleight, A. W. Negative thermal expansion in Y2W3O12. Int. J. Inorg. Mater. 1, 123–127 (1999).
Li, J., A. Yokochi, A., Amos, T. G. & Sleight, A. W. Strong Negative Thermal expansion along the O-Cu-O Linkage in CuScO2. Chem. Mater. 14, 2602–2606 (2002).
Mary, T. A., Evans, J. S. O., Vogt, T. & Sleight, A. W. Negative Thermal Expansion from 0.3 to 1050 Kelvin in ZrW2O8. Science 272, 90–92 (1996).
Tsutsui, S. et al. Lattice instability coupled with valence degrees of freedom in valence fluctuation compound YbPd. Phys. Rev. B 102, 245150 (2020).
Mazzone, D. G. et al. Kondo induced giant isotropic negative thermal expansion. Phys. Rev. Lett. 124, 125701 (2020).
van Schilfgaarde, M., Abrikosov, I. A. & Johansson, B. Origin of the Invar effect in iron nickel alloys. Nature 400, 46–49 (1999).
Acknowledgements
D.M. and A.C. thank the France-Taiwan (CNRS-MOST) bilateral project for financially supporting this research under Contract No. 109-2911-I-213-501. A.C. thanks the Ministry of Science and Technology (MOST) of the Republic of China, Taiwan, for financially supporting this research under Contract No. MOST 108-2112-M-213-001-MY3.
Author information
Authors and Affiliations
Contributions
The work was planned and supervised by D.M. and A.C. Samples were prepared and characterized by T.C. and T.M. Y.F.L carried out the HAXPES experiments. K.D.T. and A.C. contributed to HAXPES experiments. Experimental data analysis was carried out by Y.F.L. and A.C. The SIAM calculations were performed by B.T.C. and D.M. The manuscript was written by D.M. and A.C with inputs from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks George Martins, Jia-Xin Yin and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary handling editor: Aldo Isidori. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, YF., Tegomo Chiogo, B., Clausse, T. et al. Dual Kondo effect charge ordering and zero thermal expansion in a correlated intermetallic. Commun Mater 3, 23 (2022). https://doi.org/10.1038/s43246-022-00243-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-022-00243-5
